


Abstract
Many synthetic compounds to which we attribute specific activities are produced as racemic mixtures of stereoisomers, and it may be that all the desired activity comes from a single enantiomer. We have previously shown this to be the case with the α7 nicotinic acetylcholine receptor positive allosteric modulator (PAM) 3a,4,5,9b-Tetrahydro-4-(1-naphthalenyl)-3H-cyclopentan[c]quinoline-8-sulfonamide (TQS) and the α7 ago-PAM 4BP-TQS. Cis-trans-4-(2,3,5,6-tetramethylphenyl)-3a,4,5,9b-te-trahydro-3H-cyclopenta[c]quinoline-8-sulfonamide (2,3,5,6TMP-TQS), previously published as a “silent allosteric modulator” and an antagonist of α7 allosteric activation, shares the same scaffold with three chiral centers as the aforementioned compounds. We isolated the enantiomers of 2,3,5,6TMP-TQS and determined that the (−) isomer was a significantly better antagonist than the (+) isomer of the allosteric activation of both wild-type α7 and the nonorthosterically activatible C190A α7 mutant by the ago-PAM GAT107 (the active isomer of 4BP-TQS). In contrast, (+)2,3,5,6TMP-TQS proved to be an α7 PAM. (−)2,3,5,6TMP-TQS was shown to antagonize the allosteric activation of α7 by the structurally unrelated ago-PAM B-973B as well as the allosteric activation of the TQS-sensitive α4β2L15′M mutant. In silico docking of 2,3,5,6TMP-TQS in the putative allosteric activation binding site suggested a specific interaction of the (−) enantiomer with α7T106, and allosteric activation of α7T106 mutants was not inhibited by (−)2,3,5,6TMP-TQS, confirming the importance of this interaction and supporting the model of the allosteric binding site. Comparisons and contrasts between 2,3,5,6TMP-TQS isomers and active and inactive enantiomers of other TQS-related compounds identify the orientation of the cyclopentenyl ring to the plane of the core quinoline to be a crucial determinate of PAM activity.
SIGNIFICANCE STATEMENT Many synthetic ligands are in use as racemic preparations. We show that one enantiomer of the TQS analog Cis-trans-4-(2,3,5,6-tetramethylphenyl)-3a,4,5,9b-te-trahydro-3H-cyclopenta[c]quinoline-8-sulfonamide, originally reported to lack activity when used as a racemic preparation, is an α7 nicotinic acetylcholine receptor positive allosteric modulator (PAM). The other enantiomer is not a PAM, but it is an effective allosteric antagonist. In silico studies and structural comparisons identify essential elements of both the allosteric ligands and receptor binding sites important for these allosteric activities.
Introduction
Nicotinic acetylcholine receptors (nAChR) are pentameric assemblies of subunits that function as ligand-gated ion channels (Papke, 2014). In the peripheral nervous system, heteromeric nAChR mediate synaptic transmission at neuromuscular junctions and autonomic ganglia. In the brain, heteromeric nAChR mediate both cognitive effects of ACh and addictive effects of nicotine. A third class of nAChR are those that can form functional receptors as homopentamers, and the best studied of these are composed of α7 subunits. Several features distinguish homomeric α7 receptors from heteromeric nAChR. Although all nAChR subtypes desensitize in the continued presence of agonist, the desensitization of α7 nAChR to high concentrations of agonist is especially rapid but also readily reversible. As a consequence of α7’s unique mode of desensitization, α7 receptors have an intrinsically low probability of channel activation (Williams et al., 2012).
All nAChR are allosteric proteins, such that their functional properties are regulated by sites other than the orthosteric sites where typical agonists bind (Changeux, 1981; Bertrand and Gopalakrishnan, 2007; Williams et al., 2011). Some of the most profound effects of allosteric ligands have been described for α7 nAChR, and some positive allosteric modulators (PAMS) reverse intrinsic limitations on channel activation (Williams et al., 2012). The binding site for α7 PAMs is within the receptor’s transmembrane domains (Young et al., 2008) and requires the presence of a methionine residue that is unique to α7 in the pore-forming second transmembrane domain (Stokes et al., 2019).
One large class of PAMs is based on TQS, a sulfonamide-containing multiring molecule (Gronlien et al., 2007; Gill et al., 2012). In addition to acting as a PAM, one analog, 4BP-TQS, produces direct allosteric activation of α7 receptors as well as long-lived potentiation of orthosteric agonist responses, identifying the compound as an “ago-PAM.” GAT107 is the active isomer of 4BP-TQS (Papke et al., 2014), and both allosteric activation and positive modulation of α7 by GAT107 require the specific sequence in the α7 transmembrane domains. Although it was initially hypothesized that this sequence alone was sufficient for allosteric activation (Gill et al., 2011), we have provided evidence for a second allosteric activation binding site (AA site) in the receptor’s extracellular domain (Papke et al., 2014; Horenstein et al., 2016; Gulsevin et al., 2019).
Cis-trans-4-(2,3,5,6-tetramethylphenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide (2,3,5,6TMP-TQS) was previously identified as a “silent allosteric modulator” of α7 nAChR (Gill-Thind et al., 2015) based on its apparent lack of positive or negative effect on activation by 100 µM of ACh. Although apparently silent in regard to activation by 100 µM ACh, 2,3,5,6TMP-TQS blocked allosteric activation of α7 by the ago-PAM 2,4,MP-TQS. Subsequently, we used 2,3,5,6TMP-TQS [previously identified as 2,3,5,6MP-TQS (Gill-Thind et al., 2015)] as a tool to characterize the allosteric agonist activity of GAT107 (Thakur et al., 2013; Papke et al., 2014).
The allosteric activation of α7 produced by an application of GAT107 persists only as long as the drug is free in solution. Repeated applications of GAT107 produce additional episodes of allosteric activation, suggesting that there is rapid binding and dissociation of GAT107 at the AA site. In contrast to the rapid transient effects of GAT107 as an allosteric agonist, after a single application of GAT107, subsequent applications of ACh alone are increased in amplitude and duration. This “primed potentiation” can persist for at least an hour (Papke et al., 2018). We have observed that the coapplication of GAT107 with 2,3,5,6TMP-TQS could specifically reduce direct allosteric activation without apparent effects on primed potentiation, suggesting that 2,3,5,6TMP-TQS is a selective antagonist of the extracellular AA site with little or no effect at the transmembrane site required for allosteric potentiation.
Although allosteric activation by GAT107 does require positive allosteric modulation at the transmembrane PAM binding site, it does not require a functional binding site for orthosteric agonists (Horenstein et al., 2016). We recently identified ligands previously classified as silent agonists that produce PAM-dependent activation of α7 by binding to the same AA site as GAT107 (Gulsevin et al., 2019). Racemic 2,3,5,6TMP-TQS blocks this activity, but during our experiments, we also noted that 2,3,5,6TMP-TQS could additionally potentiate the responses to low concentrations of ACh, contrary to initial reports. Because we have previously documented large differences in the activity profiles of the stereoisomers of 4BP-TQS and TQS (Stokes et al., 2019), we hypothesized that the two stereoisomers of 2,3,5,6TMP-TQS might discriminate between the AA and PAM binding sites. Our results confirm that only (+)2,3,5,6TMP-TQS is a PAM and that although (−)2,3,5,6TMP-TQS is not a PAM, it is a more potent allosteric antagonist than (+)2,3,5,6TMP-TQS.
Materials and Methods
Chemicals and Reagents.
Acetylcholine chloride (ACh) and buffer chemicals were purchased from Sigma-Aldrich Chemical Company (St. Louis, MO). GAT107 and racemic 2,3,5,6TMP-TQS were synthesized in Thakur laboratory by Dr. Sumanta Garai (Northeastern University, Boston, MA), following the published procedures (Kulkarni and Thakur, 2013; Thakur et al., 2013; Gill-Thind et al., 2015). PNU-120596 was synthesized in the Horenstein laboratory by Dr. Kinga Chojnacka following the published procedure (Hurst et al., 2005).
The 2,3,5,6TMP-TQS isomers were synthesized and isolated as described in the Supplemental Data.
Expression in Xenopus Oocytes.
The human α7 nAChR clone was obtained from Dr. J. Lindstrom (University of Pennsylvania, Philadelphia, PA). The human resistance-to-cholinesterase 3 clone was obtained from Dr. M. Treinin (Hebrew University, Jerusalem, Israel) and coinjected with α7 to improve the level and speed of α7 receptor expression without affecting the pharmacological properties of the receptors (Halevi et al., 2003). After linearization and purification of the plasmid cDNAs, RNAs were prepared using the mMessage mMachine in vitro RNA transcription kit (Ambion, Austin, TX). The α7C190A mutant was made as previously described with a C116S double mutation to prevent spurious disulfide bond formation with the free cysteine (Papke et al., 2011). The β2L15′M mutant was prepared as previously described (Stokes et al., 2019) and coexpressed with a β2-α4 concatamer (Zhou et al., 2003) to obtain receptors with a single mutant β subunit in the accessory subunit position outside the ACh binding sites.
Oocytes were surgically removed from mature female Xenopus laevis frogs (Nasco, Ft. Atkinson, WI). Frogs were maintained in the Animal Care Service facility of the University of Florida, and all procedures were approved by the University of Florida Institutional Animal Care and Use Committee. In brief, the frog was first anesthetized for 15–20 minutes in 1.5 l frog tank water containing 1 g of Ethyl 3-aminobenzoate methanesulfonate buffered with sodium bicarbonate. The harvested oocytes were treated with 1.4 mg/ml Type 1 collagenase (Worthington Biochemicals, Freehold, NJ) for 2–4 hours at room temperature in calcium-free Barth’s solution (88 mM NaCl, 1 mM KCl, 2.38 mM NaHCO3, 0.82 mM MgSO4, 15 mM HEPES, and 12 mg/l tetracycline, pH 7.6) to remove the ovarian tissue and the follicular layers. Stage V oocytes were subsequently isolated and injected with 50 nl of 5–20 ng nAChR subunit RNA. Oocytes were maintained in Barth’s solution with calcium [additional 0.32 mM Ca(NO3)2 and 0.41 mM CaCl2], and recordings were carried out 1–14 days after injection.
Two-Electrode Voltage Clamp Electrophysiology.
Experiments were conducted using OpusXpress 6000A (Molecular Devices, Union City, CA) (Papke and Stokes, 2010). Both the voltage and current electrodes were filled with 3 M KCl. Oocytes were voltage-clamped at −60 mV at room temperature (24°C). The oocytes were bath-perfused with Ringer’s solution (115 mM NaCl, 2.5 mM KCl, 1.8 mM CaCl2, 10 mM HEPES, and 1 μM atropine, pH 7.2) at 2 ml/min for α7 and at 4 ml/min for α4β2. For wild-type α7 and α4β2 mutant receptors, to evaluate the effects of experimental compounds compared with ACh-evoked responses of various nAChR subtypes expressed in oocytes, control responses were defined as the average of two initial applications of ACh made before test applications. The control ACh concentrations were 60 μM for the wild-type α7 receptor experiments and 10 µM ACh for the α4β2 mutants. The average of independent 1 μM GAT107 responses were used as the control for the α7C190A experiments.
Solutions were applied from 96-well plates via disposable tips. Drug applications were 12 seconds in duration followed by 181-second washout periods for α7 and 6 seconds in duration followed by 241-second washout periods for α4β2. The responses were calculated as both peak current amplitudes and net charge, as previously described (Papke and Porter Papke, 2002). Data were collected at 50 Hz, filtered at 20 Hz, and analyzed by Clampfit 9.2 or 10.0 (Molecular Devices) and Excel (Microsoft, Redmond, WA). Data were expressed as means ± S.E.M. from at least four oocytes for each experiment and plotted with Kaleidagraph 4.5.2 (Abelbeck Software, Reading, PA). Multicell averages were calculated for comparisons of complex responses. Averages of the normalized data were calculated for each of the 10,322 points in each of the 206.44-second traces (acquired at 50 Hz) as well as the S.E.s for those averages.
Data and Statistical Analysis.
The curve fits to concentration-response studies were generated using the Levenberg-Marquardt algorithm to obtain the best Chi-Square fit to the Hill equation using the Kalidagraph plotting program (V4.5.2). The errors reported in the Supplemental Data are the calculated S.E.s of the fit parameters based on the goodness of fit (chi square and r values provided).
For comparisons of results containing three or more groups, one-way and two-way ANOVAs were used. For data for which variances were different, as measured using the Brown-Forsythe test, Brown-Forsythe ANOVAs were performed with Dunnett’s T3 multiple comparisons test. For all other data requiring an ANOVA, ordinary one-way and two-way ANOVAs were performed with Tukey’s post hoc multiple comparisons test. GraphPad Prism 8 software (San Diego, CA) was used to perform these statistical measures. GraphPad Prism 8 software was also used to generate the difference between group means or “Gardner-Altman plot” (Fig. 7B) (Ho et al., 2019). Differences of mean and confidence interval (CI) values were generated from the Tukey’s post hoc multiple comparisons test.
Comparisons of all other results were made using two-tailed t tests between the pairs of experimental measurements. A value of P < 0.05 was used to constitute a minimum level of significance. These statistics were calculated using an Excel template provided in Microsoft Office.
Results
Properties of Racemic 2,3,5,6TMP-TQS.
Shown in Fig. 1 are the selective effects of racemic 2,3,5,6TMP-TQS on the direct allosteric activation of α7 receptors by 10 µM GAT107 (P < 0.001). With this coapplication protocol, there was no effect on the long-lived potentiation of subsequent ACh responses.

Coapplication of 100 µM racemic 2,3,5,6TMP-TQS selectively inhibits the direct allosteric activation of α7 nAChR by 10 µM of the ago-PAM GAT107 (P < 0.00001) with no effect on subsequent potentiation of responses to ACh. Averaged raw data traces for cells (see Materials and Methods) were normalized to the control responses to 60 µM ACh shown. The S.E.M. of the averaged normalized responses are represented by the tan colored areas.
Note that the previous work (Gill-Thind et al., 2015) that reported that 2,3,5,6TMP-TQS had no effect on responses to “a submaximal concentration of ACh” relied on peak currents as measurements of α7 responses to ACh applications. However, the α7 peak currents responses to applications of ACh at concentrations ≥60 µM occur in advance of full solution delivery (Papke and Thinschmidt, 1998), making them poor reporters of the concentration dependence of receptor activation compared with measurements of the integrated net charge of agonist-evoked responses (Papke and Porter Papke, 2002). The concentration of 100 µM ACh used by Gill and coworkers is, in fact, sufficient to produce maximal net charge responses. Although 2,3,5,6TMP-TQS had no effect when applied alone to cells expressing α7 nAChR (Gill et al., 2012), in the course of our work with racemic 2,3,5,6TMP-TQS, we saw clear indications of PAM activity when the ACh concentrations were lower (Fig. 2).

Coapplication of racemic 100 µM 2,3,5,6TMP-TQS potentiates α7 nAChR responses to a low (10 µM) concentration of ACh. (A) Averaged raw data traces for cells responding to 10 µM ACh alone or coapplied with 100 µM racemic 2,3,5,6TMP-TQS (n = 8 cells for each condition). Data were normalized to the control responses to 60 µM ACh shown, and the normalized peak current amplitudes were compared with a t test (P < 0.001) (see Materials and Methods). The S.E.M. of the averaged normalized responses are represented by the tan colored areas. (B) Superimposed responses to 10 µM ACh in (A), normalized to 60 µM ACh controls. The data in blue are those obtained with coapplication of 100 µM 2,3,5,6TMP-TQS.
Properties of 2,3,5,6TMP-TQS Isomers on GAT107 Activity with Wild-Type and Mutant α7.
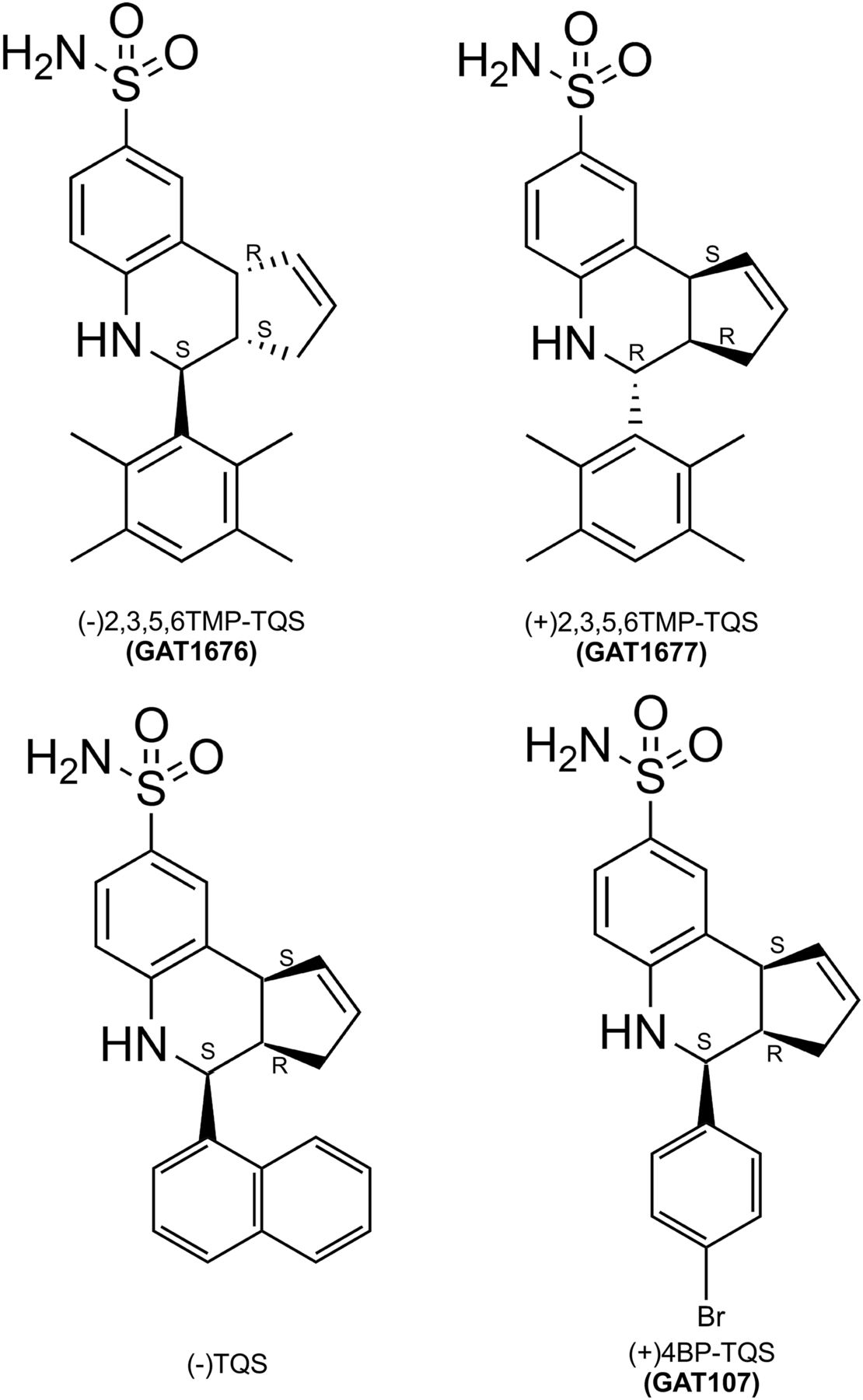
When synthesized, 2,3,5,6TMP-TQS is a racemic mixture of isomers. To test the hypothesis that PAM and allosteric antagonist activities of 2,3,5,6TMP-TQS might be associated with specific enantiomers, we separated the (+) and (−) enantiomers and resolved their crystal structures (Fig. 3). Also shown in Fig. 3 are the structures of the enantiomers of TQS and 4BP-TQS that we have previously shown to be the active α7 PAMs (Thakur et al., 2013; Stokes et al., 2019). All of these compounds have three chiral centers: two that join the cyclopentenyl ring to the quinoline and a third that connects to the aromatic rings at the base. Whereas the two that are part of the cyclopentenyl ring are constrained to orient in concert, the lower group may be either cis or trans relative to the cyclopentenyl ring of the molecule. As previously hypothesized (Gill et al., 2012), because of the steric hindrances of two ortho methyl groups, through the Povarov reaction, we only obtained trans isomers of 2,3,5,6TMP-TQS and not cis diastereomers. It was also originally hypothesized (Gill et al., 2012) that the likely transconfiguration accounted for the apparent lack of PAM activity for 2,3,5,6TMP-TQS.
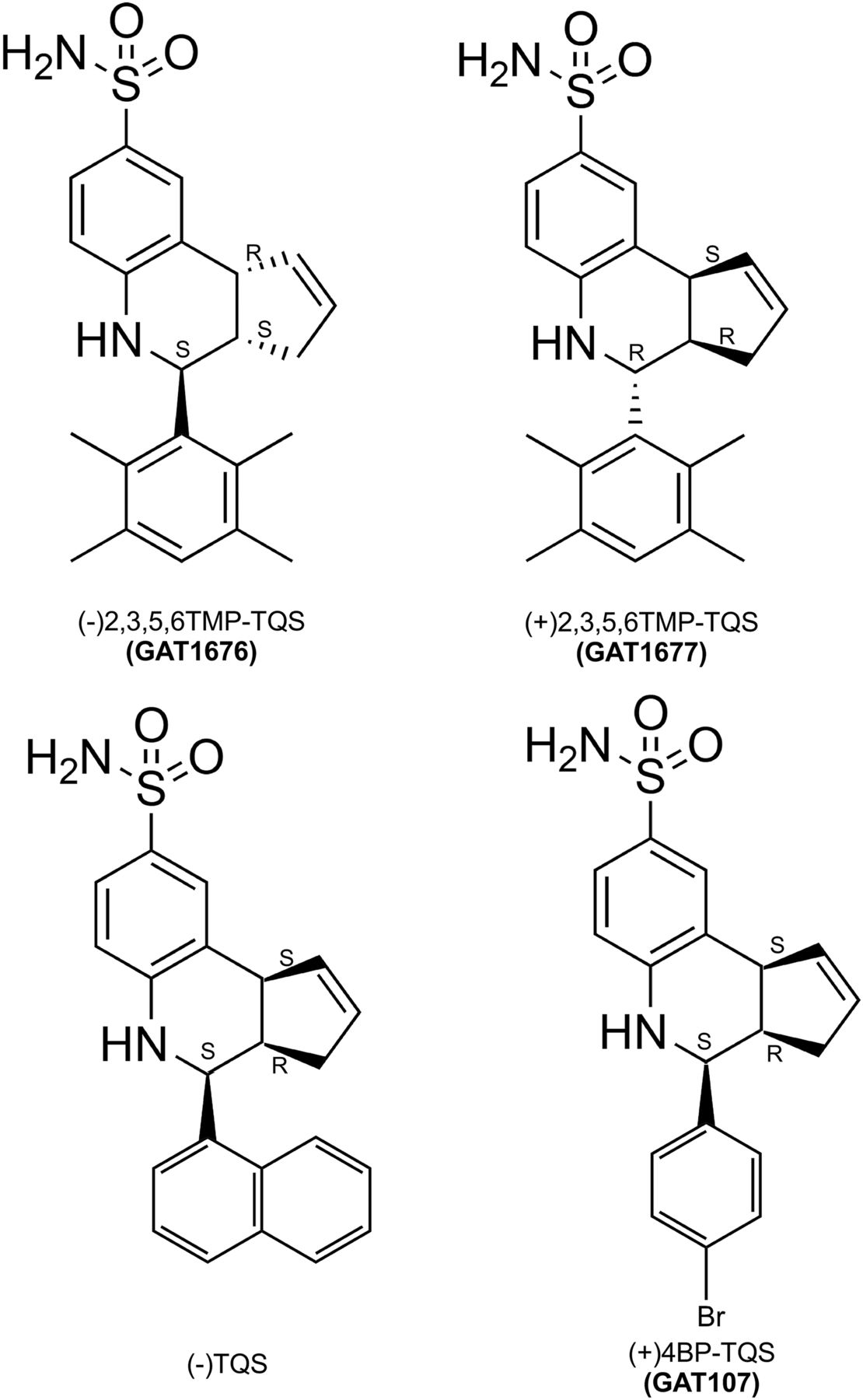
Structures of the 2,3,5,6TMP-TQS isomers. (−)2,3,5,6TMP-TQS has 3aS, 4S, 9bR absolute stereochemistry. (+)2,3,5,6TMP-TQS has 3aR, 4R, 9bS absolute stereochemistry. Also shown are the resolved structures of the active enantiomers of TQS (Stokes et al., 2019) and 4BP-TQS (Thakur et al., 2013).
We compared the effects of racemic 2,3,5,6TMP-TQS and the two isolated isomers on the two phases of GAT107 activity using a protocol with a 30-second preapplication of 100 µM 2,3,5,6TMP-TQS prior to the coapplication of 2,3,5,6TMP-TQS with 10 µM GAT107. The racemic 2,3,5,6TMP-TQS and each isomer inhibited the direct activation by GAT107 as determined by a Brown-Forsythe ANOVA [F(3, 27) = 15.17, P < 0.0001]. The racemic 2,3,5,6TMP-TQS inhibited activation by GAT107 by 98%, whereas (+)2,3,5,6TMP-TQS and (−)2,3,5,6TMP-TQS are inhibited GAT107 by 93% and 99.8%, respectively. Therefore, the inhibition by (+)2,3,5,6TMP-TQS was less than by the racemic (95% CI, −7.33 to −0.38, P = 0.01; Fig. 4A).
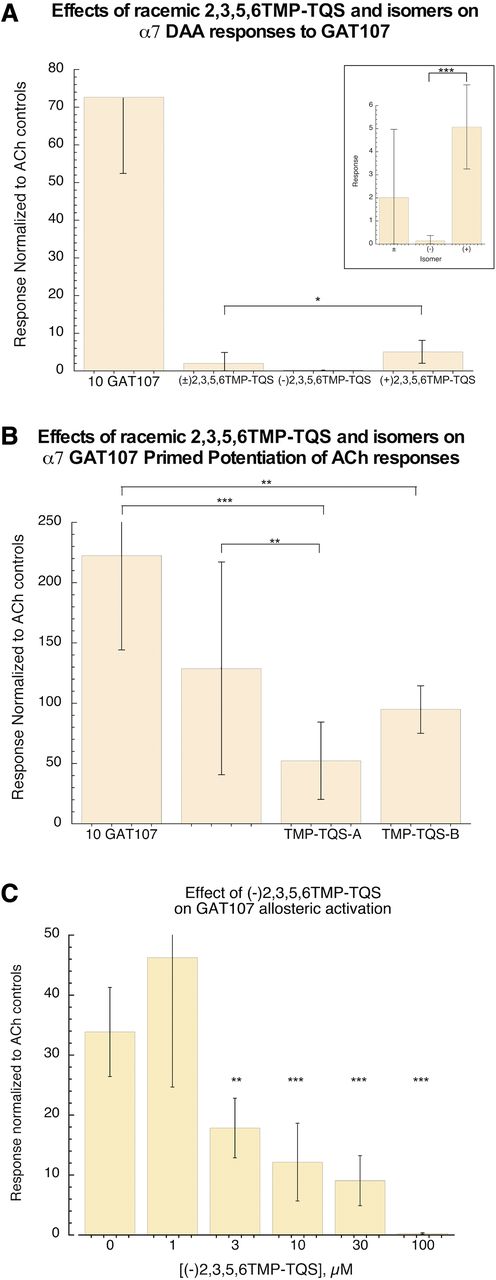
2,3,5,6TMP-TQS racemic and isomer effects on GAT107 with a preapplication/coapplication protocol. Cells were treated with Ringer’s solution or 100 µM 2,3,5,6TMP-TQS for 30 seconds prior to application of 10 µM GAT107 alone or coapplied with 100 µM 2,3,5,6TMP-TQS. Data presented are averages (±S.D.) of the responses normalized to the initial ACh controls. (A) The inhibition by (+)2,3,5,6TMP-TQS was less than by the racemic (P = 0.01). The n values were 8, 7, 8, and 8. (B) Inhibition of primed potentiation of subsequent ACh-evoked response with preapplication/coapplications. (C) Effect of (−)2,3,5,6TMP-TQS concentration of inhibition of the net charge of 10 µM GAT107-evoked α7 allosteric activation with a simple coapplication protocol. The n values were 7 for GAT107 alone and 8 for the coapplications. See Supplemental Information for detailed results of the statistical analysis. *P < 0.05, **P < 0.01, ***P < 0.001.
Note that with this preincubation protocol, there was also an inhibition of the potentiated ACh response after the GAT107 [F(3, 27) = 9.11, P = 0.0002]. Both (+)2,3,5,6TMP-TQS (95% CI, 34.65–220.2; P = 0.004; Fig. 4B) and (−)2,3,5,6TMP-TQS (95% CI, 77.40–262.9; P = 0.0002; Fig. 4B) produced an inhibition of the potentiated ACh response after the GAT107. The racemic 2,3,5,6TMP-TQS also produced a small inhibition of the potentiated ACh response after the GAT107 (95% CI, −3.13 to 188.9; P = 0.06; Fig. 4B), and the inhibition with (−)2,3,5,6TMP-TQS or (+)2,3,5,6TMP-TQS was equivalent to that produced by the racemic.
We tested the potency of (−)2,3,5,6TMP-TQS for inhibiting the direct activation of wild-type α7 produced by 10 µM GAT107 with a coapplication protocol (Fig. 4C). There was inhibition at all concentrations tested ≥3 µM using a Brown-Forsythe ANOVA [F(5, 11.55) = 22.79, P < 0.0001] with Dunnett’s T3 post hoc multiple comparisons test (3 µM: 95% CI, 2.33–29.70; P = 0.009; 10 µM: 95% CI, 7.41–36.00; P = 0.0009; 30 µM: 95% CI, 11.27–38.31; P = 0.0003; 100 µM: 95% CI, 19.63–47.58; P = 0.0002).
Key elements for the function of any nAChR are the disulfide-coupled vicinal cysteines (C190 and C191 in the α7 sequence) at the tip of the alpha subunit C-loops. Disruption of this link causes loss of function (Papke, 2014). We have previously shown that although α7C190A mutants are insensitive to ACh, even when coapplied with PNU-120596, they are effectively activated by GAT107 and that racemic 2,3,5,6TMP-TQS blocked that activation (Gulsevin et al., 2019). In basic coapplication experiments, we observed using a Brown-Forsythe ANOVA that both isomers reduced GAT107-evoked currents of α7C190A [F(2, 9.09) = 11.25, P = 0.0035; Fig. 5], although the inhibition by (−)2,3,5,6TMP-TQS was greater than by (+)2,3,5,6TMP-TQS (95% CI, −0.58 to 11.64; P = 0.04).

2,3,5,6TMP-TQS isomer effects on allosteric activation of the nonorthosterically activatible C190A α7 mutant by 10 µM GAT107 (GAT107 alone, n = 7). Coapplications of 10 µM GAT107 with (−)2,3,5,6TMP-TQS isomer (n = 7) decreased peak current responses by 93% (P = 0.015), and coapplications with (+)2,3,5,6TMP-TQS (n = 8) decreased peak current responses by 54% (P = 0.14). In addition, the inhibition by (−)2,3,5,6TMP-TQS was greater than that by (+)2,3,5,6TMP-TQS (P = 0.04), and (+)2,3,5,6TMP-TQS did not reduce the net charge of the 10-µM GAT107 responses. The data plotted are the individual values and the average normalized (see Materials and Methods) net charge responses ± S.D. See Supplemental Information for detailed results of the statistical analysis. * P < 0.05.
We also tested the two 2,3,5,6TMP-TQS enantiomers for their ability to inhibit α7 allosteric activation by the structurally unrelated ago-PAM B-973B (Quadri et al., 2019). Like GAT107, it is able to produce allosteric activation of α7 receptors and potentiate the responses to ACh. We determined that (−)2,3,5,6TMP-TQS (95% CI, −0.45 to 58.67; P = 0.05), but not (+)2,3,5,6TMP-TQS (95% CI, −24.38 to 36.68; P = 0.87), was also an effective antagonist of allosteric activation by B-973B [F(2, 19) = 3.55, P = 0.049; Fig. 6]. Neither enantiomer had any effect on the residual potentiation produced by this agent [F(2, 9) = 0.42, P = 0.67].

Effects of 2,3,5,6TMP-TQS enantiomers on the activation and potentiation of α7 nAChR by B-973B. Subsequent to the acquisition of two control responses to 60 µM ACh, cells expressing human α7 were treated with 30 µM B-973B alone or coapplied with 100 µM of the 2,3,5,6TMP-TQS enantiomers. Direct allosteric activation was measured as the response to B-973B ± 2,3,5,6TMP-TQS. Primed potentiation was measured by comparing the response to 60 µM ACh after the B-973B applications. The data shown are individual values with the average net charge responses ± S.D. (n = 4–8). (−)2,3,5,6TMP-TQS reduced (P = 0.05) the B-973B allosteric activation. See Supplemental Information for n values and detailed results of the statistical analysis.
PAM Activity of 2,3,5,6TMP-TQS Isomers.
We conducted ACh concentration-response studies with or without coapplication of the 2,3,5,6TMP-TQS isomers at 30 µM (Fig. 7). Visual inspection of the curves suggests that (+)2,3,5,6TMP-TQS functions as a PAM and (−)2,3,5,6TMP-TQS does not. We use several statistical approaches to confirm this using both the averaged data (Fig. 7) and an analysis of the concentration-response data of individual cells (n = 5 under each condition, see Supplemental Data). Specifically, using two-way ANOVA, we found an interaction effect between ACh concentration and coapplication [F(12, 91) = 10.21, P < 0.0001; Fig. 7A]. Effects of (+)2,3,5,6TMP-TQS were seen across almost the entire concentration range (10–1000 µM; Fig. 7B), including maximally effective concentrations ≥100 µM. When analyzing curve fit parameters by Brown-Forsythe ANOVA [EC50: F(2, 12) = 6.97, P = 0.05); Imax: F(2, 12) = 11.69, P = 0.01], the concentration-response function for net charge responses (Papke and Porter Papke, 2002) was relatively unaffected by addition of (−)2,3,5,6TMP-TQS (EC50: P = 0.17; Imax: P = 0.53); however, (+)2,3,5,6TMP-TQS increased ACh potency by 65% (P = 0.0006) and efficacy by 91% (P < 0.0001) (Table 1). We then ran an ANOVA for the logEC50 values with responses normalized as a percentage of the average responses across five replicates [F(2, 12) = 26.10, P < 0.0001]. We saw that addition of (−)2,3,5,6TMP-TQS caused a similar decrease in potency (95% CI, −0.67 to −0.08; P = 0.015) and an increase in potency with addition of (+)2,3,5,6TMP-TQS (95% CI, 0.18–0.77; P = 0.003). When calculating the inverse log from logEC50, values for EC50 using this method were consistent, as ACh alone was 20.61 µM with the addition of (−)2,3,5,6TMP-TQS at 48.52 µM and (+)2,3,5,6TMP-TQS at 6.87 µM.
Curve fit values of the data presented in Fig. 7
These are the averages of the fits of five cells under each condition ± S.D. See Supplemental Fig. 1 for the multiple plots and Supplemental Table 1 for individual curve fits.
Parameters of the fit shown in Fig. 7 to the averaged data at each concentration compared with average values (±S.D.) of the fits of the replicates shown in Table 1 (see Supplemental Data)
The errors reported for the fit parameters to the averaged data are the calculated S.E.s of the fit parameters based on the goodness of fit (chi square and r values, see Materials and Methods).

Effect of 2,3,5,6TMP-TQS isomers on ACh responses of wild-type α7. (A) Plotted are α7 net charge responses across a range of ACh concentrations, with or without the coapplication of 30 µM of the 2,3,5,6TMP-TQS isomers, normalized to the maximum response obtained with the application of ACh alone. Data are the averages ± S.D. of five cells under each condition at each concentration. The curves displayed are fits to the averaged data. See Table 1 for average curve-fit values (±S.D.) from the data from the individual cells and Supplemental Fig. 1 for the fit data from the single cells. ANOVA of curve fit parameters from the individual replicates are discussed in the text. Two-way ANOVAs indicate that responses to ACh plus (+)2,3,5,6TMP-TQS were greater than to ACh alone across most of the concentration range (P > 0.99, = 0.91, < 0.0001, < 0.0001, < 0.0001, < 0.0001, and < 0.0001 for 1, 3, 10, 30, 100, 300, and 1000 µM ACh, respectively). Note that the parameters of the curve fits to the averaged data shown in the figure compared with average fits of the replicates (±S.D.) are given in Table 2. (B) Differences between group means are expressed as the differences between means of ACh alone and ACh plus (−)2,3,5,6TMP-TQS or ACh plus (+)2,3,5,6TMP-TQS with 95% confidence intervals at each concentration.
Although all of these analyses confirm the PAM activity of (+)2,3,5,6TMP-TQS, it should be noted that the magnitude of the (+)2,3,5,6TMP-TQS potentiation was at least an order of magnitude less than previously reported for (−)TQS (Stokes et al., 2019) or GAT107 [(+)4BP-TQS] (Papke et al., 2014).
2,3,5,6TMP-TQS Isomer Effects on Mutant α4β2 Receptors Sensitive to α7 PAMs.
We have recently described a point mutation (L15′M) in the second transmembrane domain of neuronal β subunits that results in heteromeric neuronal nAChR that are sensitive to select α7 PAMs in the TQS family (Stokes et al., 2019). In α7, this residue is a methionine, and the opposite mutation results in a loss of PAM sensitivity.
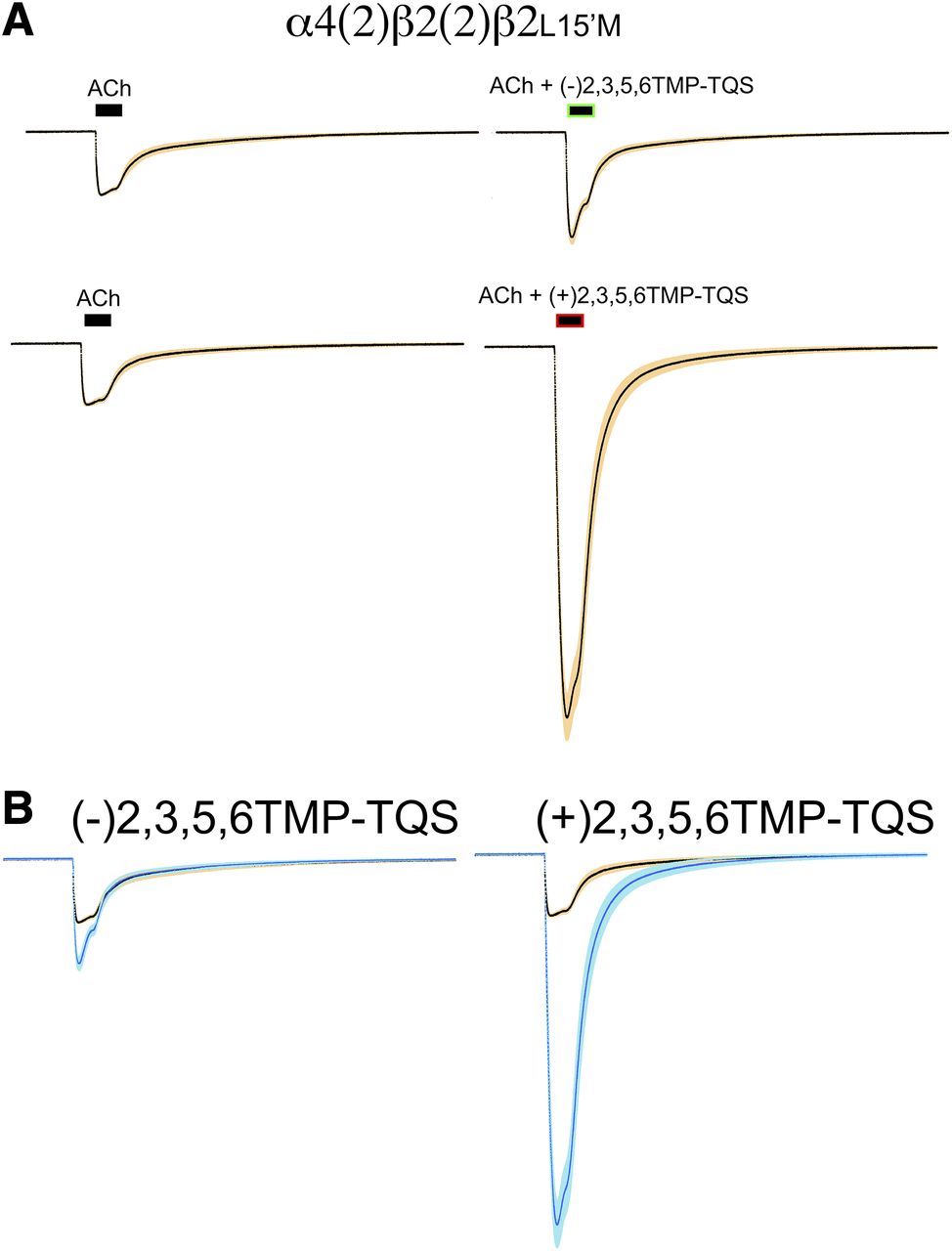
We have previously shown that the inclusion of a single mutant subunit is sufficient to produce sensitivity to α7 PAMs and that the presence of additional mutant subunits reduces receptor function (Stokes et al., 2019). To obtain α4β2 receptors with a single β2L15′M mutant subunit, we coexpressed a β2-α4 concatamer (Zhou et al., 2003) with monomeric β2L15′M. The coapplication of (−)2,3,5,6TMP-TQS with ACh gave a small increase in peak current (P < 0.01) but had no effect on net charge (Fig. 8). In contrast, coapplication of (+)2,3,5,6TMP-TQS with ACh produced large increases in both peak current and net charge responses (P < 0.001), as would be expected for a Type II PAM (Gronlien et al., 2007). The coapplication of (+)2,3,5,6TMP-TQS increased peak current and net charge by an average 5.7 ± 0.3- and 4.6 ± 0.6-fold, respectively (n = 8, P < 0.001).

The sensitivity of α4β2 receptors containing a single β2L15′M subunit to coapplications of 2,3,5,6TMP-TQS isomers. (A) Averaged raw data traces normalized to 10 µM control ACh responses from the same cells (n = 8) to ACh alone or ACh coapplied with 30 µM of the 2,3,5,6TMP-TQS isomers. (B) Superimposed traces from (A). The data in blue are those obtained with coapplication of 30 µM 2,3,5,6TMP-TQS isomers.
2,3,5,6TMP-TQS Isomer Effects on the Allosteric Activation of Mutant α4β2 Receptors.
In our initial characterization of heteromeric neuronal nAChR sensitive to α7 PAMs (Stokes et al., 2019), we identified GAT927 as an extremely potent and efficacious PAM for these mutant heteromeric receptors. In addition to strongly potentiating ACh-evoked responses, we noted that, when applied alone, high concentrations of GAT927 produced direct allosteric activation, analogous to GAT107’s effects on α7 (Fig. 9A). We tested the hypothesis that (−)2,3,5,6TMP-TQS, the isomer better for antagonizing α7 allosteric activation, would also more effectively antagonize the allosteric activation of the α4β2 mutant receptor by GAT927. Whereas (−)2,3,5,6TMP-TQS did decrease GAT927 allosteric activation (P < 0.05, Fig. 9B), activation by GAT927 was totally eliminated by (+)2,3,5,6TMP-TQS (Fig. 9C). Interestingly, the effects of the 2,3,5,6TMP-TQS isomers were restricted to the period of direct allosteric activation by GAT927, and the primed potentiation for a subsequent ACh-evoked response was unaffected (Fig. 9D). As noted above, neither of the isomers had an effect on the net charge or peak current amplitude of these responses. However, although the responses shown in Fig. 9, A and B had not decayed fully to baseline after the GAT927 applications (baselines were 82% ± 27% and 107% ± 22% the amplitude of the ACh controls, respectively), the trace in Fig. 9C (magenta in Fig. 9D) actually began with a lower baseline current than the initial control (P < 0.001).

The allosteric activation of α4(2)β2(2)β2L15′M receptors by GAT927 and sensitivity of GAT927 responses to coapplications of 2,3,5,6TMP-TQS isomers. (A) Application of the TQS analog GAT927 at 30 µM to cells expressing α4β2 receptors with a mutant β2 subunit production of allosteric activation and primed potentiation of subsequent ACh responses (n = 6). (B) The coapplication of 100 µM (−)2,3,5,6TMP-TQS with 30 µM GAT927 (n = 7) reduced the allosteric activation net charge (P < 0.005), with no effect on the primed potentiation of subsequent ACh responses. (C) The coapplication of 100 µM (+)2,3,5,6TMP-TQS with 30 µM GAT927 (n = 7) eliminated the allosteric activation net charge (P < 0.0001), also with no effect on the primed potentiation of subsequent ACh responses. (D) An overlay of the three post-GAT927 application ACh responses.
In Silico Model of (−)2,3,5,6TMP-TQS in the Putative Allosteric Activation Site of α7.
We (Horenstein et al., 2016; Gulsevin et al., 2019) and others (Spurny et al., 2015) have proposed models for an allosteric ligand binding site in the extracellular vestibule of α7 receptors. We compared the docking of GAT107 and the most active antagonist (−)2,3,5,6TMP-TQS into that site (Fig. 10) and found that (−)2,3,5,6TMP-TQS was notably different from GAT107 in that, despite an overall similar binding mode, it presented its fused cyclopentenyl ring to T106, whereas the other compound did not.
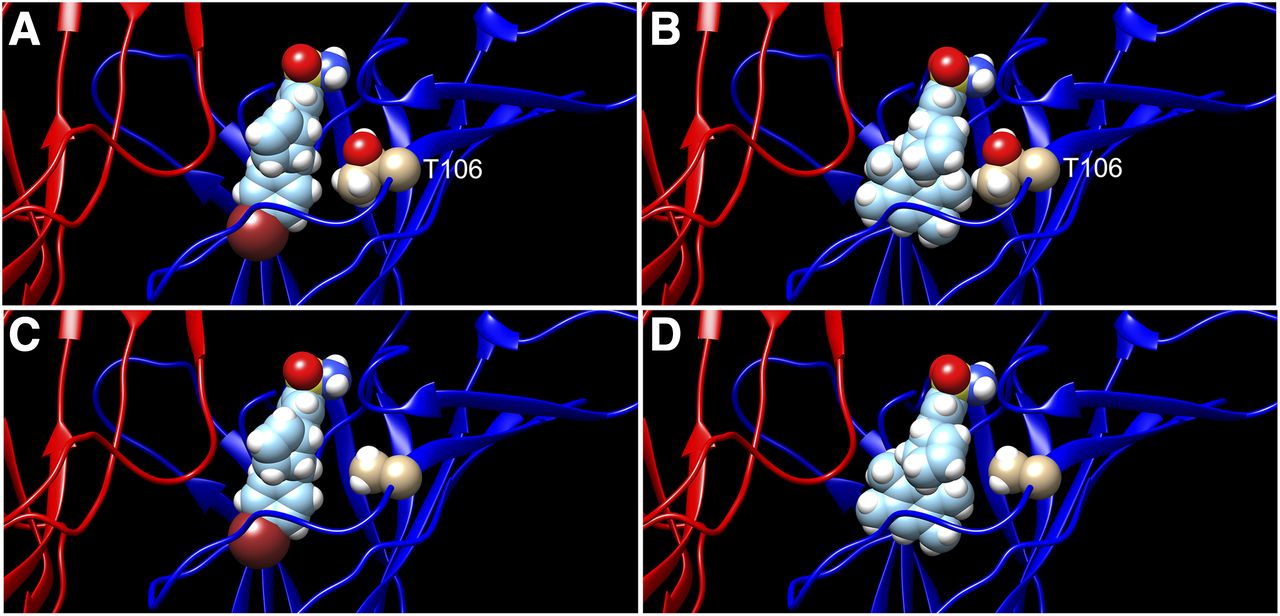
Using methods previously described (Gulsevin et al., 2019), GAT107 (A) and (−)2,3,5,6TMP-TQS (B) were docked into a homology model of the α7 extracellular domain. The orientation of (−)2,3,5,6TMP-TQS in the putative allosteric activation site suggested a close association with the T106 residue (shown) that was not present with GAT107. (C and D) The same models with the threonine residue converted to alanine.
We hypothesized that this interaction would be lost with a T106A mutation (Fig. 9D). Using a coapplication protocol, we confirmed that 30 µM (−)2,3,5,6TMP-TQS produced good inhibition of GAT107 allosteric activation of wild-type α7 (P < 0.001) with no effect on the potentiation of subsequent ACh responses (Fig. 11A). Using the same protocol with α7T106A, we saw no changes with 30 µM (−)2,3,5,6TMP-TQS on either GAT107 allosteric activation or subsequent potentiation (Fig. 11B).
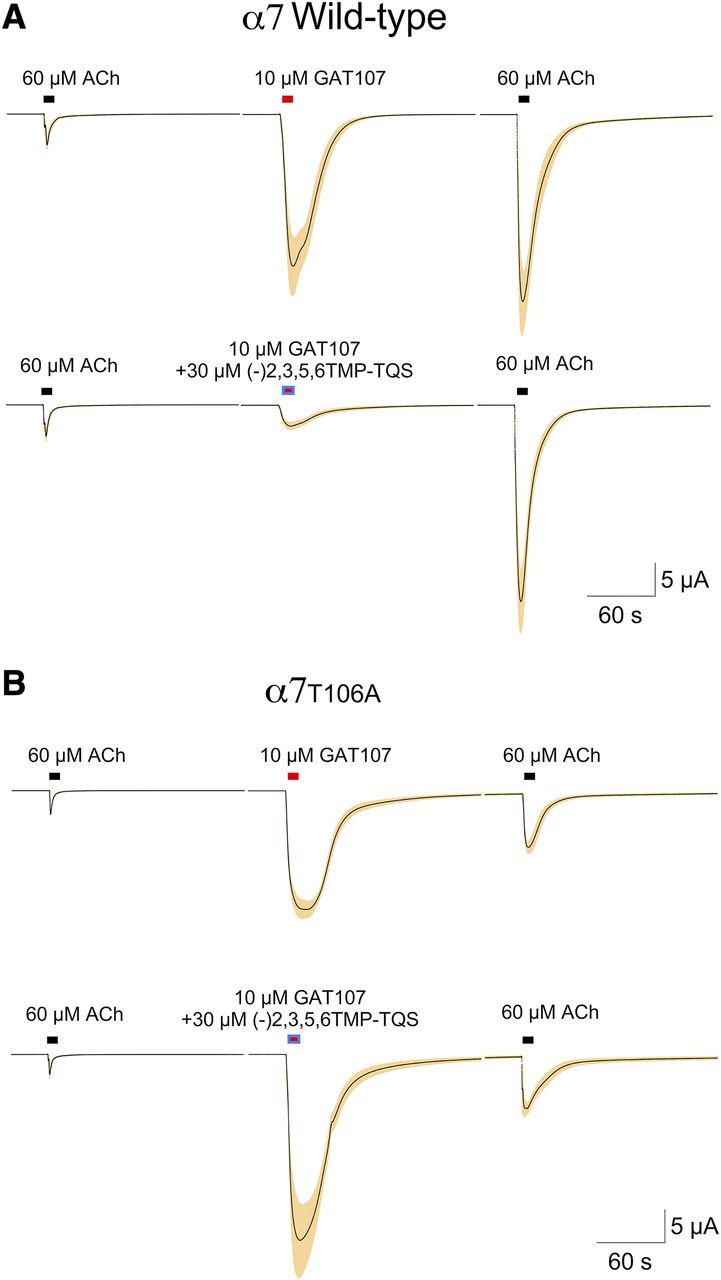
The effects of (−)2,3,5,6TMP-TQS on GAT107 responses of wild-type and T106A mutant α7 receptors. (A) Wild-type receptors (n = 6) show a selective inhibition (P ≤ 0.001) of allosteric activation by 30 µM (−)2,3,5,6TMP-TQS. (B) GAT107 responses of cells expressing α7T106A were unaffected by coapplication of 30 µM (−)2,3,5,6TMP-TQS. Averaged raw data traces for cells (n = 4, see Materials and Methods) were normalized to the control responses to 60 µM ACh shown. The S.E.M. of the averaged normalized responses are represented by the tan colored areas.
Discussion
Stereoisomerism is a critical concern when designing drugs for enzymes and receptors because stereoisomers can and often do display different pharmacology. Whereas molecules with a single chiral center will be capable of existing as a pair of enantiomers, molecules with multiple chiral centers N will have 2N maximum possible stereoisomers, as exemplified by the ring system of 2,3,5,6TMP-TQS, which has three chiral centers and eight theoretically possible stereoisomers, including enantiomers and diastereomers. However, as noted previously, constraints on the cyclopentenyl ring and steric hindrances associated with two ortho methyl groups resulted in exclusive formation and isolation of the trans diastereomer. Although most biologic processes that form organic molecules with chiral centers tend to produce single stereoisomers, such as D-sugars and L-amino acids, test tube reactions commonly produce racemic mixtures of isomers. When these reaction products are then used experimentally, the biologic response may or may not be deferentially sensitive to the component isomers.
In biologic systems, the requirement for isomer specificity is a function of the receptor and the flexibility of the ligand. Our right foot will readily accept either of a pair of socks but shows a strong preference for only one of a pair of shoes. In the case of nAChR, although the ACh binding site of muscle-type receptors shows a preference for the naturally occurring (−) isomer of nicotine, both isomers are equally effective as low potency blockers of the ion channel (Rozental et al., 1989). Likewise, both isomers of the neuronal nAChR noncompetitive antagonist mecamylamine have similar efficacy for blocking channel activity of a wide variety of heteromeric neuronal nAChR (Papke et al., 2001, 2013).
The amino acid residues in neuronal β subunits that determine sensitivity to mecamylamine face into the ion permeation pathway and are present in at least two subunits per heteromeric pentamer (Webster et al., 1999). The dimensions of the open pore and the presence of multiple binding sites may both be permissive factors for the lack of mecamylamine’s stereoselectivity. In contrast, although the α7 PAM binding site is likely present in all subunits and requires a specific residue in the pore-forming second transmembrane domain, the actual binding orientation of the modulator is likely to be within a hydrophobic cavity within the transmembrane helices (Young et al., 2008; Collins and Millar, 2010; Newcombe et al., 2018). In this tight pocket, stereochemical constraints appear to be very important. This was shown to be the case for three functionally diverse analogs in the TQS family, TQS itself (Stokes et al., 2019), the ago-PAM 4BP-TQS (Thakur et al., 2013; Papke et al., 2014), and the allosteric antagonist presented in this work, 2,3,5,6TMP-TQS, as well as for the structurally unrelated ago-PAM B-973B (Garai et al., 2018; Quadri et al., 2019). Additionally, a recently published study of a family of sulfonamide-containing PAMs showed that the stereochemical orientation of side groups around a critical central cyclopropyl ring determine whether enantiomers function as Type I or Type II α7 PAMs (Harvey et al., 2019; Wang et al., 2020).
To better understand the important structural elements of TQS-related PAMs, we made a structural comparison of (−)TQS and (+)2,3,5,6TMP-TQS (Fig. 12). The comparison reveals considerable similarity in the overall three-dimensional shape of the fused ring system, consisting of the arylsulfonamide, piperidine, and cyclopentenyl rings. It is notable that, although TQS is cis with regard to the cyclopentenyl and napthyl rings, (+)2,3,5,6TMP-TQS has a trans relationship between the cyclopentenyl and tetramethylbenzene rings. Despite this, the superposition revealed that the bulky napthyl and tetramethylphenyl rings occupied the region in space in the overlay. This comparative analysis suggests that the basis for the PAM activity of (+)2,3,5,6TMP-TQS arises from its overall similarity to the shape of TQS despite the difference in stereochemistry of the ring fusions. Further comparison with (+)4BP-TQS (GAT107) (Fig. 3) suggests that possibly the most crucial feature for PAM activity is the orientation of cyclopentenyl ring away from the plane of napthyl ring.

Ball and stick figures and molecular superposition of the PAMs (−)TQS and (+)2,3,5,6TMP-TQS. (A) (−)TQS, (B) superposition, and (C) (+)2,3,5,6TMP-TQS.
Structural models for the transmembrane domains of α7 have been proposed based on the relatively low-resolution images of the muscle-type Torpedo receptor and suggestions made for how PAMs may bind in these domains (Gill et al., 2011; Newcombe et al., 2018). However, not only are muscle-type receptors insensitive to α7 PAMs, but also the key L15′M mutation that permits heteromeric neuronal nAChR to respond to TQS-type PAMs are ineffective in muscle nAChR, so the predictive values of these models may be extremely limited. We are optimistic that in the future, structures of PAM-sensitive mutant heteromeric receptors will become available, which will allow further insights into the critical differences between the binding of active and inactive isomers (Stokes et al., 2019), as both α7 and α4β2 L15′M receptors discriminate the same way between the isomers of both TQS and 2,3,5,6TMP-TQS.
Although the PAM binding sites of both α7 and α4β2 L15′M receptors may be similar, the mechanism of allosteric activation of these receptors by GAT107 and GAT927, respectively, seems to be different (Stokes et al., 2019). Although we can suggest a specific allosteric agonist site on α7 (Gulsevin et al., 2019), there is limited evidence for a similar site on α4β2 L15′M receptors. However, whereas GAT107 does not produce allosteric activation of these receptors (Stokes et al., 2019), both isomers of 2,3,5,6TMP-TQS do antagonize allosteric activation by GAT927, albeit with reverse stereoselectivity than for allosteric antagonism of α7. The observation that neither 2,3,5,6TMP-TQS isomer reduced the potentiation of subsequent ACh response would suggest that the antagonism of GAT927 allosteric activation is not due to binding at the transmembrane PAM site.
An important consideration is that previous work that relied on the use of racemic mixtures of compounds from the TQS family (Gill et al., 2012; Gill-Thind et al., 2015; Horenstein et al., 2016; Newcombe et al., 2018) should be viewed with circumspection. Whereas racemic 4BP-TQS is a mixture of active and inactive compounds that merely has the impact of lowering the effective concentration, racemic TQS is, in fact, a mixture of a PAM and a molecule that antagonizes that activity (Stokes et al., 2019). As we show in this study, racemic 2,3,5,6TMP-TQS carries a mixture of two different activities. 2,3,5,6TMP-TQS was 1 of a total of 17 TQS analogs with varying activity that were reported in a single study (Gill et al., 2012), and all were characterized as racemic mixtures, where one isomer may have obscured the properties of another. We have also evaluated many additional TQS analogs as racemic mixtures, including GAT154, GAT155, GAT904, and GAT193 (Horenstein et al., 2016). Likewise, the ago-PAM of mutant heteromeric receptors, GAT927, was used as a racemic mixture in these and previous experiments (Stokes et al., 2019). It seems likely that the efforts required to isolate the isomers of various compounds would be rewarded by the revelation of pharmacological diversity richer than we might imagine, with the identification of relatively selective compounds that would not only inform further structural studies but also have improved therapeutic potential.
The results of docking (Fig. 10) suggested that the antagonist activity of (−)2,3,5,6TMP-TQS at the allosteric activation site would, at least in part, arise from a unique interaction with T106. The electrophysiological evaluation of mutation T106A provided support for this hypothesis via the observation that, relative to wild-type, (−)2,3,5,6TMP-TQS lost the ability to antagonize the allosteric activation by GAT107. The mechanism by which T106 is required for (−)2,3,5,6TMP-TQS antagonism remains unclear. It is possible that the interaction with T106 is required for (−)2,3,5,6TMP-TQS to bind with high affinity as an antagonist. Alternatively, T106 may cause (−)2,3,5,6TMP-TQS to adopt a conformation that is intrinsically inactive. Possibly both of these things are factors. We have confirmed that (−)2,3,5,6TMP-TQS does not become an allosteric agonist for α7T106A, as coapplication with PNU-120596 does not evoke a response (data not shown).
An important conclusion that emerges from the combination of docking and mutagenesis studies is that they provide strong support for the location of the AA binding site. It is instructive to learn that subtle changes in the interactions of a bound ligand can have such a divergent impact on receptor function, confirming that the AA site should be a continued target of interest for development of new modulators of α7 function.
Acknowledgments
We thank Lu Wenchi Corrie for conducting oocyte recordings.
Authorship Contributions
Participated in research design: Papke, Stokes.
Conducted experiments: Garai, Stokes, Abboud.
Contributed new reagents or analytic tools: Garai, Thakur.
Performed data analysis: Papke, Horenstein, Zimmerman.
Wrote or contributed to the writing of the manuscript: Papke, Stokes, Horenstein, Zimmerman, Abboud, Thakur.
Footnotes
- Received March 3, 2020.
- Accepted July 13, 2020.
This research was supported by National Institutes of Health National Institute of General Medical Sciences [Grant R01-GM57481] (to R.L.P., C.S., N.A.H.) and National Institutes of Health National Eye Institute [Grant R01-EY024717] (to S.G., G.A.T.).
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- 2,3,5,6TMP-TQS
- Cis-trans-4-(2,3,5,6-tetramethylphenyl)-3a,4,5,9b-te-trahydro-3H-cyclopenta[c]quinoline-8-sulfonamide
- 2,4,MP-TQS
- cis-cis-4-(2,4-dimethylphenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide
- AA site
- allosteric activation binding site
- ACh
- acetylcholine
- B-973B
- (S)-3-(3,4-difluorophenyl)-N-(1-(6-(4-(pyridin-2-yl)piperazin-1-yl)pyrazin-2-yl)ethyl)propanamide
- CI
- confidence interval
- GAT107
- (3aR,4S,9bS)-4-(4-bromophenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide
- GAT927
- 4-(4,5,6,7-tetrahydrobenzo[b]thiophen-3-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide
- nAChR
- nicotinic acetylcholine receptor
- PAM
- positive allosteric modulator
- TQS
- 3a,4,5,9b-Tetrahydro-4-(1-naphthalenyl)-3H-cyclopentan[c]quinoline-8-sulfonamide
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}