


Abstract
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) induces hepatic oxidative stress following activation of the aryl hydrocarbon receptor (AhR). Our recent studies showed TCDD induced pyruvate kinase muscle isoform 2 (Pkm2) as a novel antioxidant response in normal differentiated hepatocytes. To investigate cooperative regulation between nuclear factor, erythroid derived 2, like 2 (Nrf2) and the AhR in the induction of Pkm2, hepatic chromatin immunoprecipitation sequencing (ChIP-seq) analyses were integrated with RNA sequencing (RNA-seq) time-course data from mice treated with TCDD for 2–168 hours. ChIP-seq analysis 2 hours after TCDD treatment identified genome-wide NRF2 enrichment. Approximately 842 NRF2-enriched regions were located in the regulatory region of differentially expressed genes (DEGs), whereas 579 DEGs showed both NRF2 and AhR enrichment. Sequence analysis of regions with overlapping NRF2 and AhR enrichment showed over-representation of either antioxidant or dioxin response elements, although 18 possessed both motifs. NRF2 exhibited negligible enrichment within a closed Pkm chromatin region, whereas the AhR was enriched 29-fold. Furthermore, TCDD induced Pkm2 in primary hepatocytes from wild-type and Nrf2-null mice, indicating NRF2 is not required. Although NRF2 and AhR cooperate to regulate numerous antioxidant gene expression responses, the induction of Pkm2 by TCDD is independent of reactive oxygen species–mediated NRF2 activation.
Introduction
Epidemiologic and rodent studies link exposure to environmental contaminants, such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), to the development of nonalcoholic fatty liver disease (NAFLD) (Fernandez-Salguero et al., 1996; Boverhof et al., 2005; Taylor et al., 2013; Deierlein et al., 2017). Specifically, TCDD induces hepatic lipid accumulation (steatosis) and progression to steatohepatitis with fibrosis (Pierre et al., 2014; Nault et al., 2015a, 2016a,b). These effects are mostly, if not entirely, mediated by activation of the aryl hydrocarbon receptor (AhR), a ligand-activated transcription factor (Fernandez-Salguero et al., 1996; Tijet et al., 2006; Boutros et al., 2009; Denison et al., 2011) that binds structurally diverse chemicals, natural products, and endogenous metabolites. Upon ligand binding, the activated AhR dissociates from its chaperone proteins and translocates to the nucleus where it dimerizes with the AhR nuclear translocator (ARNT). The AhR-ARNT complex binds to dioxin response elements (DREs) containing the core 5′-GCGTG-3′ sequence, causing differential gene expression. Recent studies also reported differential gene expression independent of DREs (Beischlag et al., 2008; Denison et al., 2011; Dere et al., 2011; Huang and Elferink, 2012).
Phase I metabolism genes, such as cytochrome P450s and xanthine oxidase/dehydrogenase, are AhR target genes that produce reactive oxygen species (ROS) and elicit oxidative stress (Nebert et al., 1993; Tritscher et al., 1996; Sugihara et al., 2001), a key factor in NAFLD development (Day and James, 1998; Najjar, 2011). In response, cells induce antioxidant defenses, largely mediated by the master regulator, nuclear factor, erythroid derived 2, like 2 (Nfe2l2), more commonly known as Nrf2. In the presence of ROS and other electrophiles, NRF2 accumulates in the nucleus due to inactivation of Kelch-like ECH-associated protein 1 (Keap1), which directs NRF2 to proteasomal degradation (Mitsuishi et al., 2012a). In the nucleus, NRF2 acts as a transcription factor (TF), regulating gene expression associated with NADPH, glutathione, thioredoxin, and iron metabolism (Gorrini et al., 2013).
AhR- and NRF2-mediated gene expression are intimately linked with overlapping responses designated the “AhR-NRF2 gene battery” (Yeager et al., 2009). Indeed, the presence of DREs and AhR binding upstream of Nrf2 implicates the AhR in antioxidant defense regulation mediated by Nrf2 (Miao et al., 2005), whereas NRF2 regulates AhR expression (Shin et al., 2007). NRF2 also regulates lipid homeostasis (Huang et al., 2010; Chambel et al., 2015; Shimpi et al., 2017) and has been associated with NAFLD development induced by high-fat diets, developmental exposure to bisphenol A, and TCDD (Lu et al., 2011; Meakin et al., 2014; Shimpi et al., 2017). For example, TCDD-treated Nrf2-null (Nrf2−/−) mice exhibit increased weight loss and NAFLD pathologies, including more pronounced lipid accumulation and inflammation, as well as oxidative stress marker induction (Lu et al., 2011). In addition, NRF2 activation increases nuclear receptor small heterodimer partner (Nr0b2), which regulates lipogenic genes (Huang et al., 2010), and regulates sterol regulatory-element binding protein-1c expression (Shimpi et al., 2017).
AhR and NRF2 cooperation in mediating gene expression has not been systematically investigated. Studies using AhR- and/or NRF2-null models suggest both transcription factors are needed for the induction of Nqo1 and other AhR-Nrf2 battery genes (Noda et al., 2003; Ma et al., 2004; Yeager et al., 2009). We have previously reported a novel antioxidant response involving the induction of pyruvate kinase muscle isoform 2 (Pkm2) by TCDD (Nault et al., 2016b). In this study, known AhR ligands, including 2,3,7,8-tetrachlorodibenzofuran, 3,3′,4,4′,5-pentachlorobiphenyl (PCB126), and β-naphthoflavone, dose-dependently induced Pkm2 levels, whereas 2,2′,4,4′,5,5′-hexachlorobiphenyl (PCB153), a nondioxin-like polychlorinated biphenyl, did not, suggesting Pkm2 induction is AhR-dependent. However, the role of secondary responses, such as AhR-elicited production of ROS and subsequent activation of NRF2, in the induction of Pkm2 remains unclear. Pkm2 induction is typically associated with the Warburg effect, with increased expression in most cancer cells. PKM2 has a lower catalytic rate compared with PKM1, causing the accumulation of upstream glycolytic intermediates that are redirected to the pentose phosphate pathway (PPP) and serine biosynthesis pathway to produce NADPH and biomass in support of cell proliferation. This metabolic reprogramming also supports antioxidant defenses in rapidly proliferating cancer cells. Similarly, PKM2 induction by TCDD was associated with increased NADPH and the production of intermediates required for glutathione biosynthesis and recycling in normal differentiated hepatocytes (Nault et al., 2016b). However, NRF2 also promotes NADPH generation for antioxidant defenses by inducing PPP genes, suggesting multiple strategies exist to ensure cell survival in oxidative environments (Wu et al., 2011; Mitsuishi et al., 2012a,b). In this study, we integrated NRF2 chromatin immunoprecipitation sequencing (ChIP-seq) data with published AhR ChIP-seq data (Fader et al., 2017b) and incorporated differential gene expression data from a TCDD time-course study to: 1) investigate NRF2-AhR cooperation in regulating hepatic gene expression and 2) specifically examine the role of NRF2 in the TCDD-elicited induction of Pkm2. Overall, this study distinguished unique AhR and NRF2 roles in regulating gene expression in response to oxidative stress induced by TCDD.
Materials and Methods
Animal Handling and Treatment.
Animal handling and treatment were performed as previously described (Fader et al., 2017b). For in vivo treatment, male C57BL/6 mice, postnatal day 25, were obtained from Charles River Laboratories (Kingston, NY). Mice were housed at 23°C in 30%–40% humidity with a 12-hour light/dark cycle in Innocages (Innovive, San Diego, CA) with ALPHA-dri bedding (Shepherd Specialty Papers, Chicago, IL) and given Aquavive water (Innovive) and 22/5 Rodent Diet 8940 (Harlan Teklad, Madison, WI) ad libitum. On postnatal day 28 mice were orally gavaged with sesame oil vehicle (Sigma-Aldrich, St. Louis, MO) or 30 μg/kg TCDD and sacrificed by cervical dislocation at 2, 4, 8, 12, 24, 72, and 168 hours following initial exposure (N = 5). Livers were collected and immediately frozen in liquid nitrogen and stored at −80°C until analysis for ChIP-seq and RNA-sequencing (RNA-seq).
For primary hepatocyte isolations, wild-type (WT) C57BL/6 mice were obtained from Charles River Laboratories, and Nrf2-null mice (Nrf2−/−) were a generous gift from Dr. Jefferson Chan (University of California, Irvine), which were subsequently backcrossed onto the C57BL/6 background (Rockwell et al., 2012). Mice, aged 8–12 weeks, were housed at 23°C in 30%–40% humidity with a 12-hour light/dark cycle in OptiMice cages (Animal Care Systems, Inc., Centennial, CO) with Aspen wood chips (Nrf2−/−mice) or ALPHA-dri bedding (WT C57BL/6 mice) and had free access to water and Harlan Teklad 22/5 Rodent Diet 8940.
Primary Hepatocyte Isolation.
Primary hepatocytes were freshly isolated using a two-step collagenase perfusion method as previously described (Kim et al., 2006). In brief, the liver was perfused with calcium and magnesium-free Hanks’ balanced salt solution (Sigma-Aldrich) supplemented with 0.5 mM EGTA, 5.5 mM glucose, and penicillin-streptomycin (Sigma-Aldrich) by cannulation of the inferior vena cava. The liver was then perfused with Hanks’ balanced salt solution supplemented with 1.5 mM calcium chloride, 5.5 mM glucose, penicillin-streptomycin, and 0.02 g of type IV collagenase (Sigma-Aldrich). Following perfusions, the liver was excised, gently broken apart with forceps in 10 ml of William’s medium E (WME; Invitrogen, Carlsbad, CA), and centrifuged at 50g for 2 minutes. Hepatocyte pellet was washed three times with WME (Invitrogen), and cells were cultured in WME with 10% fetal bovine serum and 8% penicillin-streptomycin, then allowed to attach for 2 hours, after which the medium was replaced with fresh medium to remove unattached cells. Viability was determined by trypan blue exclusion and only used when ≥90%. Cells were treated with dimethylsulfoxide vehicle control (0.1%) or TCDD 12 hours following isolation. Cell viability following exposure to dimethylsulfoxide vehicle or TCDD was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Cayman Chemical, Ann Arbor, MI).
Protein Extraction and Quantitation.
Primary hepatocytes (2 × 106 cells) were plated in 60 × 15-mm culture dishes (n = 3) for harvesting protein. Medium was removed and plates were rinsed two times with cold Dulbecco’s phosphate-buffered saline (Thermo Fisher Scientific, Waltham, MA). While on ice, radioimmunoprecipitation buffer with protease inhibitor cocktail (Sigma-Aldrich) was added and plates were scraped. The cell suspension was collected and maintained at constant agitation for 30 minutes at 4°C using a rotary shaker, followed by centrifugation at 4°C for 20 minutes at 12,000 rpm. Supernatant was collected and stored at −80°C. Total protein was quantitated by bicinchoninic acid assay (Sigma-Aldrich). Capillary electrophoresis using the Wes system (ProteinSimple, San Jose, CA) was used to detect PKM2 (1:65; Cell Signaling, Danvers, MA), which was normalized to β-actin (ACTB) (1:65; Cell Signaling) or glyceraldehyde-3-phosphate dehydrogenase (1:10; Santa Cruz, Dallas, TX). Chemiluminescence signals were exported from Compass software (ProteinSimple) and used to calculate fold-change. A pseudoimage representing a traditional western blot was created from the raw signal intensities using the Compass software (Supplemental Figs. 1 and 2). Statistical analyses of protein levels were performed using a repeated-measures one-way analysis of variance and Sidak’s post-hoc test with GraphPad Prism (GraphPad Software, La Jolla, CA).
ChIP-Seq.
ChIP-seq analysis of NRF2 following 2-hour TCDD exposure was performed using the FactorPath services by Active Motif (Carlsbad, CA) using equal amounts of pooled liver samples from five individual mice (∼100 mg). The 2-hour time point was selected due to reported induction of ROS producing cytochrome P450s (Boverhof et al., 2005) and to match previous AhR ChIP-seq analyses (Fader et al., 2017a) to facilitate comparisons. In short, livers cut in small pieces were incubated in 1% formaldehyde for 15 minutes at room temperature. Cross-linking was halted by the addition of 0.125 M glycine, and tissue was homogenized by a Dounce homogenizer followed by shearing DNA by sonication to achieve an average length of 300–500 bp. A total of 30 μg of precleared sheared chromatin with protein A agarose beads was immunoprecipitated using 20 μl of rabbit anti-NRF2 polyclonal IgG (sc-13032; Santa Cruz) overnight at 4°C. Chromatin was eluted from the beads with SDS buffer and cross-links reversed by incubation with RNase and Proteinase K followed by an overnight incubation at 65°C. Libraries for sequencing were prepared by Active Motif and sequenced using NextSeq 500 (Illumina, San Diego, CA; 1 × 75 bp) at a depth of ∼35 M (34,804,091 untreated and 35,381,126 treated). Reads were mapped to the mouse genome (10 mm) using the Burrows-Wheeler Aligner (BWA) algorithm. Aligned reads were extended to a length of 150–250 bp on their 3′ ends, and density was determined across 32-nt bins along the genome. Peak finding was performed using model-based analysis for ChIP-seq (MACS) (Zhang et al., 2008) and compared against input control.
Previously published AhR ChIP-seq data (GSE97634) were generated and processed as previously described (Fader et al., 2017b) using five individual livers from the same experiment as the NRF2 ChIP-seq samples. In brief, following cross-linking in 1% formaldehyde for 10 minutes and shearing using a Bioruptor (Diagenode, Denville, NJ), DNA was immunoprecipitated with either rabbit IgG or rabbit anti-AhR (BML-SA210; Enzo Life Sciences, Farmingdale, NY). Libraries for sequencing were prepared using the Diagenode MicroPlex kit (Diagenode) and sequenced on an Illumina HiSeq 2500 (Illumina, San Diego, CA) at the Michigan State University Research Technology Support Facility Genomics Core (https://rtsf.natsci.msu.edu/genomics) at an average depth of 22 M. Alignment of reads to the mouse reference genome (GRCm38 release 81) was performed using Bowtie 2.0.0 (Langmead et al., 2009), then converted to Sequence Alignment Map (SAM) format using SAMTools v0.1.2 (Li et al., 2009). Normalization and peak calling were performed using CisGenome (Ji et al., 2008) by comparing AhR enrichment to IgG controls (N = 5), a bin size of 25, and boundary refinement resolution of 1 with default parameters.
NRF2 and AhR ChIP-seq data are publicly available on the Gene Expression Omnibus using accession numbers GSE109865 (NRF2) and GSE97634 (AhR). Links to the genome tracks for viewing on the University of California Santa Cruz (UCSC) genome browser are available at http://dbzach.fst.msu.edu/index.php/publications/supplementary-data/.
Functional and Binding Motif Enrichment Analyses.
Gene set enrichment analysis (GSEA) using the GSEA tool (Subramanian et al., 2005) was used to evaluate enrichment of genes demonstrating NRF2 binding within their genomic region among differentially expressed genes (DEGs) at individual time points. GSEA was performed on preranked gene expression change lists where genes were ranked by fold-change and gene sets consisted of genes with altered NRF2 binding following TCDD treatment. Normalized enrichment scores (NES) were used to assess enrichment of ChIP-seq–enriched regions among upregulated (positive NES) or downregulated (negative NES) DEGs. Statistical significance was estimated as previously described, where enrichment was considered significant when compared with a shuffled data set representing background when the nominal P value was ≤0.05 and the family-wise error rate (FWER) was ≤0.05 (Subramanian et al., 2005). Functional enrichment analysis of genes demonstrating altered NRF2 binding was performed using the Database for Annotation, Visualization, and Integrated Discovery v6.8 (Dennis et al., 2003) using only gene ontology terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. Categories meeting enrichment scores ≥1.3 (equivalent to –log of the geometric mean P value of 0.05) were considered. Motif analysis of known motifs was performed using HOMER (http://homer.ucsd.edu/homer/motif/) using default parameters.
RNA Isolation and RNA-Seq Analysis.
RNA isolation from whole liver was performed as previously described (Nault et al., 2015b). In summary, total RNA was isolated from frozen liver samples (∼100 mg) using TRIzol (Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions with an additional phenol:chloroform extraction (Sigma-Aldrich). RNA quality was assessed for purity by nanodrop (Thermo Scientific) and Bioanalyzer (Agilent Technologies, Santa Clara, CA), and quantitated using Qubit (Thermo Fisher Scientific). Library preparation using the Illumina TruSeq sample preparation kit and sequencing using a HiSeq 4000 (single-ended 50-nt; ∼30 M reads/sample) for three independent samples per time point was performed at the Michigan State University Research Technology Support Facility Genomics Core. Quality control using FastQC v0.11.3 (www.bioinformatics.babraham.ac.uk/projects/fastqc/), adaptor cleaning using Trimmomatic v0.33 (Bolger et al., 2014), and cleaning of low-complexity reads using FASTX v0.0.14 (http://hannonlab.cshl.edu/fastx_toolkit/index.html) were performed on the Michigan State University High Performance Computer (https://icer.msu.edu/hpcc). RNA-seq data are available on the Gene Expression Omnibus using accession number GSE109863.
Reads were mapped to the mouse reference genome (GRCm38 release 81) using Bowtie 2 v2.2.6 and Tophat2 v2.1.0 (Langmead et al., 2009). Gene counts were determined using HTSeq v0.6.1 in intersection-nonempty mode (Anders et al., 2015), and genes were considered detected when the number of aligned reads was greater than four. Counts were transformed by variance-stabilizing transformation using the DESeq package in R (Anders and Huber, 2010), and data were normalized using a semiparametric approach in SAS v9.3 (SAS institute Inc., Cary, NC). Posterior probability P1(t) values for each gene at each time point were calculated using an empirical Bayes method (Nault et al., 2015b).
Results
Genome-Wide Profiling of NRF2 Binding in Livers of TCDD-Treated Mice.
Hepatic ChIP-seq analyses identified 3487 NRF2-enriched regions throughout the genome in controls and 7853 peaks in samples from mice treated with 30 μg/kg TCDD for 2 hours for a combined total of 8483 unique NRF2 enrichment sites throughout the hepatic genome, of which 2857 were in common between both vehicle- and TCDD-treated samples (Fig. 1A). The majority of NRF2-bound regions (7323 peaks, 86%; Fig. 1A, pink area) were intragenic [located within 10 kb upstream of the transcription start site (TSS) to transcript end site (TES)], representing 6734 unique genes with the remaining 1160 peaks binding in intergenic regions (Fig. 1A, green area).
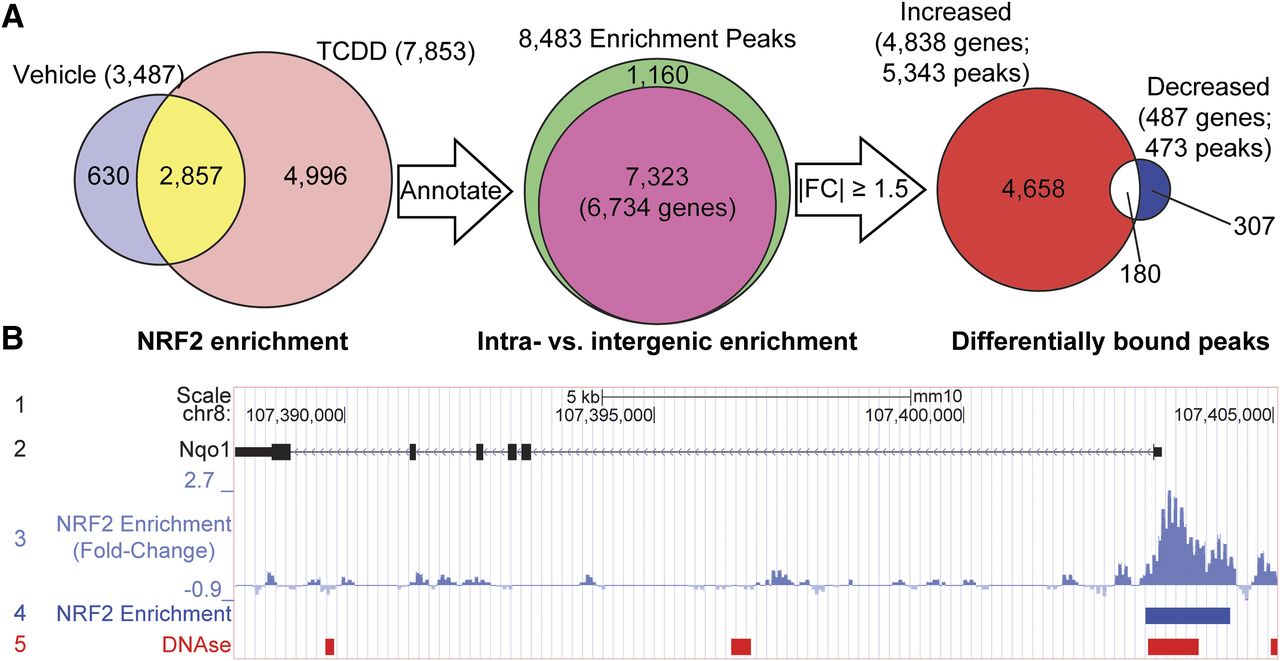
Summary of hepatic NRF2 enrichment after TCDD treatment. (A) Male hepatic NRF2-enriched regions following oral gavage with either sesame oil vehicle (control) or 30 μg/kg TCDD for 2 hours. NRF2 enrichment was assessed within 10 kb upstream of TSS to the end of 3′ region. (B) UCSC genome browser track of the known NRF2 target gene Nqo1 illustrating 1) chromosome location and scale, 2) gene details showing exons as solid boxes (black) and arrows demonstrating direction of transcription, 3) NRF2 ChIP-seq fold-changes, 4) NRF2-enriched regions (blue boxes), and 5) DNAse hypersensitive regions in adult male livers from the Encyclopedia of DNA Elements (ENCODE) project using C57BL/6 mice (red boxes; GSM1014195). FC, fold-change.
Globally, NRF2 enrichment increased following TCDD treatment, consistent with the induction of oxidative stress (Slezak et al., 2000). Some NRF peaks were only detected in treated samples, whereas others ranged from 14-fold induction to 10-fold repression. In total, 5145 genes (5816 peaks) exhibited changes in NRF2 enrichment (|fold-change| ≥ 1.5), with 5343 peaks (associated with 4838 genes; Fig. 1A, red area) showing increased enrichment and 473 (associated with 487 genes; Fig. 1A, blue area) showing decreased enrichment with treatment, whereas 180 genes with multiple peaks were represented by both increased and decreased enrichment (Fig. 1A, white area). Further examination of the prototypical NRF2 target gene Nqo1 revealed a 2.7-fold increase in NRF2 binding following TCDD treatment (Fig. 1B, tracks 3 and 4). Similarly, other target genes, including Rxra, Txnrd1, Srxn1, Sqstm1, Ephx1, and Als2 (Malhotra et al., 2010; Chorley et al., 2012; Lacher et al., 2015), also had increased NRF2 enrichment (5.9-, 2.5-, 3.1-, 4.9-, 2.2-, and 3.7-fold, respectively) (Supplemental Table 1), confirming the performance of the NRF2 antibody and ChIP-seq analysis.
Expression of Nrf2-Enriched Genes.
To evaluate the role of NRF2 binding on TCDD-elicited gene expression, a RNA-seq liver time course using male mice gavaged with 30 μg/kg TCDD for 2, 4, 8, 12, 24, 48, 72, or 168 hours was examined. TCDD elicited the differential expression of 3819 genes at at least one time point [fold-change ≥ 1.5, P1(t) ≥ 0.8], of which 842 DEGs possessed NRF2 enrichment (Fig. 2A).

Differential gene expression analysis of NRF2-bound genes. (A) Genes differentially expressed at any of the examined time points (|fold-change| ≥ 1.5, P1(t) ≥ 0.8) were compared with NRF2 ChIP-seq–enriched genomic regions (|fold-change| ≥ 1.5) and published AhR-enriched genomic regions (false detection rate ≤ 0.05) (Fader et al., 2017b). GSEA was performed on ranked gene expression at each time point using genes demonstrating reduced (B) or increased (C) NRF2 binding as gene set. Normalized enrichment scores were plotted as bars. *Statistically significant (P value ≤0.05) enrichment of the gene set (NRF2-bound genes) among either induced or repressed genes (nominal P value ≤0.05, family-wise error rate ≤0.05) (Subramanian et al., 2005). (D) All NRF2-enriched genes were examined for functional enrichment using the Database for Annotation, Visualization, and Integrated Discovery. Categories with enrichment scores ≥1.3 [dashed line, equivalent to –log(P value) of 0.05] are shown as bars.
GSEA using the 487 genes with reduced NRF2 enrichment following TCDD treatment as the gene set (Fig. 1A) only showed statistical significance, with differential gene expression at the 168-hour time point with a negative NES (Fig. 2B), suggesting negative feedback regulation of gene expression at later time points or decreasing hepatic TCDD levels which has a half-life of 6 - 12 days (Kopec et al., 2013). Conversely, using the 4838 genes showing increased NRF2 enrichment as the gene set showed positive NES at all time points with significance achieved at 4 hours, suggesting early increases in gene expression driven, at least in part, by NRF2 activation (Fig. 2C). Induced genes at 2, 4, and 8 hours with NRF2 enrichment included Cyp1a1 (64.0-, 723.1-, and 964.5-fold, respectively), Serpine1 (aka PAI-1; 55.1-, 54.2-, and 31.0-fold, respectively), Fabp12 (1.0-, 18.8-, and 15.9-fold, respectively), Cyp1a2 (3.0-, 8.8-, and 13.9-fold, respectively), and Tiparp (20.3-, 17.3-, and 8.0-fold, respectively).
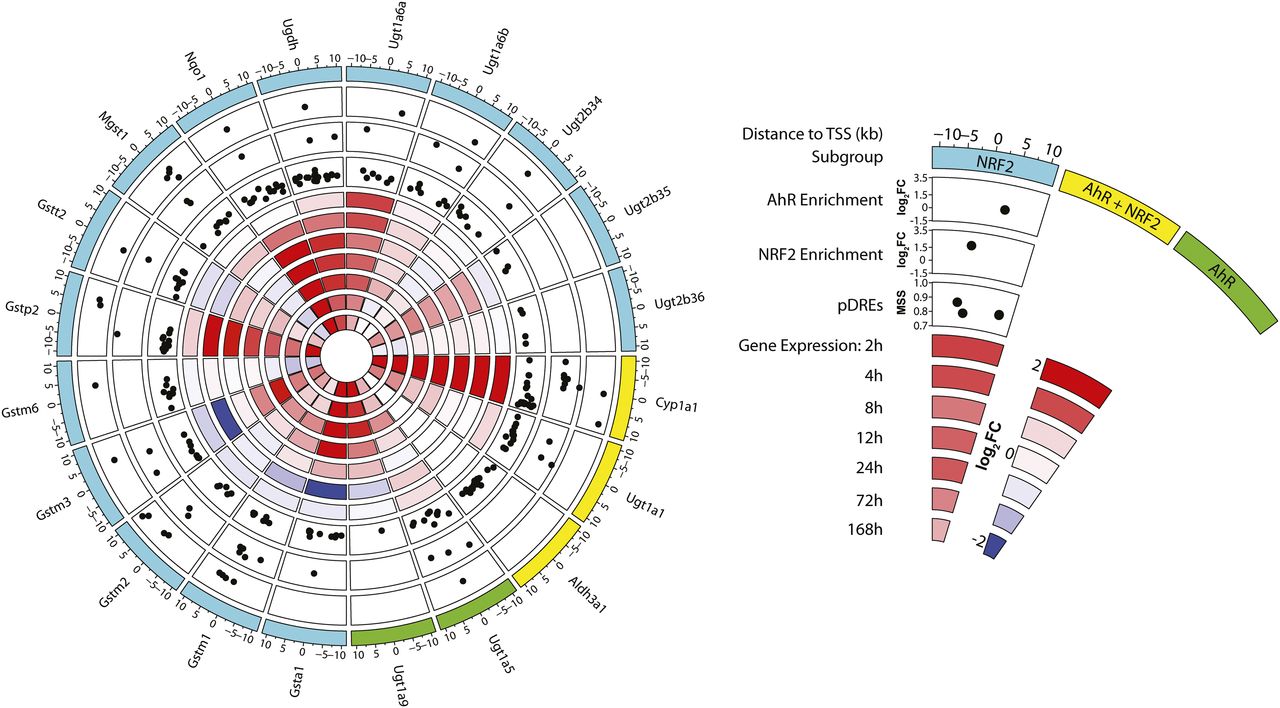
Functional enrichment analysis of all 842 NRF2-enriched DEGs found over-representation of genes involved in DNA binding and gene regulation, xenobiotic and glutathione metabolism, steroid metabolism, fatty acid metabolism, circadian rhythm, and heme metabolism (Fig. 2D), all of which have been previously linked to NRF2 signaling (Lu et al., 2011; Ma, 2013; Xu et al., 2013). The 21 repressed DEGs with reduced NRF2 binding at 168 hours included Ces1d (1.5-fold), which is directly repressed by the AhR (Matsubara et al., 2012), the TF ChREBP (Mlxipl; 1.5-fold), metabolism-related genes Aldob (1.5-fold) and Gpd2 (2.3-fold), and extracellular matrix remodelers Serpina9 (2.7-fold) and Serpina12 (4.8-fold). DEGs exhibiting increased NRF2 binding and gene induction included known NRF2 targets, including members of the AhR-NRF2 gene battery (Fig. 3). NRF2 enrichment of genes within the AhR-NRF2 battery ranged from 1.8-fold repression (Gstt2) to 10.6-fold induction (Cyp1a1), although enrichment did not correspond to reported AhR and/or NRF2 regulation (Yeager et al., 2009). For example, although Cyp1a1 is reported to be only AhR-regulated (Yeager et al., 2009), both AhR and NRF2 peaks were present in the upstream regulatory region (10 kb upstream to TSS).

Enrichment of NRF2 and AhR of AhR-NRF2 battery genes. Members of the AhR-NRF2 gene battery defined by Yeager et al. (2009) were examined for NRF2 and AhR enrichment and presence of pDREs within ±10 kb of the TSS. Time-dependent gene repression is shown in blue, whereas induction is shown in red. Colors indicate previously defined subgroups of target genes: NRF2, NRF2+AhR, and AhR. Black dots indicate: 1) the presence of putative DREs (pDREs) and their matrix similarity score (MSS), genomic enrichment, and log2fold-change (log2FC) of NRF2 peak; or 2) genomic enrichment and log2FC of AhR peak.
Coregulation of Gene Expression by AhR and Nrf2.
Previous studies have established that AhR and NRF2 coregulate gene expression via protein-protein interactions (Yeager et al., 2009; Wang et al., 2013). Comparison of the 6629 previously reported AhR-enriched genes (false detection rate ≤ 0.05) (Fader et al., 2017a) to the 5145 NRF2-enriched genes identified in this study found 2769 with both AhR and NRF2 enrichment, although putative biases in either data set due to differences in sample preparation and analysis are difficult to discern apart from validation of enrichment at expected genomic regions. AhR and NRF2 genomic binding occurred either independently at nonoverlapping genomic regions or at overlapping genomic regions. Within the 2769 genes exhibiting AhR plus NRF2 enrichment, 579 showed differential expression (Fig. 2A, yellow).
Examination of genomic coordinates of the 18,181 reported AhR ChIP peaks (Fader et al., 2017b) and 5816 NRF2 ChIP peaks with differential genomic binding (Fig. 1A) identified 1999 AhR peaks that overlapped with 1939 NRF2 peaks (Fig. 4A). These overlapping enrichment sites were present in the intragenic region of 1870 genes, 370 of which were differentially expressed (Figs. 2A and 4B). Not surprisingly, response element analysis of shared binding sites identified that AHR:ARNT (515 peaks) and NF-E2/NRF2 (89 peaks) motifs were over-represented, as well as BACH1 (76 peaks) and BACH2 (122 peaks) binding motifs compared with the background occurrence of the motif in random DNA sequences (Fig. 4C; Supplemental Table 2). In agreement with reported direct protein-protein interactions (Wang et al., 2013), only 18 (≤20%) AhR-NRF2–enriched regions, representing 25 genes, contained both AhR and NRF2 binding motifs. Only 12 of the 18 genes exhibiting AhR and NRF2 enrichment, and both binding motifs were induced, including known phase II metabolism genes Ugt1a6a, Ugt1a7c, Ugt1a9, and Ugt1a10 (Fig. 4D).

Comparison of AhR and NRF2 binding. (A) Previously identified AhR-bound genomic regions by ChIP-seq in male mice gavaged with 30 μg/kg TCDD were compared with NRF2-enriched regions. (B) AhR- and NRF2-enriched genes were compared with TCDD-elicited DEGs. (C) For all enriched regions showing both AhR and NRF2 genomic binding, motif analysis was performed using HOMER. The five most highly represented motifs are shown. (D) DEGs demonstrating the presence of ARNT:AHR and NF-E2/NRF2 motifs are shown in a heat map where fold-changes are shown as repressed (blue) or induced (red).
Role of Nrf2 in Regulating Pkm2 Isoform Expression.
Pkm2 regulates the fine balance between cell proliferation and antioxidant responses by redirecting intermediate flux through central carbon metabolism (Harris et al., 2012). We have previously reported that AhR activation by TCDD induces “Warburg-like” metabolic reprogramming in normal differentiated hepatocytes and liver tissue (Nault et al., 2016b), characterized by increased Pkm2 expression. Pkm2 was induced by other AhR activators, including 2,3,7,8-tetrachlorodibenzofuran (TCDF) and PCB126 but not PCB153, which does not bind or activate the AhR (Nault et al., 2016b). Dose-dependent Pkm2 induction was also suggested to be AhR-dependent based on the 29.0-fold increase in AhR enrichment found within an open chromatin region, indicative of enhancer activity (Fig. 5A, tracks 5, 6, and 7) (Thurman et al., 2012; Nault et al., 2016b). However, the potential involvement of the canonical antioxidant defense mediator NRF2 as a link between AhR activation and induction was not investigated at that time. The presented ChIP-seq analysis identified a modest 1.6-fold increase in NRF2 enrichment within Pkm within a closed chromatin region (Fig. 5A, tracks 3, 4, and 7), indicative of an inactive genome region and distinct from the 29.0-fold AhR-enriched region (Fig. 5A, tracks 5 and 6), which was consistent with negligible NRF2 regulation yet warranted further investigation.

TCDD-induced PKM2 expression does not require NRF2. (A) UCSC genome browser track of the Pkm loci illustrating: 1) location and scale, 2) gene details showing exons as solid boxes, 3) NRF2 enrichment, 4) identified NRF2 enrichment peaks, 5) AhR enrichment, 6) identified AhR enrichment peaks, and 7) DNAse hypersensitive regions in adult male livers from the mouse Encyclopedia of DNA Elements (ENCODE) project (GSM1014195). Tracks 3 and 5 are shown on equivalent scales. Time- (B) and dose-dependent (C) induction of PKM2 protein levels in WT or Nrf2−/− primary hepatocytes treated with 0.1% dimethylsulfoxide vehicle or TCDD (10 nM for time course or 0.01–100 nM for dose response). PKM2 protein signal was normalized to β-actin or glyceraldehyde-3-phosphate dehydrogenase, and bars represent mean + S.E.M. fold-change relative to vehicle control for three animals (N = 3). Graphical representation as standard western blot for individual samples, including housekeeping signal, are shown in Supplemental Figs. 1 and 2. *Significant difference (P ≤ 0.05) compared with vehicle control determined by repeated-measures one-way analysis of variance and Sidak’s post-hoc test.
To confirm Pkm2 induction by TCDD was independent of NRF2 activation, primary hepatocytes from WT and Nrf2−/− mice were isolated, and PKM2 protein levels were measured in cells treated with 10 nM TCDD for 2, 4, 12, and 24 hours (Fig. 5B) or 0.01, 0.1, 1, 10, or 100 nM TCDD for 24 hours (Fig. 5C). No cytotoxicity was observed at concentrations below 100 nM in either genotype. An EC50 of 2500 nM was estimated for the cytotoxicity of TCDD in Nrf2−/− primary hepatocytes (Supplemental Fig. 1). PKM2 exhibited time- and dose-dependent induction following treatment with TCDD that was closely matched in both genotypes (1.7 ± 0.2- and 2.0 ± 0.4-fold in WT and Nrf2−/− mice, respectively, at 24 hours with 10 nM TCDD) (Fig. 5B). Similarly, PKM2 induction by 100 nM TCDD at 24 hours showed no significant difference between WT and Nrf2−/− primary hepatocytes, increasing 2.3 ± 0.3- and 2.9 ± 0.6-fold, respectively (Fig. 5C), indicating NRF2 is not required for the induction of Pkm by TCDD.
Discussion
Several members of what was classically known as the “AhR gene battery” have been shown to require NRF2 activation, resulting in the establishment of the AhR-NRF2 gene battery (Yeager et al., 2009). These genes may possess DREs and antioxidant response elements (AREs), and tended to be associated with detoxification mechanisms involving the induction of xenobiotic metabolism and/or Warburg-like metabolic reprogramming to support cell survival from toxic insult (Wu et al., 2011; Mitsuishi et al., 2012a,b; Nault et al., 2016b; Bortoli et al., 2018). To further investigate their complementary roles, TCDD-elicited changes in AhR and NRF2 genomic DNA binding were integrated with time-dependent differential gene expression RNA-seq data to further investigate the composition and regulation of the AhR-NRF2 gene battery. Furthermore, Nrf2-null mice were used to specifically investigate the role of NRF2 as a putative intermediate between AhR activation and Pkm2 induction, and the induction of Warburg-like metabolic reprogramming in support of antioxidant responses in response to TCDD treatment.
To our knowledge, this is the first study examining genome-wide NRF2 binding following AhR activation by TCDD. Previous NRF2 ChIP-seq analyses in isothiocyanate sulforaphane–treated lymphoid cells, diethyl maleate (DEM)–treated Hepa1c1c7 cells, as well as Keap1−/− and Nrf2−/− mouse embryonic fibroblasts reported ∼100–15,000 NRF2 enrichment peaks (Malhotra et al., 2010; Chorley et al., 2012; Hirotsu et al., 2012). We found 7853 NRF2 enrichment peaks elicited by TCDD were comparable to the 15,534 enriched sites reported in mouse Hepa1c1c7 hepatoma cells treated with DEM for 4 hours (Hirotsu et al., 2012). Indeed, whereas DEM increased NRF2 binding, only 2340 (15%) of the 15,534 sites were found within genomic regions (±10 kb upstream of a TSS to the TES). In contrast, 7323 NRF2 sites (93%) were present within the genomic region in TCDD-treated mice. This difference could be due to differences in the model, analysis, and duration of exposure.
The AhR-NRF2 gene battery was initially established using AhR- and NRF2-null mice, resulting in the classification 20 AhR ligand-responsive genes as AhR-regulated, NRF2-regulated, and AhR+NRF2-regulated. Examination of NRF2 binding within the genomic region of these genes (±10 kb of TSS) showed little correspondence between binding of either TF and their AhR-NRF2 gene battery classification. In fact, most NRF2-regulated genes exhibited binding of both TFs following TCDD treatment, whereas the two AhR-regulated genes, Ugt1a5 and Ugt1a9, showed binding of AhR+NRF2, although enrichment of both TFs within Ugt1a9 was much closer to the TES (10 kb beyond the TSS). Similarly, AhR+NRF2-regulated genes did not demonstrate a clear TF enrichment pattern associated with the AhR-NRF2 gene battery subclassification. These results indicate that TF binding alone does not appear to define membership within the gene battery, but that gene expression patterns, context, and possibly other factors not yet evident are also involved. Overlapping regions of AhR and NRF2 enrichment found throughout the genome also suggest more complex mechanisms of recruitment and gene activation, such as tethering, as only a small subset of these sites possessed both DREs and AREs. Indeed, tethering of the AhR as a noncanonical gene-regulation mechanism has been proposed for COUP-TFII and NRF2 (Klinge et al., 2000; Dere et al., 2011; Wang et al., 2013).
We hypothesized that NRF2, the master antioxidant defense regulator, would be involved in AhR ligand-elicited PKM2 induction and Warburg-like metabolic reprogramming in support of hepatic defense mechanisms responding to TCDD-induced oxidative stress (Nault et al., 2016b). NRF2 activation is associated with metabolic changes that support glutathione biosynthesis and NADPH production consistent with Warburg-like metabolic reprogramming involving Pkm2 induction (Wu et al., 2011; Mitsuishi et al., 2012a,b; Hayes and Dinkova-Kostova, 2014). However, to our knowledge, NRF2 regulation of Pkm2 has not been previously demonstrated. Although NRF2 binding was detected in the Pkm genomic region, it was within a closed chromatin region, and the signal was modest relative to other NRF2-regulated genes and dwarfed in comparison with AhR enrichment. While chromatin remodeling with subsequent binding of NRF2 at later time points may underlie Pkm2 regulation, studies in Nrf2−/− primary hepatocytes confirmed that NRF2 has a minor, if any, role in the induction of PKM2 by TCDD. This suggests the AhR serves a complementary defensive role by inducing PKM2 to provide glycolytic intermediates for serine/glycine biosynthesis, whereas NRF2 upregulates other factors required for glutathione biosynthesis, such as the influx of cystine via the xCT (also known as Slc7a11) gene (e.g., dose-dependent induction of Slc7a11, ∼900-fold in male mice; GSE87519). Therefore, AhR and NRF2 cooperate to increase PPP gene expression and NADPH production to support cell survival from TCDD-induced oxidative stress (Wu et al., 2011; Mitsuishi et al., 2012a,b; Hayes and Dinkova-Kostova, 2014; Nault et al., 2016b; Bortoli et al., 2018).
In summary, AhR and NRF2 serve complementary and cooperative roles following exposure to environmental contaminants to support xenobiotic metabolism, antioxidant defenses, and cell survival. We also provide further evidence of AhR-NRF2 tethering to regulate gene expression (Wang et al., 2013). Notably, our studies showed that the induction of Pkm2 is independent of NRF2. Therefore, induction of Pkm2 represents a novel antioxidant defense response that complements NRF2-regulated defenses which appears to be AhR-mediated (Nault et al., 2016b). Interestingly, recent studies have demonstrated that AhR ligands also alter Pkm2 expression and activity in human cells (Matsuda et al., 2016; Song et al., 2017). Furthermore, our studies suggest that the regulation of PKM2 expression in off-target tissues may be a treatment strategy to enhance cell survival following toxic insult or treatment with cytotoxic therapeutics.
Acknowledgments
We thank Holly Cline-Fedewa for her assistance in the management of the mouse colony. We thank Dr. Jefferson Chan for generously providing Nrf2−/− mice.
Authorship Contributions
Participated in research design: Nault, Doskey, Fader, Zacharewski.
Conducted experiments: Nault, Doskey, Fader.
Contributed new reagents or analytic tools: Rockwell, Zacharewski.
Performed data analysis: Nault, Doskey, Fader.
Wrote or contributed to the writing of the manuscript: Nault, Doskey, Fader, Rockwell, Zacharewski.
Footnotes
- Received February 13, 2018.
- Accepted May 4, 2018.
This work was supported by the National Institutes of Health (NIH) National Institute of Environmental Health Sciences Superfund Research Program [NIEHS SRP P42ES04911]. T.Z. is supported by AgBioResearch at Michigan State University (MSU). C.E.R. is supported by NIEHS [R01 ES024966]. R.N. was supported by the MSU Barnett Rosenberg Endowed Assistantship and Integrative Training in the Pharmacological Sciences Grant [NIH 5T32GM092715]. C.M.D. is supported by the NIEHS Training Grant in Environmental Toxicology [5T32ES007255-27]. K.A.F. is supported by the Canadian Institutes of Health Research Doctoral Foreign Study Award [DFS140386].
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- AhR
- aryl hydrocarbon receptor
- ARNT
- AhR nuclear translocator
- ChIP-seq
- chromatin immunoprecipitation sequencing
- DEG
- differentially expressed gene
- DEM
- diethyl maleate
- DRE
- dioxin response element
- GSEA
- gene set enrichment analysis
- NAFLD
- nonalcoholic fatty liver disease
- NES
- normalized enrichment score
- NRF2
- nuclear factor, erythroid derived 2, like 2
- PCB126
- 3,3′,4,4′,5-pentachlorobiphenyl
- PCB153
- 2,2′,4,4′,5,5′-hexachlorobiphenyl
- PKM2
- pyruvate kinase muscle isoform 2
- PPP
- pentose phosphate pathway
- RNA-seq
- RNA-sequencing
- ROS
- reactive oxygen species
- TCDD
- 2,3,7,8-tetrachlorodibenzo-p-dioxin
- TES
- transcription end site
- TF
- transcription factor
- TSS
- transcription start site
- WME
- William’s medium E
- WT
- wild-type
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}