


Abstract
2,5-Dimethyl-celecoxib (DMC) is a derivative of celecoxib, a cyclooxygenase-2 (COX-2) inhibitor with anticancer activity in both preclinical studies and clinical practice, and lacks COX-2-inhibitory activity. Several preclinical studies have demonstrated that DMC has better apoptosis-inducing activity than celecoxib, albeit with undefined mechanisms, and exhibits anticancer activity in animal models. In this study, we primarily investigated DMC's cooperative effect with tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) on the induction of apoptosis and the underlying mechanisms in human non–small-cell lung cancer (NSCLC) cells. We found that DMC was more potent than celecoxib in decreasing the survival and inducing apoptosis of NSCLC cells. When combined with TRAIL, DMC exerted enhanced or synergistic effects on the induction of apoptosis, indicating that DMC cooperates with TRAIL to augment the induction of apoptosis. To determine the underlying mechanism of the synergy between DMC and TRAIL, we have demonstrated that DMC induces a CCAAT/enhancer binding protein homologous protein-dependent expression of DR5, a major TRAIL receptor, and reduces the levels of cellular FLICE-inhibitory protein (c-FLIP) (both the long and short forms), key inhibitors of death receptor-mediated apoptosis, by facilitating c-FLIP degradation through a ubiquitin/proteasome-dependent mechanism. It is noteworthy that enforced expression of c-FLIP or silencing of DR5 expression using DR5 small interfering RNA abrogated the enhanced effects on induction of apoptosis by the combination of DMC and TRAIL, indicating that both DR5 up-regulation and c-FLIP reduction contribute to cooperative induction of apoptosis by the combination of DMC and TRAIL. Together, we conclude that DMC sensitizes human NSCLC cells to TRAIL-induced apoptosis via induction of DR5 and down-regulation of c-FLIP.
Celecoxib (Celebrex, Pfizer Inc., New York, NY) is an approved drug for adjuvant treatment of patients with familial adenomatous polyposis. In addition, celecoxib is being tested in various clinical trials for its chemopreventive and therapeutic efficacy against a broad spectrum of epithelial malignancies, including lung cancers, either as a single agent or in combination with other agents (Koki and Masferrer, 2002; Grösch et al., 2006). Although celecoxib is a cyclooxygenase 2 (COX-2) inhibitor, it also exerts antitumor activity in tumor cells and tissues that lack the COX-2 enzyme (Grösch et al., 2006). Therefore, celecoxib seems to be able to inhibit tumor growth independently of its COX-2-inhibitory activity.
The major concern for celecoxib as a cancer therapeutic agent is that it induces apoptosis only at high concentrations (>50 μM) in cell culture systems, which exceed clinically achievable plasma levels (<10 μM) (Davies et al., 2000). In addition, a more practical issue is the potential cardiovascular side effects of celecoxib, which probably are associated with its COX-2-inhibitory activity (Dogné et al., 2006; Marwali and Mehta, 2006). Given that celecoxib has been developed and marketed mainly for the treatment of arthritis and pain but not primarily for anticancer purposes, it is conceivable that this drug might be suboptimal for inclusion in the treatment of advanced cancers, such as non–small-cell lung cancer (NSCLC). To this end, efforts have been made recently to synthesize celecoxib derivatives optimized for anticancer applications, and novel non-COX-2 inhibitory celecoxib derivatives have been identified that show better activity than celecoxib in inducing apoptosis and inhibiting the growth of tumors (Zhu et al., 2002, 2004; Kardosh et al., 2005; Pyrko et al., 2006).
Among these derivatives, 2,5-dimethyl-celecoxib (DMC) mimicked celecoxib's numerous antitumor effects, including the induction of apoptosis, the reduction of neovascularization, and the inhibition of experimental tumor growth in some in vivo tumor models with increased efficacy (Schönthal, 2006). Thus, this compound exhibits cancer therapeutic potential in the absence of COX-2 inhibitory activity. Recently, it has been shown that DMC down-regulates survivin expression in human cancer cells; this effect seems to correlate with DMC's ability to induce apoptosis (Pyrko et al., 2006).
There are two major apoptotic signaling pathways: the intrinsic mitochondria-mediated pathway, and the extrinsic death receptor-induced pathway, and cross-talk between these pathways is mediated by the truncation of the proapoptotic protein Bid (Hengartner, 2000). It is well known that the extrinsic apoptotic pathway is negatively regulated by the cellular FLICE-inhibitory protein (c-FLIP), including both long (FLIPL) and short (FLIPS) forms, through inhibition of caspase-8 activation (Krueger et al., 2001). The tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) binds to its receptors: death receptor 4 (DR4, also called TRAIL-R1), and death receptor 5 (DR5, also named Apo2, TRAIL-R2, or Killer/DR5) to activate the extrinsic apoptotic pathway (Kelley and Ashkenazi, 2004). Recently, TRAIL has received much attention because it preferentially induces apoptosis in transformed or malignant cells, demonstrating potential as a tumor-selective apoptosis-inducing cytokine for cancer treatment (Kelley and Ashkenazi, 2004). Currently, TRAIL is being tested in phase I clinical trials. It is noteworthy that certain cancer therapeutic agents including celecoxib sensitize various types of cancer cells to TRAIL-induced apoptosis (Wang and El-Deiry, 2003; Kelley and Ashkenazi, 2004; Liu et al., 2004b). Thus, these agents are useful in combination with TRAIL to overcome TRAIL resistance as demonstrated in some types of cancer cells (Wang and El-Deiry, 2003).
We have shown previously that celecoxib increases DR5 expression, down-regulates c-FLIP levels, and hence enhances TRAIL-induced apoptosis in human NSCLC cells (Liu et al., 2004b, 2006). The present study determined whether DMC exerted similar effects on induction of apoptosis, modulation of DR5 and c-FLIP, and sensitization of TRAIL-induced apoptosis in human NSCLC cells. We show that DMC potently stimulates the expression of DR5, which is dependent on the transcription factor CCAAT/enhancer binding protein homologous protein (CHOP)/growth arrest and DNA damage gene 153. Furthermore, the drug stimulates ubiquitin/proteasome-mediated degradation of c-FLIP, and both of these events contribute to DMC-mediated enhancement of TRAIL-induced apoptosis.
Materials and Methods
Reagents. DMC was synthesized as described previously (Kardosh et al., 2005). Celecoxib was purchased from LKT Laboratories (St. Paul, MN). Both drugs were dissolved in DMSO at the concentration of 100 mM, and aliquots were stored at –80°C. Stock solutions were diluted to the appropriate concentrations with growth medium immediately before use. The soluble recombinant human TRAIL was purchased from PeproTech, Inc. (Rocky Hill, NJ). The specific JNK inhibitor SP600125 was purchased from BIOMOL Research Laboratories (Plymouth Meeting, PA). The proteasome inhibitor MG132 was purchased from Sigma Chemical Co. (St. Louis, MO). Rabbit polyclonal anti-DR5 antibody was purchased from ProSci Inc. (Poway, CA). Mouse monoclonal anti-DR4 antibody (B-N28) was purchased from Diaclone (Stamford, CT). Mouse monoclonal anti-FLIP antibody (NF6) was purchased from Alexis Biochemicals (San Diego, CA). Mouse monoclonal anti-CHOP antibody (B-3) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse monoclonal anticaspase-3 antibody was purchased from Imgenex (San Diego, CA). Rabbit polyclonal anticaspase-8, anticaspase-9, and anti-PARP antibodies were purchased from Cell Signaling Technology, Inc. (Danvers, MA). Rabbit polyclonal anti-β-actin antibody was purchased from Sigma Chemical Co.
Cell Lines and Cell Culture. Human NSCLC cell lines used in this study were purchased from the American Type Culture Collection (Manassas, VA). The stable H157-FLIPL-5 and H157-FLIPL-21 transfectants that express ectopic FLIPL, and H157-FLIPL-16 transfectant that were infected with lentiviral FLIPL but did not express ectopic FLIPL, were established as described previously (Liu et al., 2006). Both H157-lac Z-5 (Liu et al., 2006) and H157-FLIPL-16 transfectants were used as control cell lines. These cell lines were cultured in RPMI 1640 medium containing 5% fetal bovine serum at 37°C in a humidified atmosphere of 5% CO2/95% air.
Cell Survival Assay. Cells were seeded in 96-well cell culture plates and were treated the next day with the agents indicated. The viable cell number was determined using the sulforhodamine B assay as described previously (Sun et al., 1997).
Detection of Apoptosis. Apoptosis was evaluated by Annexin V staining using Annexin V-PE apoptosis detection kit purchased from BD Biosciences (San Jose, CA) following the manufacturer's instructions. We also detected caspase activation by Western blotting (as described below) as an additional indicator of apoptosis.
Western Blot Analysis. Whole-cell protein lysates were prepared and analyzed by Western blotting as described previously (Sun et al., 1999; Liu et al., 2004b).
Immunoprecipitation for Detection of Ubiquitinated c-FLIP. H157-FLIPL-21 cells, which stably express FLIPL, were transfected with HA-ubiquitin plasmid using the FuGENE 6 transfection reagent (Roche Diagnostics, Indianapolis, IN) following the manufacturer's instructions. After 24 h, the cells were treated with DMC or MG132 plus DMC for 4 h and then were lysed for immunoprecipitation of Flag-FLIPL using Flag M2 monoclonal antibody (Sigma) as described previously (Chen et al., 2005) followed by the detection of ubiquitinated FLIPL with Western blotting using anti-HA antibody (Abgent, San Diego, CA).
Silencing of DR5 Expression Using Small Interfering RNA. The nonsilencing control small interfering RNA (siRNA) and DR5 siRNA duplexes were described previously (Liu et al., 2004b). Transfection of these siRNA duplexes was conducted in six-well plates using the HiPerFect transfection reagent (QIAGEN, Valencia, CA) following the manufacturer's manual. Gene-silencing effect was evaluated by Western blot analysis.
Transient Transfection and Luciferase Activity Assay. pGL3-DR5(–552) containing a wild-type CHOP binding site and pGL3-DR5(–552)CHOPm, in which the CHOP binding site was mutated, were generously provided by H. G. Wang (University of South Florida College of Medicine, Tampa, FL) (Yamaguchi and Wang, 2004). To examine the effects of DMC on DR5 promoter activity, cells were cotransfected with 0.35 μg of reporter plasmids and 0.25 μgof β-galactosidase expression plasmid (Pharmacia Biotech, Piscataway, NJ) using FuGENE 6 transfection reagent (Roche Molecular Biochemicals, Indianapolis, IN) following the manufacturer's protocol. Twenty-four hours later, the cells were treated with DMC. After 12 h, the cells were lysed and subjected to luciferase activity assay using Luciferase Assay System (Promega, Madison, WI) in a luminometer. Relative luciferase activity was normalized to β-galactosidase activity, which was measured as described previously (Pfahl et al., 1990).
Chromatin Immunoprecipitation Assay. Chromatin immunoprecipitation (ChIP) assay was conducted using the ChIP assay kit purchased from Upstate Biotechnology (Charlottesville, VA) following the manufacturer's instruction and was described previously (Liu et al., 2004a). CHOP antibody for immunoprecipitation in this assay was the same used in the Western blot analysis. Mouse IgG2α isotype antibody was purchased from EMD Bioscience, Inc. (La Jolla, CA). Polymerase chain reaction amplification of a 111-bp fragment of DR5 promoter containing a CHOP binding site on immunoprecipitated chromatin was conducted using the primers 5′-AGGTTAGTTCCGGTCCCTTC-3′ (forward) and 5′-CAACTGCAAATTCCACCACA-3′ (reverse) as described previously (Abdelrahim et al., 2006).
Results
DMC Was More Potent than Celecoxib in Inhibiting the Growth and Inducing Apoptosis of Human NSCLC Cells. We first compared the effects of DMC with celecoxib on the growth of a panel of human NSCLC cell lines. In this experiment, six NSCLC cell lines including H157, H460, H1792, A549, Calu-1, and H226 were treated with increasing concentrations of DMC or celecoxib for 3 days, and then cell growth inhibition was determined. As presented in Fig. 1, A and B, both compounds inhibited the growth of the tested cell lines in a dose-dependent manner. DMC inhibited cell growth with IC50 values ranging from 10 to 20 μM, whereas celecoxib did so with IC50 values ranging between 20 and 35 μM, indicating that DMC is more effective than celecoxib in inhibiting the growth of NSCLC cells. Second, we examined the effects of DMC and celecoxib on the induction of apoptosis in four NSCLC cell lines. At the concentration of 50 μM, DMC induced more than 60% apoptosis in all of the cell lines tested, whereas celecoxib caused only up to 35% apoptosis (Fig. 1C). Together, these results indicate that DMC is more potent than celecoxib in inhibiting cell growth and inducing apoptosis in human NSCLC cells.
DMC Cooperated with TRAIL to Augment the Induction of Apoptosis in Human NSCLC Cells. Our previous work has shown that celecoxib enhances TRAIL-induced apoptosis in NSCLC cells (Liu et al., 2004b). Therefore, we determined whether DMC also augmented TRAIL-induced apoptosis in human NSCLC cells. To this end, we treated four NSCLC cell lines (i.e., H157, Calu-1, H460, and A549) with TRAIL alone, DMC alone, or both drugs combined and then assessed cell survival and apoptosis. As presented in Fig. 2A, the combination of DMC at concentrations of 10 to 30 μM with either dose of TRAIL (10 or 20 nM) was much more effective in decreasing tumor cell survival than either single agent alone. For example, in Calu-1 cells, DMC alone at 20 μM decreased cell survival by 33%, and TRAIL (10 ng/ml) alone decreased cell survival by 10%, but the combination of the two agents reduced cell survival by almost 70%, which is greater than the sum of the effects of each agent alone. Moreover, we detected apoptosis in two NSCLC cell lines (H157 and H460) exposed to the combination of DMC and TRAIL. During a 24-h treatment, DMC at 25 μM did not increase apoptosis in either cell line, and 10 ng/ml TRAIL alone induced only 24 (H460) and 15% (H157) apoptosis. However, the combination of DMC and TRAIL caused 63 and 30% of cells to undergo apoptosis (Fig. 2B). In addition, the combination of DMC and TRAIL was much more potent than each single agent alone in inducing cleavage of caspase-8, caspase-9, caspase-3, and PARP (Fig. 2C). Together, these results clearly indicate that DMC cooperates with TRAIL to augment the induction of apoptosis in human NSCLC cells.
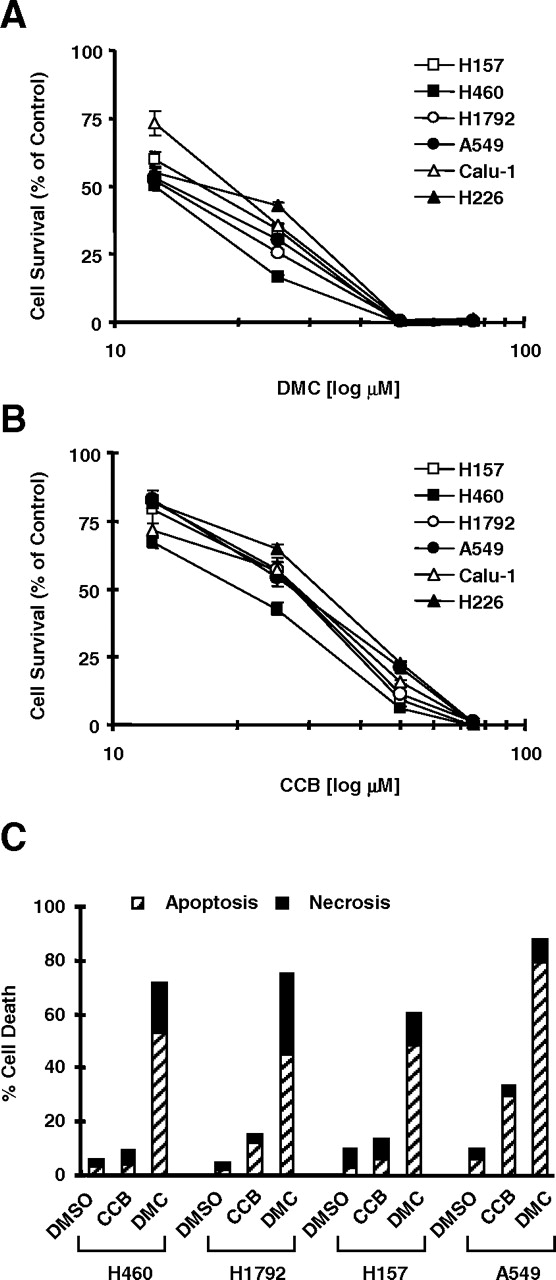
DMC exhibits more potent efficacy than celecoxib in decreasing the survival (A and B) and inducing apoptosis (C) of human NSCLC cells. A and B, the indicated NSCLC cell lines were seeded in 96-well cell culture plates and were treated the next day with the given concentrations of DMC (A) or celecoxib (CCB) (B). After 3 days, cell number was estimated using the SRB assay. Cell survival was expressed as the percentage of control (DMSO-treated) cells. Data are the means of four replicate determinations; bars, ± S.D. C, the indicated NSCLC cell lines were treated with DMSO, 50 μM DMC, or 50 μM celecoxib for 24 h. Cell death, including apoptosis and necrosis from these cell lines, was then determined by Annexin V staining.
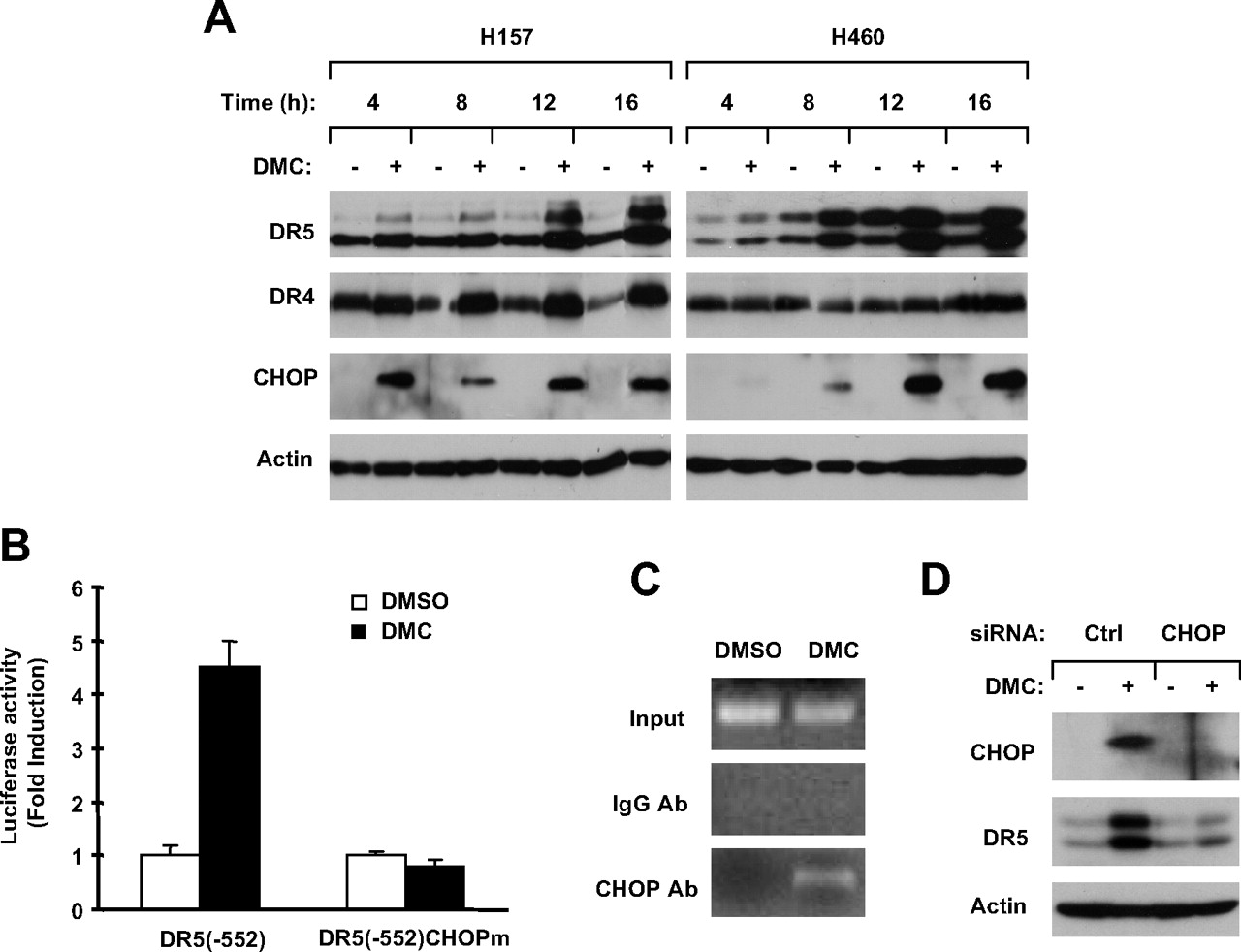
DMC Induced CHOP-Dependent DR5 Expression. DR5 induction is one mechanism accounting for the sensitization of TRAIL-induced apoptosis by some small-molecule drugs, including celecoxib (Liu et al., 2004b; Sun, 2005). Thus, we next determined whether DMC modulated DR5 expression in human NSCLC cells. By Western blot analysis, we detected a time-dependent induction of DR5 expression in both H157 and H460 cell lines exposed to DMC, which occurred early at 4 h and was sustained for at least 16 h (Fig. 3A). In addition, we found that DMC at a concentration as low as 10 μM was able to increase DR5 expression in both H157 and H460 cell lines (data not shown). Because DR4 is another TRAIL death receptor, we also examined DR4 expression in cells exposed to DMC and found that DMC increased DR4 levels in H157 cells but not in H460 cells (Fig. 3A). Thus, we focused on DR5 in the subsequent studies.
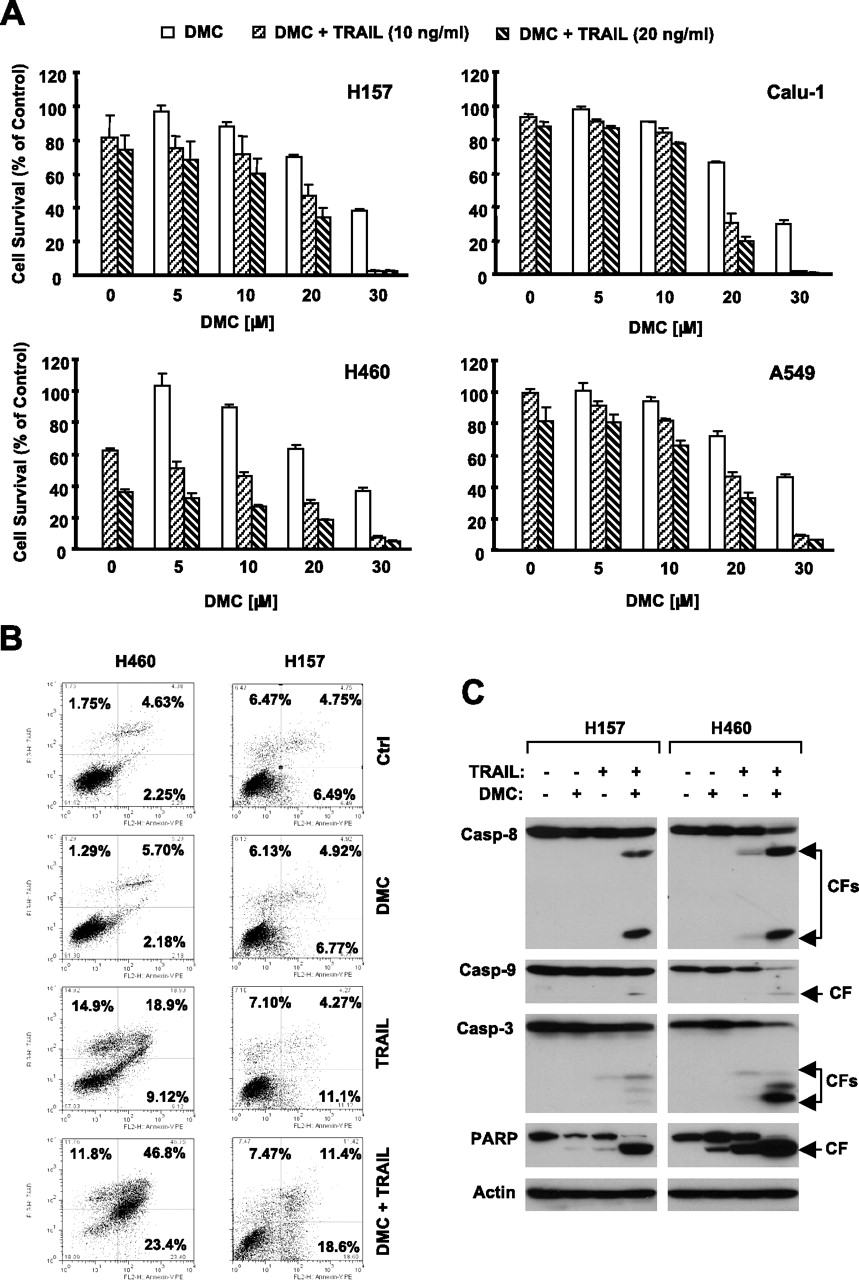
DMC enhances TRAIL's effects on decreasing cell survival (A), inducing apoptosis (B), and activating caspases (C) in human NSCLC cells. A, the indicated cell lines were treated with the given concentrations of TRAIL alone, DMC alone, and their combination as indicated. After 24 h, cell number was estimated using SRB assay for the calculation of cell survival. Data are the means of four replicate determinations. Bars, S.D. B, the indicated cell lines were treated with 10 ng/ml TRAIL alone, 25 μM DMC alone, and their respective combination as indicated. After 24 h, the cells were subjected to measurement of apoptosis using Annexin V staining. The percentage of positive cells in the upper right and lower right quadrants were added to yield the total of apoptotic cells. C, the indicated cell lines were treated with 25 μM DMC alone, 10 ng/ml TRAIL alone, and their combination. After 8 h, the cells were harvested for the preparation of whole-cell protein lysates and subsequent Western blot analysis for detecting cleavage of caspases and their substrates. Casp, caspase; CF, cleaved fragment.
To understand the mechanism by which DMC induces DR5 expression, we analyzed the expression of CHOP, a transcriptional factor that is known to regulate DR5 expression via binding to its promoter region (Yamaguchi and Wang, 2004; Yoshida et al., 2005; Abdelrahim et al., 2006). As shown in Fig. 3A, DMC increased CHOP levels in both H157 and H460 cell lines, and the kinetics of induction were similar to those of DR5. To determine whether CHOP mediates DMC-induced DR5 expression, we compared the effects of DMC on transactivation of DR5 promoters with and without the wild-type CHOP binding site. As shown in Fig. 3B, DMC significantly increased the luciferase activity in cells transfected with pGL3-DR5(–552) carrying the wild-type promoter, but this effect was completely inhibited in cells transfected with the mutant construct pGL3-DR5(–552)CHOPm, in which the CHOP binding site was inactivated. Moreover, DMC facilitated CHOP binding to DR5 promoter evaluated by ChIP assay (Fig. 3C). Together, these results indicate that the CHOP binding site is essential for DMC-mediated transactivation of the DR5 promoter. To firmly prove that DMC-induced DR5 expression is CHOP-dependent, we knocked down CHOP expression using CHOP siRNA and then examined its impact on DMC-induced DR5 expression. As presented in Fig. 3D, DMC increased levels of CHOP in control siRNA-transfected H460 cells but not in CHOP siRNA-transfected cells, indicating the successful blockage of CHOP induction by DMC. Therefore, DMC failed to increase DR5 expression in cells transfected with CHOP siRNA compared with cells transfected with control siRNA, indicating that DR5 induction by DMC is CHOP-dependent. Together, we conclude that DMC induces CHOP-dependent DR5 expression in human NSCLC cells.

DMC induces DR5 expression (A) through a CHOP-mediated mechanism (B–D). A, time-dependent modulatory effects of DMC on the expression of DR5, DR4, and CHOP. The given cell lines were treated with 25 μM DMC for various times from 4 to 16 h as indicated and then subjected to the preparation of whole-cell protein lysates and subsequent Western blot analysis for the given proteins. B, effects of DMC on the transactivation of DR5 promoters with and without wild-type CHOP binding sites. The given reporter constructs were cotransfected with pCH110 plasmid into H460 cells. After 24 h, the cells were treated with DMSO or 25 μM DMC for 12 h and then subjected to luciferase assay. Data are means of triplicate determinations. Bars, ± S.D. C, DMC facilitates CHOP binding to DR5 promoter. H460 cells were treated with 25 μM DMC for 8 h and then subjected to ChIP assay for detecting CHOP binding in the DR5 promoter. The amplified DNA fragment by polymerase chain reaction was 111 bp. Ab, antibody. D, effect of CHOP knockdown on DMC-induced DR5 expression. H460 cells were transfected with control (Ctrl) or CHOP siRNA. After 48 h, the cells were treated with 25 μM DMC for 12 h and then subjected to the preparation of whole-cell protein lysates and subsequent Western blot analysis.
DMC Down-Regulated c-FLIP Levels through Ubiquitin/Proteasome-Mediated Degradation. In addition to DR5 induction, c-FLIP down-regulation is another important mechanism underlying enhancement of TRAIL-induced apoptosis by some anticancer drugs, including celecoxib (Liu et al., 2006; Zou et al., 2007). Therefore, we further determined whether DMC also down-regulates c-FLIP levels. DMC decreased the levels of both FLIPL and FLIPS in H157 and H460 cells at 4 h after treatment. It is interesting that prolonged treatment with DMC generated cell line-dependent results on c-FLIP modulation. The decrease of c-FLIP (FLIPL and FLIPS) by DMC was maintained for up to 16 h in H157 cells. However, FLIPL was actually increased by DMC at late times (e.g., 12 and 16 h) in H460 cells, although FLIPS was still decreased at up to 16 h (Fig. 4A). For a fixed 16-h treatment experiment, we found that DMC at concentrations ranging from 10 to 30 μM decreased levels of FLIPL and FLIPS in H157 cells; however, it increased levels of FLIPL while still reducing the levels of FLIPS in H460 cells (Fig. 3B).
Because c-FLIP proteins are known to be regulated by ubiquitin/proteasome-mediated degradation (Kim et al., 2002; Chang et al., 2006), we investigated whether the observed down-regulation of c-FLIP by DMC would be mediated via this process. To this end, we found that DMC-induced down-regulation of c-FLIP was abrogated by the presence of the proteasome inhibitor MG132 (Fig. 4C), indicating that DMC-induced c-FLIP reduction is proteasome-dependent. By immunoprecipitation/Western blotting, we detected the highest levels of ubiquitinated FLIPL in cells treated with DMC plus MG132 compared with cells exposed to DMC alone or MG132 alone (Fig. 4D), indicating that DMC increases c-FLIP ubiquitination. Taken together, we conclude that DMC facilitates ubiquitin/proteasome-mediated c-FLIP degradation, leading to down-regulation of c-FLIP in human NSCLC cells.

DMC down-regulates c-FLIP levels (A and B) through ubiquitin/proteasome-mediated protein degradation (C and D). A and B, the given cell lines were treated with 25 μM DMC for the indicated times (A) or with the indicated concentrations of DMC for 16 h (B). The cell lines were then subjected to preparation of whole-cell protein lysates and subsequent Western blot analysis. C, H157 cells were pretreated with 20 μM MG132 for 30 min and then cotreated with 25 μM DMC for another 8 h. The cells were then harvested for preparation of whole-cell protein lysates and subsequent Western blot analysis. D, H157-FLIPL-21 cells that stably express ectopic flag-FLIPL were transfected with HA-ubiquitin plasmid using FuGENE 6 transfection reagent for 24 h. The cells were then pretreated with 20 μM MG132 for 30 min and then cotreated with 25 μM DMC for 4 h. Whole-cell protein lysates were then prepared for immunoprecipitation using anti-Flag antibody followed by Western blotting (WB) using anti-HA antibody for the detection of ubiquitinated FLIPL (Ub-FLIPL) and anti-Flag antibody for the detection of ectopic FLIPL.
DMC Exerted Minimal Modulatory Effects on the Expression of Bax, Bcl-XL, Bcl-2, Bad, and Survivin. Bcl-2 family members such as Bax, Bcl-2, and Bcl-XL and inhibitor of apoptosis proteins such as survivin are know to be involved in the regulation of the intrinsic apoptotic pathway (Hengartner, 2000; Harada and Grant, 2003). To further understand the molecular mechanism underlying DMC enhancement of TRAIL-induced apoptosis, we also analyzed the modulatory effects of DMC on Bax, Bcl-XL, Bcl-2, Bad, and survivin in H157 and H460 NSCLC cell lines. Our results showed that DMC only decreased the levels of survivin but had minimal effects on the expression of Bax, Bcl-XL, Bcl-2, and Bad (see Supplemental Fig. S1). The down-regulation of survivin by DMC in human NSCLC cells is in agreement with the finding by Pyrko et al. (2006) in other types of cancer cell lines. However, we found that DMC reduced the levels of survivin in H460 cells only starting at 8 h, indicating that survivin modulation is not as early an event as the modulation of DR5 and c-FLIP in DMC-treated cells.
DMC Modulated DR5 Expression and c-FLIP Levels Independently of JNK Activation. Several studies have demonstrated that JNK activation regulates DR5 expression (Higuchi et al., 2004; Zou et al., 2004). Recently, JNK has also been demonstrated to be responsible for tumor necrosis factor-induced, ubiquitin/proteasome-mediated FLIPL degradation (Chang et al., 2006). Therefore, we wanted to determine whether DMC induces JNK activation in human NSCLC cells and, if so, whether JNK activation is responsible for DR5 induction and c-FLIP down-regulation by DMC. By Western blot analysis, we detected that DMC increased levels of phosphorylated c-Jun (p-c-Jun), a well known substrate and indicator of JNK activation, in a time- and dose-dependent manner in H157 cells. In parallel, a comparatively smaller and later increase of total c-Jun protein levels was noted as well. However, such an increase in p-c-Jun and c-Jun was not detected in H460 cells (Fig. 5, A and B), primarily because of the absence of basal level c-Jun expression. The stimulation of c-Jun phosphorylation in H157 cells occurred early at 2 h and was sustained for up to 16 h after DMC treatment (Fig. 5A). Moreover, p-c-Jun levels were increased even by 5 μM DMC (Fig. 5B).

Effects of DMC on JNK activation (A and B) by DMC in human NSCLC cells and the impact of JNK activation on DMC-induced DR5 expression and c-FLIP down-regulation (C). A and B, the indicated cell lines were treated with 25 μM DMC for the given times (A) or the indicated concentrations of DMC for 16 h (B). C, H157 cells were treated with 25 μM DMC alone, DMC plus the indicated concentrations of SP600125, and SP600125 alone for 8 h (B). Whole-cell protein lysates were then prepared from the aforementioned treatments for detection of the indicated proteins by Western blot analysis.
To further address the involvement of JNK activation in the modulation of DR5 and c-FLIP by DMC, we examined the effects of DMC on DR5 and c-FLIP expression in the presence of the JNK-specific inhibitor SP600125 in H157 cells. SP600125 at the concentrations of 10 to 20 μM inhibited the phosphorylation of c-Jun (Fig. 5C), confirming that SP600125 worked as expected in our cell system. However, SP600125 did not block DMC-induced DR5 up-regulation and did not prevent the down-regulation of c-FLIP by DMC (Fig. 5C). Together, our results suggest that JNK does not play a significant role in DMC-mediated modulation of either DR5 or c-FLIP in human NSCLC cells.
DMC Enhanced TRAIL-Induced Apoptosis through DR5 Induction and c-FLIP Down-Regulation. It is well known that both DR5 and c-FLIP are key components in the TRAIL/death receptor-mediated apoptotic pathway (Wang and El-Deiry, 2003; Kelley and Ashkenazi, 2004). We wanted to know whether DR5 induction and c-FLIP down-regulation were required for DMC-mediated enhancement of TRAIL-induced apoptosis. To demonstrate the involvement of DR5 induction in cooperative augmentation of apoptosis by the combination of DMC and TRAIL, we used DR5 siRNA to block DMC-induced DR5 up-regulation and then examined its impact on the induction of apoptosis by the combination of DMC and TRAIL. In control siRNA-transfected H460 cells, we detected basal levels of DR5, which were increased by DMC treatment as expected. In contrast, in DR5 siRNA-transfected cells, the basal levels of DR5 were substantially reduced and were not induced upon treatment with DMC (Fig. 6A). These results clearly indicate the successful knock-down of DR5 expression and blockade of DR5 induction. As a result, we detected cooperative induction of apoptosis by the combination of DMC and TRAIL in control siRNA-transfected cells but not in DR5 siRNA-transfected cells (Fig. 6B), indicating that DR5 up-regulation is required for the enhancement of TRAIL-induced apoptosis by DMC.
To determine the involvement of c-FLIP down-regulation in the induction of apoptosis by the DMC and TRAIL combination, we used a lentiviral expression system to enforce c-FLIP overexpression in NSCLC cells and then analyzed its effect on induction of apoptosis by the DMC and TRAIL combination. Taking advantage of a lentiviral expression system that achieves stable gene expression, we established H157 cell lines that stably expressed high levels of ectopic FLIPL (i.e., H157-FLIPL-5 and H157-FLIPL-21). As controls, we also used H157-Lac Z-5, which expressed irrelevant Lac Z protein and H157-FLIPL-16, which did not express ectopic FLIPL albeit being infected with lentiviral FLIPL (Fig. 7A). Treatment of H157-Lac Z-5 and H157-FLIPL-16 with TRAIL, a death ligand known to activate the extrinsic death receptor-mediated apoptotic pathway, induced substantial cleavage of caspase-8, caspase-3, and PARP; however, these effects were only minimally observed in H157-FLIPL-5 and H157-FLIPL-21 cells (Fig. 7A). Thus, enforced expression of ectopic FLIPL indeed confers cell resistance to TRAIL-induced apoptosis.

Blockage of DR5 induction (A) attenuates the induction of apoptosis by DMC and TRAIL combination (B). H460 cells were cultured in a six-well plate and the next day were transfected with control (Ctrl) or DR5 siRNA. Forty-eight hours after transfection, cells were treated with 25 μM DMC alone or DMC plus 10 ng/ml for 8 h and then subjected to preparation of whole-cell lysates for Western blot analysis (A) or harvested for detection of apoptosis by Annexin V staining (B). In Annexin V assay, the percentage of positive cells in the upper right and lower right quadrants were added to yield the total of apoptotic cells.
By measuring cell survival, we found that the combination of DMC and TRAIL very effectively decreased cell survival in H157-Lac Z-5 and H157-FLIPL-16 cell lines but had only a weak effect in H157-FLIPL-5 and H157-FLIPL-21 cells (Fig. 7B), indicating that enforced overexpression of ectopic FLIPL confers cell resistance to the combination of DMC and TRAIL. By directly measuring apoptosis using annexin V staining after combination drug treatment, we detected much less apoptotic cell death in H157-FLIPL-5 or H157-FLIPL-21 cell lines than in H157-Lac Z-5 and H157-FLIPL-16 control cell lines. Specifically, the combination of DMC and TRAIL caused 50% apoptosis in H157-Lac Z-5 cells and 97% apoptosis in H157-FLIPL-16 control cells but only 34% apoptosis in H157-FLIPL-5 and 11% apoptosis in H157-FLIPL-21 cells (Fig. 7C). Thus, these results collectively show that down-regulation of c-FLIP contributes to DMC-mediated enhancement of TRAIL-induced apoptosis.
Discussion
In the present study, we demonstrated that DMC is more active than its parental compound celecoxib in decreasing the survival and inducing apoptosis of human NSCLC cells and that DMC sensitizes human NSCLC cells to TRAIL-induced apoptosis. Given that TRAIL is a potential cancer therapeutic protein and is being tested in phase I clinical trials, our finding on the enhancement of TRAIL-induced apoptosis by DMC is of clinical significance in terms of its potential application in combination with TRAIL in cancer treatment. Our previous work has shown that celecoxib enhances TRAIL-induced apoptosis in human NSCLC cells (Liu et al., 2004b). Thus, our current finding implies that DMC, which lacks COX-2 inhibitory activity, retains celecoxib's ability to enhance TRAIL-induced apoptosis and thereby indicates that the inhibition of COX-2 is not involved in these processes.
It is generally recognized that both the death receptor-mediated extrinsic and the mitochondrial intrinsic apoptotic pathways, respectively, are regulated by multiple proteins; the former involves proteins like DR5, DR4, and c-FLIP, and the latter includes Bcl-2 family members such as Bax, Bcl-2, Bcl-XL, and inhibitor of apoptosis proteins such as survivin (Hengartner, 2000; Harada and Grant, 2003). Our results showed that DMC only decreased the levels of survivin but had minimal effect on the expression of Bax, Bcl-XL, Bcl-2, and Bad (Supplemental Fig. S1). We noted that DMC also induced DR4 expression; however, this effect is cell line-dependent because DMC induced DR4 in H157 cells but not in H460 cells. Given the important roles of DR5 and c-FLIP in the regulation of the TRAIL-mediated extrinsic apoptotic pathway, and considering that both proteins are quickly and strongly modulated by DMC in all tested NSCLC cell lines, we believe that DR5 induction and c-FLIP down-regulation are two major key events that mediate cooperative induction of apoptosis by the combination of DMC and TRAIL. This view is further supported by our findings that blockade of DR5 induction by knocking down DR5 expression using DR5 siRNA or enforced overexpression of ectopic c-FLIP confers cell resistance to induction of apoptosis by the combination of DMC and TRAIL. In this study, we do not exclude the possibility that DR4 and survivin modulation also contribute to DMC-mediated enhancement of TRAIL-induced apoptosis in certain cell lines (e.g., H157).
Our study further demonstrates that DMC down-regulates c-FLIP levels by facilitating ubiquitin/proteasome-mediated degradation of c-FLIP. This is evidenced by the prevention of DMC-induced c-FLIP reduction using the proteasome inhibitor MG132 and by increased levels of ubiquitinated c-FLIP, which are detected in cells cotreated with MG132 and DMC using immunoprecipitation/Western blotting (Fig. 4). This mechanism is consistent with the one by which celecoxib decreases c-FLIP levels, as we have demonstrated previously (Liu et al., 2006), furthering the notion that DMC retains some major biological activities similar to celecoxib, even at molecular levels. A recent study has shown that JNK activation is involved in regulating ubiquitin/proteasome-dependent degradation of FLIPL (Chang et al., 2006). In our study, however, we did not detect a role of JNK in mediating DMC-induced c-FLIP degradation, and this conclusion is based on the following observations. First, DMC decreases both forms of c-FLIP (i.e., FLIPL and FLIPS), whereas JNK regulates the degradation of only FLIPL (Chang et al., 2006). Second, DMC increases JNK activation in one cell line (i.e., H157) but not in another cell line (i.e., H460), in which c-FLIP was still down-regulated. Third, the JNK inhibitor SP600125 inhibited DMC-induced JNK activation but at the same time failed to prevent DMC-induced down-regulation of c-FLIP. It therefore seems that JNK does not play a major role in these DMC-induced processes.
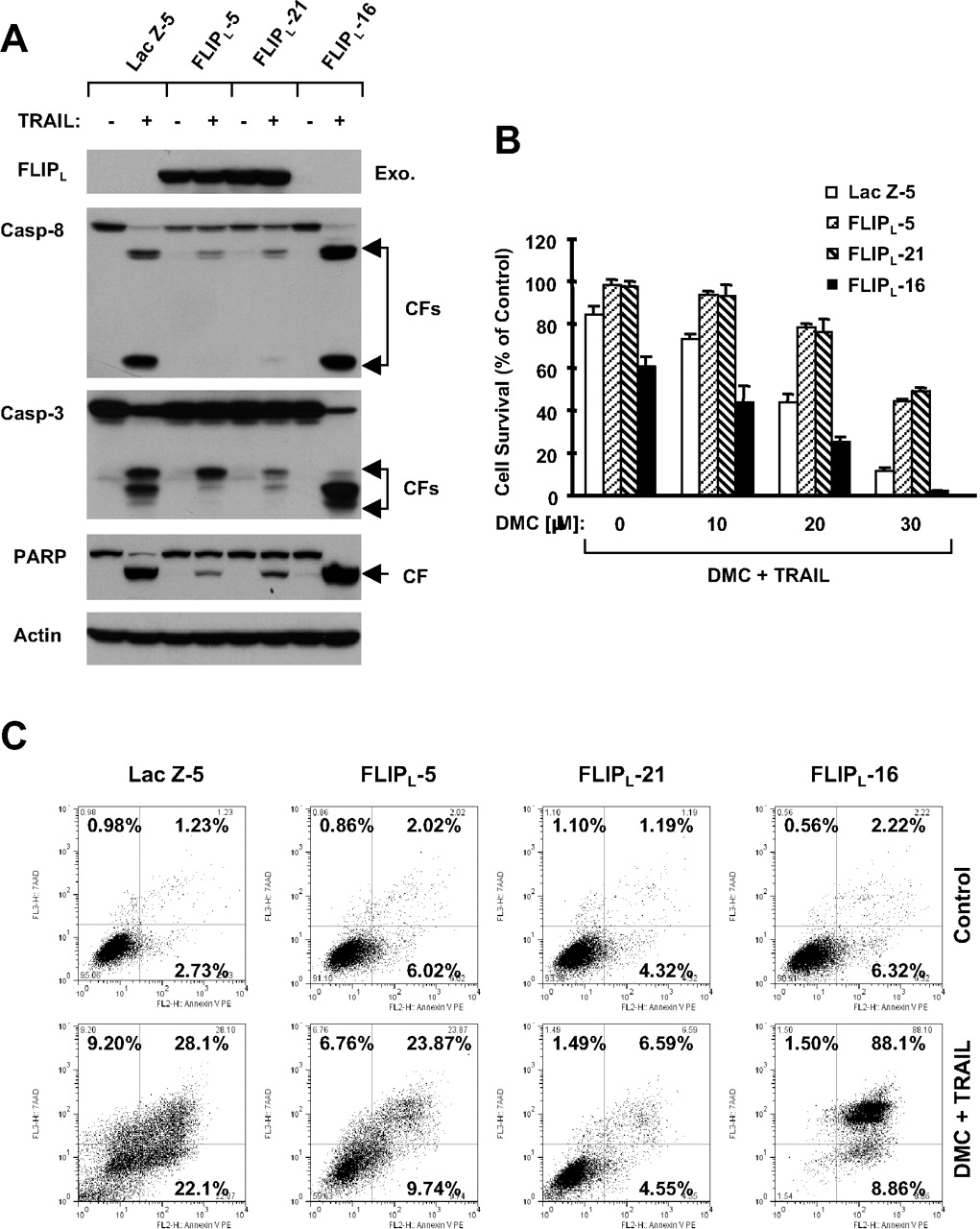
Enforced expression of ectopic FLIPL confers resistance to induction of apoptosis by TRAIL (A) or the combination of DMC and TRAIL (B and C). A, the indicated transfectants were exposed to 20 ng/ml TRAIL. After 4 h, the cells were harvested and subjected to preparation of whole-cell lysates for the detection of exogenous (Exo.) FLIPL and caspase cleavage by Western blot analysis. B, the indicated transfectants were seeded in 96-well plates and treated with the indicated concentrations of DMC combined with 10 ng/ml TRAIL. After 24 h, the cells were subjected to the SRB assay for measurement of cell survival. Data are means of four replicate determinations. Bars, ± S.D. C, the indicated transfectants were treated with DMSO and 25 μM DMC plus 10 ng/ml TRAIL for 24 h and then subjected to detection of apoptosis by Annexin V staining. In Annexin V assay, the percentage of positive cells in the upper right and lower right quadrants were added to yield the total of apoptotic cells.
We noted that FLIPL levels were decreased by DMC at early times (e.g., 4 and 8 h) and then increased after prolonged treatment (e.g., 12 and 16 h), whereas FLIPS levels were still reduced in H460 cells (Fig. 4A). The mechanism and biological significance of DMC-induced later increase in FLIPL are currently unclear. It is possible that the later increase in FLIPL may represent a survival mechanism for cells trying to escape DMC-induced cell death. Although DR5 can be induced through a JNK-dependent mechanism, as demonstrated in some studies (Higuchi et al., 2004; Zou et al., 2004), our results demonstrate that DMC induces DR5 expression in NSCLC cells independently of JNK activation because DMC did not induce JNK activation in H460 cells in which DR5 was induced and DMC could still induce DR5 expression in the presence of the JNK inhibitor SP600125 in H157 cells in which JNK was activated (Fig. 5).
DR5 is known to be regulated by p53 (Wu et al., 1997), nuclear factor-κB (Shetty et al., 2005), and CHOP (Yamaguchi and Wang, 2004; Yoshida et al., 2005). Because DMC induced DR5 expression in H157 cells carrying mutant p53, it is likely that DMC induced a p53-independent DR5 expression. In an effort to reveal the mechanism by which DMC induces DR5 expression, we found that CHOP was induced by DMC and accompanied with DR5 up-regulation. By analyzing the DR5 promoter, we found that the presence of the CHOP binding site is required for transactivation of the DR5 promoter by DMC. ChIP assay demonstrated that DMC also enhanced the binding of CHOP to DR5 promoter. Moreover, blockade of CHOP induction by silencing CHOP expression with CHOP siRNA abolished DMC's ability to induce DR5 expression. Thus, we conclude that DMC induces a CHOP-dependent DR5 up-regulation. Because CHOP is a typical protein associated with endoplasmic reticulum (ER) stress-induced apoptosis (Oyadomari and Mori, 2004), it is possible that DMC-induced up-regulation of CHOP and DR5 is caused by ER stress. Indeed, a recent study has demonstrated that DMC indeed induces ER stress of cancer cells in both cell culture and xenograft models in vivo, including CHOP up-regulation (Pyrko et al., 2007). Future studies need to demonstrate how DMC increases CHOP expression and whether nuclear factor-κB is activated and involved in DMC-induced DR5 expression.
It has been documented in the literature that activation of the TRAIL apoptotic pathway such as up-regulation of DR5 contributes to the induction of apoptosis by certain anticancer agents, including celecoxib (Huang et al., 2001; LaVallee et al., 2003; Wagner et al., 2003; Liu et al., 2004b; Kabore et al., 2006). The current work does not address whether DR5 up-regulation participated in the induction of apoptosis by DMC only, although we assume that it may contribute to DMC-induced apoptosis because celecoxib induces apoptosis requiring DR5 up-regulation. The ongoing work is to test whether DR5 induction is indeed involved in DMC-induced apoptosis.
In summary, the present study has shown for the first time that DMC, a celecoxib derivative lacking COX-2 inhibitory activity, induces CHOP-mediated DR5 up-regulation and ubiquitin/proteasome-mediated down-regulation of c-FLIP, leading to the enhancement of TRAIL-induced apoptosis in human NSCLC cells. Our results clearly indicate that DMC possesses more potent anticancer activity than celecoxib, although the underlying mechanisms seem to be quite similar. Given the concern over cardiovascular side effects of celecoxib, which is associated with its anti-COX-2 activity and is also noted with other COX-2 inhibitors (Dogné et al., 2006; Marwali and Mehta, 2006), DMC might be a better candidate or alternative to celecoxib for cancer chemoprevention and therapy.
Acknowledgments
We are grateful to H.-G. Wang (University of South Florida College of Medicine, Tampa, FL) for providing DR5 reporter constructs with a wild-type and mutant CHOP binding site, respectively, and H. A. Elrod in our lab for editing of the manuscript.
Footnotes
-
This study was supported by Georgia Cancer Coalition Distinguished Cancer Scholar award (to S.-Y.S.) and Department of Defense VITAL grant W81XWH-04-1-0142 (to S.-Y.S. for Project 4). F.R.K. and S.-Y.S. are Georgia Cancer Coalition Distinguished Cancer Scholars.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.107.037465.
-
ABBREVIATIONS: COX-2, cyclooxygenase-2; DMC, 2,5-dimethyl-celecoxib; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; NSCLC, non–small-cell lung cancer; c-FLIP, cellular FLICE-inhibitory protein; c-FLIPL, long form of cellular FLICE-inhibitory protein; c-FLIPS, short form of cellular FLICE-inhibitory protein; DR, death receptor; CHOP, CCAAT/enhancer binding protein homologous protein; siRNA, small interfering RNA; JNK, c-Jun N-terminal kinase; p-c-Jun, phospho-c-Jun; ER, endoplasmic reticulum; DMSO, dimethyl sulfoxide; PARP, poly(ADP-ribose) polymerase; HA, hemagglutinin; ChIP, chromatin immunoprecipitation; SRB, sulforhodamine B; SP600125, anthra(1,9-cd)pyrazol-6(2H)-one 1,9-pyrazoloanthrone; MG132, N-benzoyloxycarbonyl (Z)-Leu-Leu-leucinal.
-
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material. - Received April 25, 2007.
- Accepted August 7, 2007.

- The American Society for Pharmacology and Experimental Therapeutics
References
MolPharm articles become freely available 12 months after publication, and remain freely available for 5 years.Non-open access articles that fall outside this five year window are available only to institutional subscribers and current ASPET members, or through the article purchase feature at the bottom of the page.
|





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}