


Abstract
Organic cation transporters OCT1 (SLC22A1) and OCT2 (SLC22A2) are critically involved in absorption and excretion of diverse cationic drugs. Because drug-drug interactions at these transporters may induce adverse drug effects in patients, in vitro testing during drug development for interaction with the human transporters is mandatory. Recent data performed with rat OCT1 (rOCT1) suggest that currently performed in vitro tests assuming one polyspecific binding site are insufficient. Here we measured the binding and transport of model substrate 1-methyl-4-phenylpyridinium+ (MPP+) by cell-free-expressed fusion proteins of rOCT1 and rOCT1 mutants with green fluorescent protein that had been reconstituted into nanodiscs or proteoliposomes. The nanodiscs were formed with major scaffold protein (MSP) and different phospholipids, whereas the proteoliposomes were formed with a mixture of cholesterol, phosphatidylserine, and phosphatidylcholine. In nanodiscs formed with 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine or cholesterol, phosphatidylserine, and phosphatidylcholine, two low-affinity MPP+ binding sites and one high-affinity MPP+ binding site per transporter monomer were determined. Mutagenesis revealed that tryptophan 218 and aspartate 475 in neighboring positions in the modeled outward-open cleft contribute to one low-affinity binding site, whereas arginine 440 located distantly in the cleft is critical for MPP+ binding to another low-affinity site. Comparing MPP+ binding with MPP+ transport suggests that the low-affinity sites are involved in MPP+ transport, whereas high-affinity MPP+ binding influences transport allosterically. The data will be helpful in the interpretation of future crystal structures and provides a rationale for future in vitro testing that is more sophisticated and reliable, leading to the generation of pharmacophore models with high predictive power.
Introduction
The polyspecific organic cation transporters OCT1 (SLC22A1), OCT2 (SLC22A2), and OCT3 (SLC22A3) play a pivotal role in absorption, tissue distribution and elimination of cationic drugs including psychopharmaca, cytostatics, antidiabetics, and antiviral drugs (Amphoux et al., 2006; Koepsell et al., 2007; Minuesa et al., 2011; Nies et al., 2011). These transporters also have important physiologic functions because they translocate various endogenous cations. To anticipate drug-drug interactions at the level of individual organic cation transporters (OCTs), in vitro evaluation of novel drugs for interaction with human OCT1 (hOCT1) and OCT2 (hOCT2) has been recommended (Giacomini et al., 2010, Zamek-Gliszczynski et al., 2018) and is demanded by the American Food and Drug Administration (https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidance/UCM581965.pdf) and the European Medicine Agencies (http://www.ema.europa.eu/en/docs/enGB/documentlibrary/Scientificguideline/2012/07/WC500129606.pdf). Currently, novel molecular entities (NMEs) are tested for interaction with hOCT1 and hOCT2 expressed in epithelial cells to determine whether they inhibit uptake of a cationic model substrate that is applied at a micromolar concentration (Ahlin et al., 2008, 2011; Chen et al., 2017). This procedure has turned out to be insufficient because it was observed that the efficacy of inhibitors was dependent on the molecular structure of the employed substrate and was different when substrate concentrations far below their respective Michaelis-Menten constant (Km) values were used for uptake measurements (Belzer et al., 2013; Thévenod et al., 2013; Yin et al., 2016; Minuesa et al., 2017; Gorboulev et al., 2018; Sandoval et al., 2018). For example, high-affinity inhibition of uptake of 1-methyl-4-phenylpyridinium+ (MPP+) by hOCT1, hOCT2, and human OCT3 (hOCT3) by the nucleoside reverse transcriptase inhibitor tenofovir disoproxil fumarate was observed when uptake was measured with 12.5 nM MPP+ but did not show up when uptake was performed with 1 μM MPP+ (Minuesa et al., 2009, 2017). Such observations indicate the necessity for more sophisticated in vitro testing of NMEs during drug development (Koepsell, 2018). To establish appropriate test protocols and to enforce their application by pharmaceutical companies, a basic experimentally supported concept of how different drugs can interact at individual OCTs is imperative.
The current knowledge about function and substrate recognition by OCTs is mainly derived from studies performed with rat OCT1 (rOCT1) and rat OCT2 (rOCT2). Evidence has been provided that rOCT1 and rOCT2 are facilitated diffusion systems that may operate as electrogenic cation uniporters or electroneutral cation exchangers (Busch et al., 1996; Budiman et al., 2000; Keller et al., 2005; Schmitt and Koepsell, 2005). The apparent Km values of OCTs for different substrates are in the micromolar and millimolar concentration range. In addition, high-affinity cation interaction sites with half-maximal effective concentrations (EC50) in the picomolar and nanomolar ranges have been determined and may be inhibitory (Gorbunov et al., 2008; Minuesa et al., 2009). Homo-oligomerization of OCT1 and OCT2 has been demonstrated (Keller et al., 2011; Brast et al., 2012), however, it remained unclear whether high-affinity binding is dependent on oligomerization. To obtain molecular insight into polyspecific binding and transport, extensive mutagenesis was performed in rOCT1. Effects on apparent Km values and on half-maximal concentration values of inhibitors were interpreted with the help of homology models derived from crystal structures of bacterial transporters of the same major facilitator superfamily (MFS) (Gorboulev et al., 1999, 2005, 2018; Popp et al., 2005; Volk et al., 2009; Egenberger et al., 2012). The data support the hypothesis that rOCT1 contains clefts with cation binding domains in the outward- or inward-open conformation, which contain overlapping binding sites for structurally different cations. However, these interpretations remained speculative because changes in Km and IC50 values observed upon mutagenesis do not directly reflect effects on binding of substrates and/or inhibitors.
In the present study we measured the binding of radioactively labeled 1-methyl-4-phenylpyridinium+ (MPP+) to cell-free-expressed rOCT1 and rOCT1 mutants fused with green fluorscent protein (GFP) that had been reconstituted into nanodiscs (NDs). We analyzed apparent dissociation constant (KD) values for MPP+ binding to reconstituted NDs and the amounts of MPP+ per rOCT1 monomer in the preparation. In addition, we reconstituted the proteins into proteoliposomes and determined the effects of the mutations on the apparent Km values and the turnover numbers. We observed that each rOCT1 monomer contained two low-affinity MPP+ binding sites that are directly engaged in transport and one high-affinity MPP+ binding site that exhibits allosteric effects on the low-affinity sites. These data provide a basis for understanding interactions of cationic ligands at the highly polyspecific organic cation transporters.
Materials and Methods
Materials.
[3H]-1-methyl-4-phenylpyridinium+ (MPP+) (3.1 TBq/mmol) was obtained from ART Chemicals (Amsterdam, Holland). 1-Myristoyl-2-hydroxy-sn-glycero-3(phospho-rac-(1-glycerol)) (cat. no. 858120), 1,2-dimyristoyl-sn-glycero-3-phospho-(1-rac-glycerol) (DMPG) (cat. no. 840445P), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) (cat. no. 850355P), and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) (cat. no. 850457P) were purchased from Avanti Polar Lipids (Alabaster, AL). Cholesterol (cat. no. C8667), 1,2-diacyl-sn-glycero-3-phosphocholine from egg yolk (PC) (cat. no. P3556), 1,2-diacyl-sn-glycero-3-phospho-L-serine from bovine brain (PS) (cat. no. P7769), and goat anti-rabbit IgG antibody coupled to horse radish peroxidase (cat. no.12-348) were delivered by Sigma-Aldrich (Schnelldorf, Germany). Dodecyl-phosphocholine (cat. no. F308S) was obtained from Anatrace (Maumee, OH) and 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) (cat. no. A1099) from AppliChem GmbH (Darmstadt, Germany). Escherichia coli strain BL21 Star(D3) (cat. no. C6010-03) was purchased from Invitrogen/Thermo Fisher Scientific (Carlsbad, CA) and E. coli strain A19 (cat. no.5997) from Coli Genetic Stock Center (New Haven, CT). Plasmid EGFP-C1 (cat. no. 6084-1) was obtained from Takara Bio Inc. (Kusatsu, Japan), plasmid pIVEX2.3-MCS (cat. no. 3 253 538) from Roche Diagnostics (Mannheim, Germany), plasmid pET21a (cat. no. 69740-3) from Merck Life Science GmbH (Eppelheim, Germany), and plasmid pcDNA3.1 (cat. no. V790-20) from Invitrogen/Thermo Fisher Scientific. Other materials were obtained as described (Keller et al., 2008).
Cloning.
For cloning of the fusion proteins, we employed the GFP mutant A206K, which does not form dimers (Zacharias et al., 2002). The GFP mutant A206K was created by polymerase chain reaction using EGFP-C1 plasmid as template. A forward flanking primer with the NdeI recognition site and a reverse flanking primer with the BamHI recognition sequence followed by an XhoI site were employed. The amplificate was cut with NdeI/XhoI and cloned into the respective sites of the pIVEX2.3-MCS vector.
To generate GFP(A206K)-rOCT1 with a His-tag at the C-terminus, GFP(A206K) cDNA was cloned upstream of rOCT1 into NdeI/BamHI sites of the rOCT1-His/pET21a plasmid (Keller et al., 2011). The resulting plasmid was cut with NdeI/XhoI, and the cDNA of GFP(A206K)-rOCT1 was transferred into the NdeI/XhoI sites of the vector pIVEX2.3-MCS.
Mutants GFP(A206K)-rOCT1(F160Y), GFP(A206K)-rOCT1(W218L), and GFP(A206K)-rOCT1(W218Y) were cloned by replacing the Eco147I/Eco81I fragment of the rOCT1 by the fragments of the respective rOCT1 mutants prepared previously (Popp et al., 2005; Volk et al., 2009).
Mutants GFP(A206K)-rOCT1(R440K), GFP(A206K)-rOCT1(L447Y), GFP(A206K)-rOCT1(L447F), GFP(A206K)-rOCT1(Q448E), and GFP(A206K)-rOCT1(D475E) were generated by replacing the Eco81I/XhoI fragment of the rOCT1 by the respective fragments of rOCT1 mutants described earlier (Gorboulev et al., 1999, 2005; Volk et al., 2009). To construct the rOCT1 double mutant GFP(A206K)-rOCT1(W218Y/D475E), the Eco81I/XhoI fragment of GFP(A206K)-rOCT1(W218Y) was replaced by the respective fragment of the GFP(A206K)-rOCT1(D475E). The rOCT1 double mutant GFP(A206K)-rOCT1(R440K/D475E) was made by polymerase chain reaction with the GFP(A206K)-rOCT1(D475E) as a template.
For cloning of GFP(A206K)-rOCT1(6∆C) the pIVEX2.3-MCS vector was used after removal of the unique BamHI site. Therefore, the plasmid was linearized with BamHI, the ends were filled using DNA polymerase I, and the plasmid was ligated. The GFP(A206K)-rOCT1 cDNA was cloned first into the NdeI/XhoI sites of this vector. Then the BamHI/Eco81I fragment of the rOCT1 WT was replaced by the respective fragment of the rOCT1(6∆C), which had been prepared from the previously described GFP-rOCT1(6∆C)/pcDNA3.1 plasmid (Keller et al., 2011).
Expression and Purification of Major Scaffold Protein 1E3D1.
A His-tagged elongated construct of membrane scaffold protein 1 which contains a truncated first α-helix (MSP1E3D1) (Denisov et al., 2004; Roos et al., 2012) was expressed in E. coli. Bacteria (E. coli strain BL21(DE3)Star) were transformed with pET28a plasmid encoding His-tagged MSP1E3D1 and grown to mid-log phase. Protein expression was induced by isopropyl-β-d-thiogalactopyranoside and bacteria were grown for 3 hours at 30°C. After a 15-minute centrifugation at 6000g, bacteria were washed, suspended in 20 mM Tris-HCl pH 8.0 containing 500 mM NaCl and 50 mM imidazole, lysed by sonication at 4°C, and cellular debris was removed by a 1-hour centrifugation at 100,000g. For protein purification, the supernatant was mixed with Ni2+-nitriloacetic acid (Ni2+-NTA)-agarose, incubated for 1 hour under rotation, and poured into an empty gravity flow column. After extensive washing with 20 mM Tris-HCl pH 8.0 containing 500 mM NaCl and 50 mM imidazole, protein was eluted with the same buffer containing 500 mM imidazole. Fractions containing purified protein were pooled. Aliquots were snap frozen in liquid nitrogen and stored at −80°C.
Formation of Empty Nanodiscs.
The following were incubated for 1 hour at room temperature: 0.1% (w/v) dodecyl-phosphocholine containing 20 μM MSP1E3D1 plus either 2.2 mM DMPG or 2.2 mM DPPC or 2.2 mM POPC or 0.6 mg/ml cholesterol, 0.2 mg/ml 1,2-diacyl-sn-glycero-3-phosphocholine (PC) and 1 mg/ml 1,2-diacyl-sn-glycero-3-phospho-L-serine (PS). For nanodisc formation, the mixture was dialyzed for 48 hours at 0°C against excess of dialysis buffer (40 mM Tris-HCl pH 8.0, 100 mM NaCl) employing three buffer changes. Residual aggregates were separated from soluble NDs by a 20-minute centrifugation at 22,000g. The supernatant was transferred to Centriprep concentrator devices (10-kDa MWCO; Millipore, Merck, Darmstadt, Germany). After equilibration with 3 ml of dialysis buffer, the concentrator devices were centrifuged at 2000g (4°C) and NDs were concentrated up to final MSP1E3D1 concentrations of 2.4 mM corresponding to a concentration of NDs of 1.2 mM. After another 20-minute centrifugation at 22,000g, the NDs in the supernatant were kept on ice before usage. For long-term storage, NDs were snap-frozen in liquid nitrogen and stored at −80°C.
Cell-Free Expression and Insertion into Nanodiscs.
Extracts for cell-free expression were isolated from E. coli strain A19 and reaction mixtures containing vectors and cell-free extracts as well as feeding mixtures were prepared as described (Klammt et al., 2004). Cell-free expression of GFP-rOCT1 wild-type and mutants and cotranslational insertion into nanodiscs was performed in the absence of detergent in the continuous exchange mode. Dispodialysers from Spectrum Laboratories Inc. (Breda, Netherlands) with a cutoff of 25 kDa and a 1-ml reaction volume were used. For cotranslational membrane insertion of the transport proteins during cell-free synthesis, 150 μM empty NDs were added to the reaction mixtures. Cell-free reaction was performed by shaking the Dispodialysers filled with reaction mixtures for 20 hours at 30°C in glass tubes containing 17 ml of feeding mixture. Thereafter, the reaction mixture was centrifuged for 10 minutes at 10,000g (4°C) and supernatant containing empty NDs and the NDs with reconstituted transport proteins was harvested. For binding measurements, Ni2+-NTA-agarose beads were added to the supernatant and the beads with attached NDs were precipitated.
Measurement of MPP+-Binding.
NDs attached to Ni2+-NTA-agarose beads were incubated for 1 minute at 37°C or 0°C with phosphate buffered saline (PBS) pH 7.4 containing 12.5 nM MPP+ traced with [3H]MPP+ without and with different additional concentrations of nonradioactive MPP+. The samples were washed for about 10 seconds with 5 ml of ice-cold PBS on 0.22-μm cellulose acetate filters. The filters were dissolved in LUMASAFE PLUS mixture (Lumac LSC, Groningen, The Netherlands) and assayed for radioactivity by liquid scintillation counting. A one-minute incubation with MPP+ at 37°C was shown to be long enough to achieve saturation of binding at high- and low-affinity MPP+ binding sites of GFP-rOCT1 (Supplemental Fig. 1, left panel). Since MPP+ binding to GFP-rOCT1 was not decreased when washing of the filters was extended using 10 or 15 ml of washing buffer, a dissociation of bound MPP+ during routine washing with 5 ml was considered negligible (Supplemental Fig. 1, right panel). Because the slow dissociation of MPP+ could have been attributable to a structural change in the transporter, possibly induced by MPP+ binding and/or by cooling during the washing procedure, the determined dissociation constants were considered as apparent dissociation constants.
Purification of Cell-Free Expressed Transporters.
Cell-free expression was performed in the absence of detergent without addition of NDs as described above. The pellet obtained after a 10-minute centrifugation at 10,000g (4°C) was washed with Tris buffer (20 mM Tris-HCl pH 8, 500 mM NaCl) containing 10 mM imidazole. The precipitate was solubilized by a 1-hour incubation at 30°C with 1 ml of 2% (w/v) 1-myristoyl-2-hydroxy-sn-glycero-3(phospho-rac-(1-glycerol)) dissolved in Tris buffer containing 10 mM imidazole and centrifuged for 10 minutes at 10,000g (4°C). The supernatant was mixed with 11 ml of Tris buffer containing 10 mM imidazole plus 1% (w/v) CHAPS and Ni2+-NTA-agarose and incubated for 1 hour at 4°C. The suspension was poured into a column and washed with Tris buffer containing 1% CHAPS and 10 mM imidazole and Tris buffer containing 1% CHAPS and 20 mM imidazole. Proteins were eluted with 5 ml of Tris buffer containing 1% CHAPS and 100 mM imidazole.
Reconstitution of Proteoliposomes.
For freeze-thaw reconstitution protein-lipid aggregates were mixed with large multilamellar liposomes, and the mixture was frozen, thawed, pelleted, and homogenized as described (Keller et al., 2011). Protein-lipid aggregates were formed as follows: Cholesterol, PC, and PS (1 mg of each) were solubilized in 1 ml of chloroform/methanol (1:1, v/v), dried in a round-bottom flask under nitrogen, and 500 μl of the purified protein solved in CHAPS was added. The mixture was shaken for 1 hour at 4°C, and CHAPS was removed by dialysis at 4°C against 20 mM Tris/HCl, 500 mM NaCl, and 100 mM choline chloride. After a 30-minute centrifugation at 200,000g (4°C), the sediment was suspended in 2 ml of ice-cold KC buffer (20 mM imidazole pH 7.4, 0.1 mM Mg2+, 100 mM K+, 100 mM cyclamate–), again centrifuged at 200,000g, and the pellet was suspended in 150 μl of KC buffer. Large multilamellar liposomes were prepared in the following way: 2 mg of cholesterol and 4 mg of PS in 1 ml of chloroform/methanol (1:1, v/v) were dried in a round-bottom flask under nitrogen, 1 ml of KC buffer was added, and the flask was shaken for 3 hours at room temperature. Aggregates were removed by a 10-minute centrifugation at 10,000g. The multilamellar liposomes in the supernatant were pelleted by a 15-minute centrifugation at 200,000g and suspended in 150 μl of KC buffer (room temperature). For the formation of large proteoliposomes 150 μl of the protein-lipid aggregates were mixed with 150 μl of the multilamellar liposomes, incubated for 15 minutes at 41°C, and frozen in liquid nitrogen. Before transport measurements were started, the sample was thawed at 37°C in a water bath, 1.7 ml KC buffer (room temperature) was added, the proteoliposomes were pelleted by a 15-minute centrifugation at room temperature, the pellet was resuspended in 300 μl KC buffer (room temperature), and homogenized by repeated suction into and forced extrusion out of a 100-μl pipette tip.
Measurement of MPP+-Uptake into Proteoliposomes.
The proteoliposomes were preincubated for 10 minutes at 37°C with 20 μM valinomycin in the absence or presence of 100 μM quinine. Quinine is an established high-affinity inhibitor of rOCT1-mediated transport (Koepsell et al., 2007). It inhibits rOCT1-mediated transport of 12.5 μM MPP+ into proteoliposomes to a degree similar to other high-affinity inhibitors of rOCT1 such as cyanine863 and tetrapentylammonium+ (Keller et al., 2005). One-second uptake of MPP+ was measured at 37°C in the absence and presence of quinine. Ten microliters of prewarmed proteoliposomes (37°C) were mixed with 90 μl of prewarmed (37°C) sodium cyclamate buffer (20 mM imidazole pH 7.4, 0.1 μM Mg2+, 100 mM Na+, 100 mM cyclamate–), which contained different concentrations of MPP+ that were traced with [3H]MPP+. Proteoliposomes pretreated with quinine were incubated in the presence of 100 μM quinine. One-second incubation was performed as follows: The proteoliposomes were placed at the inner wall of the reaction tube just above the incubation medium. Uptake was initiated by mixing on a switched-on vortexer and stopped after 1 second by adding ice-cold stop solution (KC buffer containing 100 μM quinine) via a prepositioned pipette. A metronome was used to perform the 1-second incubation measurements. Radioactivity was determined by washing the proteoliposomes on 0.22-μm cellulose acetate filters and liquid scintillation counting of the solubilized filters. MPP+ uptake was corrected for MPP+ uptake measured in the presence of quinine.
SDS-PAGE and Western Blotting.
Samples were incubated for 30 minutes at 37°C in 60 mM Tris-HCl pH 6.8, containing 2% (w/v) SDS, 7% (v/v) glycerol, and 100 mM dithiothreitol. SDS-PAGE, staining of gels with Coomassie brilliant blue, and electroblotting to a polyvinylidene difluoride membrane were performed as described previously (Keller et al., 2008). For immunostaining of GFP-rOCT1 or mutants of GFP-rOCT1, an antibody against the large extracellular loop of rOCT1 raised in rabbit and goat anti-rabbit IgG coupled to horseradish peroxidase were used (Meyer-Wentrup et al., 1998). Binding of horseradish peroxidase-coupled IgG was visualized using enhanced chemiluminescence (ECL system; Amersham Buchler, Braunschweig, Germany). Prestained molecular weight markers (BenchMark; Invitrogen) were used to determine apparent molecular masses.
Estimation of Transporter in Nanodiscs.
In NDs coupled to Ni+-NTA-agarose beads that were used for MPP+ binding measurements the concentrations of GFP-rOCT1 wild-type or GFP-rOCT1 mutants were determined by analysis of total protein (Ptotal) in combination with densitometry of Coomassie-stained SDS polyacrylamide gels. Since the preparation consisted of empty NDs that contained only MSP1E3D1 protein or loaded NDs that contained only MSP1E3D1 and GFP-rOCT1 wild-type or one of the GFP-rOCT1 mutants (see Fig. 1B), the protein concentration of GFP-fusion (PGFP-F) protein could be calculated as follows: where Ptotal represents the total protein concentration; SGFP-F, staining of the respective GFP fusion protein; and SMSP1E3D1, staining of MSP1E3D1. Staining of proteins in Coomassie-stained gels was quantified by densitometry using the open access program ImageJ1 developed by the National Institutes of Health. It was verified that the staining intensities obtained for GFP-rOCT1 and MSP1E3D1 were correlated equally with protein concentrations (Supplemental Fig. 2).
where Ptotal represents the total protein concentration; SGFP-F, staining of the respective GFP fusion protein; and SMSP1E3D1, staining of MSP1E3D1. Staining of proteins in Coomassie-stained gels was quantified by densitometry using the open access program ImageJ1 developed by the National Institutes of Health. It was verified that the staining intensities obtained for GFP-rOCT1 and MSP1E3D1 were correlated equally with protein concentrations (Supplemental Fig. 2).

Reconstitution of GFP-rOCT1 into nanodiscs formed with MSP1E3D1 and DMPG and their isolation. (A) Cell-free expression of His-tagged GFP-rOCT1 in absence or presence of empty NDs composed of DMPG and His-tagged MSP1E3D1 (MSP). An SDS-polyacrylamide gel stained with Coomassie brilliant blue is shown. Lanes R show proteins in reaction mixtures without and with NDs at the beginning of cell-free synthesis. After completion of the synthesis, the reaction mixtures were centrifuged for 20 minutes at 22,000g. The supernatants are shown in lanes S and the pellets in lanes P. When the synthesis was performed in the presence of NDs some GFP-rOCT1 appeared in the supernatant, whereas the amount of GFP-rOCT1 in the pellet was decreased. (B) Preparation of NDs containing His-tagged GFP-rOCT1 that was employed for MPP+ binding measurements. Using Ni2+-NTA linked to agarose beads, the His-tagged NDs were precipitated from the 22,000g supernatant obtained after cell-free expression of GFP-rOCT1 in the presence of NDs. Purified GFP (pur. GFP), purified GFP-rOCT1 (pur. GFP-rOCT1), purified MSP1E3D1 (pur. MSP), empty NDs (empty NDs), and precipitated NDs with reconstituted GFP-rOCT1 (GFP-rOCT1, NDs) were separated by SDS-PAGE and stained with Coomassie brilliant blue (left panel) or visualized in a Western blot (right panel), which was developed with an antibody against rOCT1. Two micrograms of protein were applied per lane.
In Silico Docking of MPP+ to Models of rOCT1.
In silico docking was performed to outward-facing and inward-facing homology models of rOCT1 (Popp et al., 2005; Volk et al., 2009). All docking simulations were performed using the software package SYBYL version 7.1 (Tripos Inc., St. Louis, MO) and employed the force field MMFF94s. A coordinate file of the organic cation MPP+ was built in SYBYL assuming standard geometry deduced from its two-dimensional SMILES (Simplified Molecular Input Line Entry System) description, hydrogen atoms were added to the initial coordinate set and the three-dimensional (3D) structure of MPP+ was subsequently energy-minimized by a conjugate gradient algorithm. The models of rOCT1 in its inward-facing and outward-facing conformations were complemented with hydrogens, partial charges were assigned using the AMBER7 FF02 force field (SYBYL version 7.1), and the rOCT1 structures carrying all-hydrogens were subsequently energy-minimized with a conjugate gradient algorithm and the positions of all heavy atoms restrained by a strong positional harmonic potential. Docking simulations of MPP+ to both conformations of the rOCT1 model were performed using the FlexX module of the software SYBYL version 7.1. For the simulations, the side chains of lysine residues were protonated, the carboxylate groups of aspartic and glutamic acid residues were nonprotonated, and cysteine and histidine residues were treated as neutral. In a first round of docking simulations, any site in the interior of the inward-facing or outward-facing cleft was accepted and the results were sorted according to their docking energies using the corresponding total FlexX-Scores. Since many docked MPP molecules were found in close proximity to residues Trp218, Arg440, and Asp475, a second round of docking simulations was performed with the docking considered successful only when it occurred within a 7 Å radius around these three residues. Again the results were sorted according to the docking energy and the top 30 results were analyzed.
Statistics.
To determine the concentration dependence of MPP+ binding to GFP-rOCT1 wild-type or GFP-rOCT1 mutants reconstituted into NDs or transporter-mediated MPP+ uptake into proteoliposomes, three independent experiments were performed in which 8–11 different MPP+ concentrations were analyzed and three parallel measurements were performed per experimental condition. A one-site or two-site binding model was fitted to the binding data and the Michaelis-Menten equation to the uptake data of individual experiments. Mean values ± S.D. of three experiments are presented. The software package GraphPad Prism Version 4.1 (GraphPad Software, LaJolla, CA) was used to compute statistical parameters. When more than two groups were compared, the statistical significance of differences was determined by one-way analysis of variance (ANOVA) with post-hoc Dunnett comparison (effects of mutants on binding or transport) or by two-way ANOVA with Bonferroni correction (comparison between effects of mutants on binding vs. transport). When two individual groups were compared, the Student’s t test was used. P < 0.05 was considered statistically significant. The curves presented in the figures were obtained by fitting the one-site or two-site model or the Michaelis-Menten equation to the compiled data sets.
Results
Identification of Two MPP+ Binding Sites per rOCT1 Monomer in Nanodiscs Formed with 1,2-Dimyristoyl-phosphoglycerol.
For reconstitution of rOCT1 into NDs, we performed cell-free expression of His-tagged rOCT1 fusion protein with a nondimerizing mutant of green fluorescent protein (Zacharias et al., 2002) (GFP-rOCT1) in the absence of detergent and presence of empty NDs (Roos et al., 2013). The N-terminal fusion of rOCT1 with GFP did not impair the transport activity because the apparent Km value for MPP+ uptake determined after reconstitution of GFP-rOCT1 into proteoliposomes was similar to the apparent Km after reconstitution of purified rOCT1 (Keller et al., 2005, 2008). Empty NDs were formed by detergent removal from 1,2-dimyristoyl-sn-[phospho-rac-(1-glycerol)] (DMPG) and His-tagged membrane scaffold protein variant MSP1E3D1 that had been dissolved with dodecyl-phosphocholine (Denisov et al., 2004; Roos et al., 2012). When cell-free expression in the absence of detergent was performed without addition of NDs, GFP-rOCT1 and some free GFP were observed in a 22,000g pellet (Fig. 1A, see left P). After cell-free expression in the presence of empty NDs, NDs with reconstituted GFP-rOCT1 appeared in the 22,000g supernatant, and the amount of GFP-rOCT1 in the 22,000g pellet was decreased (Fig. 1A, see S and P). For binding measurements, the NDs in the supernatant were purified with Ni2+-NTA agarose beads (Fig. 1B, see GFP-rOCT1, NDs). This preparation contained two proteins, the nanodisc component MSP1E3D1 (see MSP in Fig. 1) and GFP-rOCT1. Densitometric analysis revealed that GFP-rOCT1 represented 1.8% ± 0.3% (n = 6) of the total protein (Supplemental Table 1). Assuming that each GFP-rOCT1 containing nanodisc includes two MSP1E3D1 molecules and one GFP-rOCT1 monomer (Roos et al., 2013), about 1% of the NDs contained GFP-rOCT1. Considering the low percentage of NDs containing reconstituted transporter, the absence of post-translational modifications after cell-free expression, and the reconstitution of GFP-rOCT1 into NDs in the absence of detergent, it is probable that the NDs containing GFP-rOCT1 represent a homogeneous population.
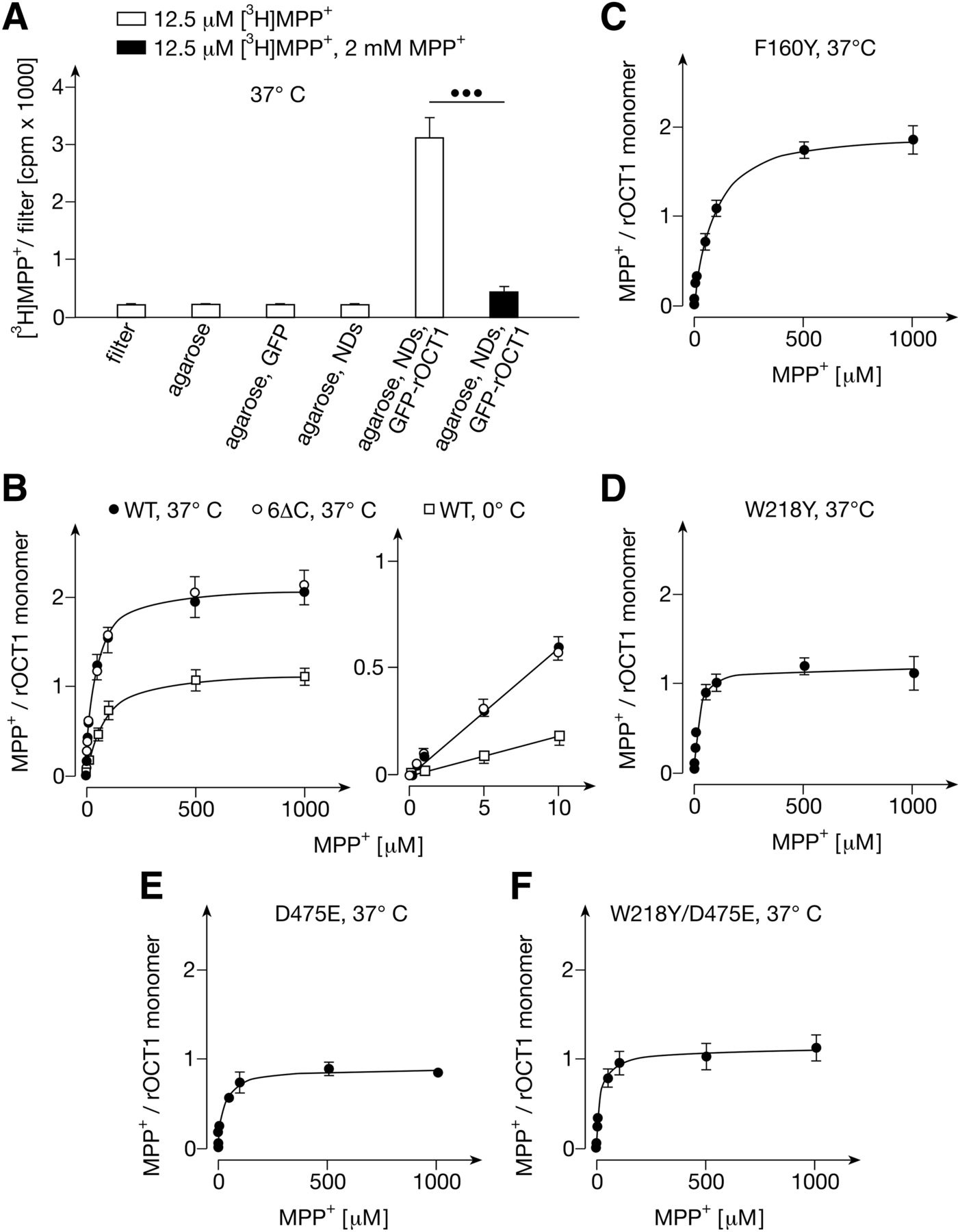
For binding measurements, samples were incubated with radioactively labeled MPP+ for 1 minute at 37°C and washed on cellulose acetate filters. Low nonspecific binding of MPP+ to filters, to agarose beads, to GFP linked to agarose beads, and to empty NDs linked to agarose beads was observed (Fig. 2A). In contrast, we observed distinct saturable MPP+ binding to agarose-linked NDs containing GFP-rOCT1 (Fig. 2, A and B). Binding of [3H]MPP+ to GFP-rOCT1 containing NDs in the presence of 2 mM nonradioactive MPP+ was not statistically significantly higher compared with [3H]MPP+ binding to filters. Per rOCT1 monomer in the NDs, 2.1 ± 0.1 MPP+ binding sites were determined that have similar apparent KD values that could not be discriminated. For these sites a common value of 32 ± 3.8 μM was determined (Fig. 2B; Table 1). For agarose-coupled NDs containing GFP fusion protein with the nonoligomerizing rOCT1 variant GFP-rOCT1(6∆C) (Keller et al., 2011), a similar common apparent KD value and the same number of MPP+ binding sites per transporter monomer were obtained (Fig. 2B; Table 1). This indicates that the determined number and affinity of MPP+ binding sites of rOCT1 reflect properties of transporter monomers. Measurements of MPP+ binding to GFP-rOCT1 at 0°C revealed 1.2 ± 0.1 MPP+ molecules per monomer and an apparent KD value of 66 ± 3.1 μM (Fig. 2B; Table 1). This suggests that a reduced conformational flexibility of the transporter at 0°C prevents access to one MPP+ binding site or impedes access to both identified MPP+ binding sites.
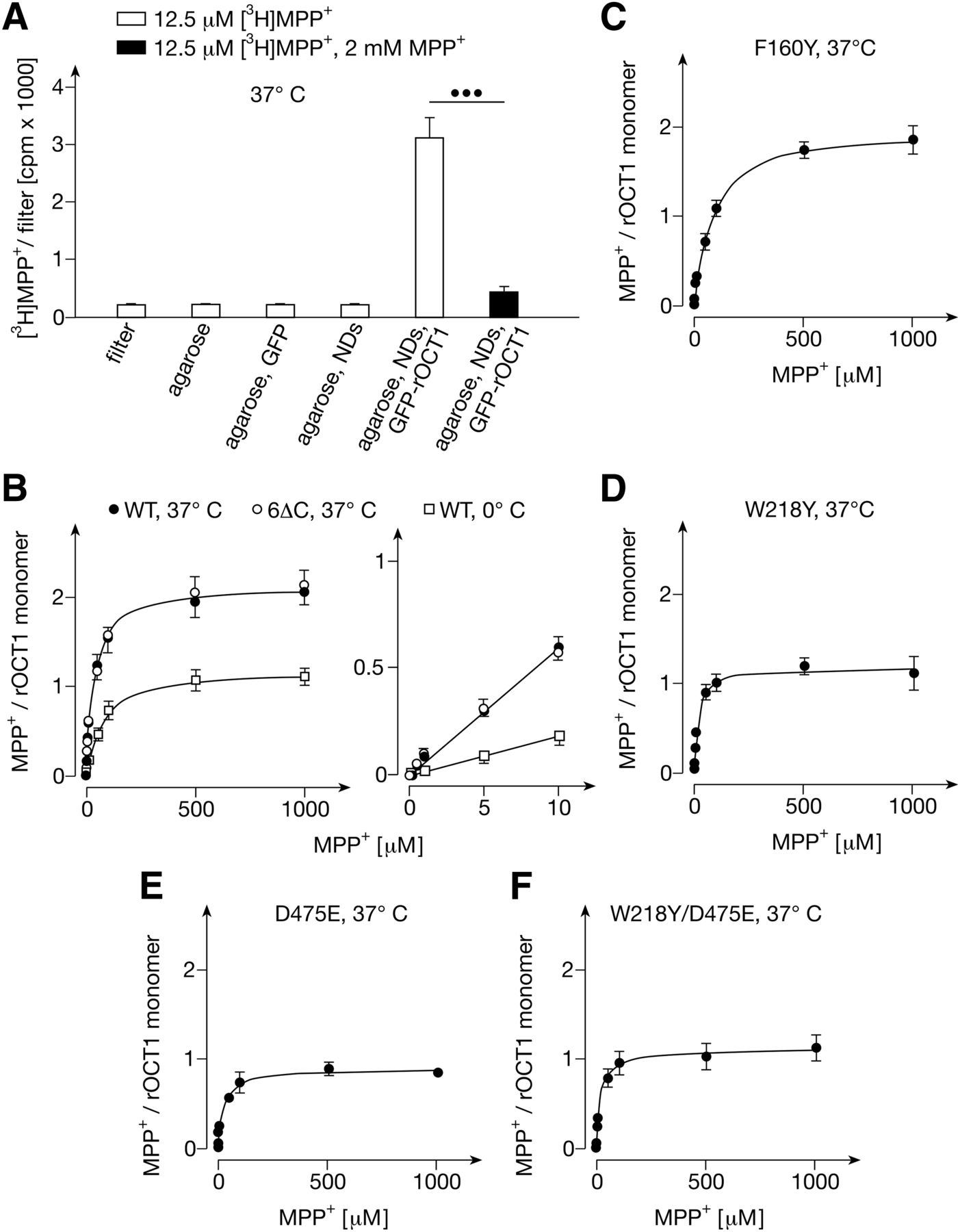
Binding of MPP+ to nanodiscs composed of DMPG and MSP1E3D1 containing wild-type GFP-rOCT1 and GFP-rOCT1 variants. (A) Characterization of MPP+ binding to nanodics (NDs). Binding of 12.5 nM [3H]MPP+ in the absence and presence of 2 mM nonradioactive MPP+ was measured at 37°C. Binding was measured to filters, Ni2+-NTA-agarose (agarose), GFP linked to Ni2+-NTA-agarose (agarose, GFP), empty NDs linked to Ni2+-NTA-agarose (agarose, NDs), and a mixture of empty NDs and GFP-rOCT1 containing NDs linked to Ni2+-NTA-agarose (agarose, NDs, GFP-rOCT1). Identical amounts of agarose beads were analyzed. Mean values ± S.D. of four measurements are shown. ●●●P < 0.001 Student’s t test. (B) Effects of temperature and capability of rOCT1 to dimerize on MPP+ binding. MPP+ binding to GFP-rOCT1 (WT) containing NDs was measured at 37°C or 0°C, and MPP+ binding to nondimerizing GFP-rOCT1(6∆C) variant was measured at 37°C. Binding to filters was subtracted. Concentrations of rOCT1 and rOCT1(6∆C) were determined by measuring total protein in combination with densitometry of stained gels. Mean values ± S.D. of three experiments are indicated. The curves in the left panel were obtained by fitting a “one site binding” model to the data. The straight lines in the right panel were obtained by linear regression. (C–F) Concentration dependence of MPP+ binding to NDs containing mutants GFP-rOCT1(F160Y) (C), GFP-rOCT1(W218Y) (D), GFP-rOCT1(D475E) (E) or GFP-rOCT1(W218Y, D475E) (F). The measurements in (C–F) were performed and the data were calculated as in Fig. 2B.
Binding of MPP+ to GFP-rOCT1 wild-type and GFP-rOCT1 mutants in nanodiscs formed with DMPG
Empty NDs were formed from DMPG and His-tagged MSP1E3D1. Fusion proteins between a nondimerizing GFP mutant and rOCT1 wild-type, rOCT1(6∆C), or single point mutants of rOCT1 were reconstituted into NDs by cell-free expression in the presence of the empty NDs. The NDs were bound to Ni2+-NTA-agarose beads and precipitated. Binding of MPP+ traced with [3H]MPP+ to the beads was measured at 37°C as described in Fig. 2B. For individual experiments, apparent KD values and Bmax values were determined by fitting a one-site binding model to the data. The amounts of GFP-fusion protein in the NDs were determined, and Bmax values per transporter monomer in the preparation were calculated. Means ± S.D. of three experiments are indicated. Significance of difference to GFP-rOCT1 was determined by one-way ANOVA with post-hoc Dunnett test (**P < 0.01).
Trp218 and Asp475 Are Involved in MPP+-Binding to the Same Low-Affinity MPP+ Binding Site.
Previously, we observed that exchange by tyrosine of Phe160 and Trp218, that were localized to outward-facing and inward-facing clefts of 3D homology models of rOCT1, increased and decreased the apparent Km value for MPP+, respectively (Popp et al., 2005; Gorboulev et al., 2018). When Asp475, the neighboring residue of Trp218 in the 3D model of the outward-facing cleft, was replaced by glutamate, the apparent Km for MPP+ remained unchanged. In the present study we introduced the F160Y, W218Y, or D475E mutation into GFP-rOCT1 and measured MPP+ binding to the mutants in NDs formed with DMPG (Fig. 2, C–F; Table 1). The mutations had no effect on incorporation of the GFP-fusion proteins into the NDs (Supplemental Table 1). As with to the apparent Km value, the apparent KD value for MPP+ binding to the GFP-rOCT1 mutants compared with wild-type was increased in mutant F160Y, whereas it was apparently decreased in mutant W218Y (see Table 1). Noteworthy, in mutants W218Y and D475E the maximal binding (Bmax) of about two MPP+ molecule per monomer observed in wild-type rOCT1 was decreased to about one. To determine whether both mutations decrease MPP+ binding at the same site, we measured MPP+ binding to the double mutant W218Y/D475E. As in both single mutants, a Bmax of about one per transporter monomer was observed, suggesting that both mutations prevent MPP+ binding to the same site.
Identification of Three MPP+ Binding Sites per rOCT1 Monomer in Nanodiscs Formed with 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine or PC, PS, and Cholesterol.
Wondering why the previously described high-affinity binding site for MPP+ (Gorbunov et al., 2008) was not detected after reconstitution of GFP-rOCT1 in NDs formed with DMPG, we measured MPP+ binding to GFP-rOCT1 in NDs formed with DPPC, and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC). With these lipids, formation of NDs and incorporation of GFP-rOCT1 into NDs was similar to that with DMPG (Supplemental Fig. 3; Supplemental Table 1). In NDs formed with DPPC, a similar apparent KD value (25.6 ± 3.1 μM vs. 31.2 ± 3.8 μM, n = 3 each) and a slightly higher Bmax per monomer were observed compared with NDs formed with DMPG (2.35 ± 0.12 vs. 2.08 ± 0.07, n = 3 each, P < 0.01). When GFP-rOCT1 was reconstituted into NDs formed with 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), one high-affinity site (Bmax (high aff.) 0.93 ± 0.05) with an apparent KD of 0.23 ± 0.02 μM (KD (high aff.)) could be identified per transporter monomer in addition to two low-affinity sites (Bmax (low aff.) 2.29 ± 0.05) (Fig. 3A; Table 2). As with NDs formed with DMPG or DPPC, the affinities of the two low-affinity sites could not be discriminated, and a common apparent KD value (36 ± 2.6 μM) was determined. The properties of GFP-rOCT1 in NDs formed with POPC are independent of dimerization because high- and low-affinity MPP+ binding sites with similar properties were obtained after reconstitution of GFP-rOCT1 and GFP-rOCT1(6∆C) (Table 2). The data indicate that an optimal lipid environment is required for accessibility of MPP+ to the high-affinity MPP+ binding site.

Binding of MPP+ to nanodiscs composed of POPC and MSP1E3D1 containing GFP-rOCT1 wild-type (A) and mutants with amino acid exchanges in positions 218 (B), 440 (C), 475 (D), 218 and 475 (E), and 440 and 475 (F). His-tagged GFP-rOCT1 (WT) or GFP-rOCT1 mutants were reconstituted by cell-free expression in the presence of empty NDs formed from POPC and His-tagged MSP1E3D1. The NDs were precipitated by Ni2+-NTA agarose, and binding of 12.5 nM [3H]MPP+ was measured at 37°C in the presence of various concentrations of unlabeled MPP+. MPP+ binding was measured and numbers of transporter monomers were determined as in Fig. 2B. Mean values ± S.D. of three experiments are indicated. A two-site binding model was fitted to the data. In the right panel, MPP+ binding and the fitted curve at low MPP+ concentrations are shown.
Comparison of MPP+ binding to GFP-rOCT1 wild-type and mutants in NDs with MPP+ uptake into proteoliposomes
NDs were prepared with POPC. The MPP+ binding measurements are shown in Figs. 3 and 5. Bmax (high aff.) and Bmax (low aff.) per rOCT1 monomer are indicated. Purified GFP-rOCT1 wild-type and mutants were reconstituted into proteoliposomes formed with cholesterol, PC, and PS. The MPP+ uptake measurements are presented in Figs. 6 and 8. Means ± S.D. of three experiments are shown.
Next, we determined whether the nontransported inhibitor tetrabutylammonium+ (TBuA+) (Gorboulev et al., 2018) blocks the identified MPP+ binding sites. TBuA+ inhibits rOCT1-mediated MPP+ uptake into proteoliposomes with an IC50 value of 19 μM (Keller et al., 2005). In NDs formed with POPC containing GFP-rOCT1, we measured the concentration dependence of MPP+ binding in the presence of 2 mM TBuA+ (Supplemental Fig. 4). In the presence of 2 mM TBuA+, 98% of MPP+ binding to the high-affinity site in the NDs and 75% of MPP+ binding to the low-affinity sites was blocked.
Previously, we characterized MPP+ uptake into proteoliposomes that were generated from cell-free-expressed GFP-rOCT1 and equal-weight mixtures of 1,2-diacyl-sn-glycero-3-phospho-L-serine from egg yolk (PS), 1,2-diacyl-sn-glycero-3-phosphocholine from bovine brain (PC), and cholesterol (Keller et al., 2008). To evaluate whether the failure to distinguish a high-affinity Km value for MPP+ uptake in proteoliposomes could be attributable to inaccessibility of the high-affinity MPP+ binding site in this lipid environment, we reconstituted GFP-rOCT1 also into NDs formed with the PS/PC/cholesterol mixture. Under these conditions, one high-affinity MPP+ binding site per transporter monomer Bmax (high aff.) 1.01 ± 0.19) and two low-affinity MPP+ binding sites per monomer (Bmax (low aff.) 2.06 ± 0.19) were determined as well (Fig. 4). The apparent KD of the high-affinity binding site (0.16 ± 0.07 μM) was similar to NDs formed from POPC (0.23 ± 0.02 μM). At variance, for binding of MPP+ to the two low-affinity sites, a lower common apparent KD value compared with NDs formed from POPC was obtained (9.9 ± 2.8 μM vs. 36 ± 2. 6 μM, P < 0.001, n = 3 each). Noteworthy, the apparent low-affinity KD value for MPP+ binding measured in NDs formed with PS/PC/cholesterol mixture was lower compared with the apparent Km value measured in proteoliposomes formed with the same lipids (see Table 2, 9.9 ± 2.8 μM vs. 19.3 ± 3.1 μM, P < 0.001, n = 3 each).
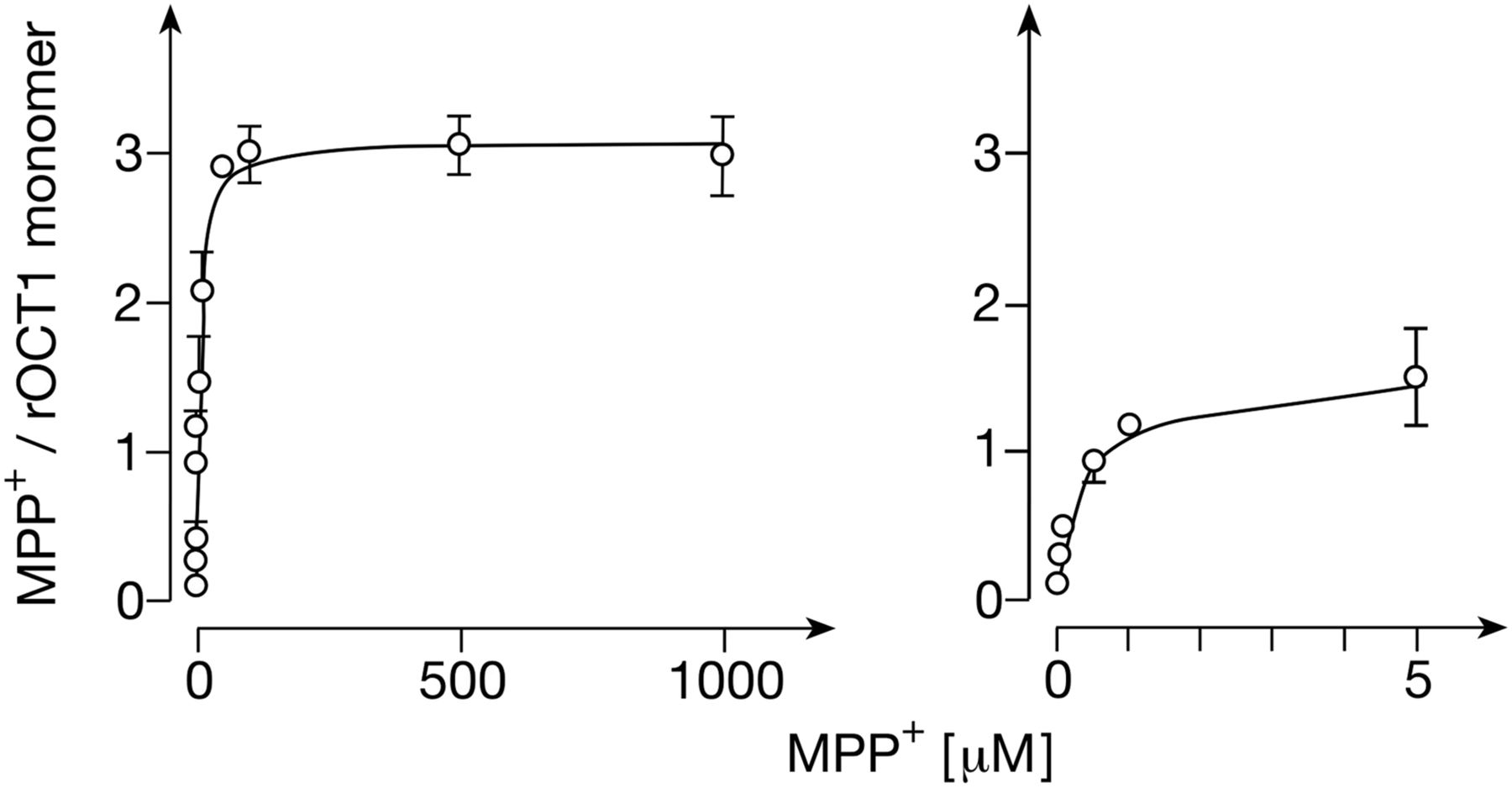
Binding of MPP+ to GFP-rOCT1 in nanodiscs formed with PC, PS, and cholesterol and MSP1E3D1. His-tagged GFP-rOCT1 was reconstituted by cell-free expression in the presence of empty NDs formed from PC, PS, and cholesterol and His-tagged MSP1E3D1. The NDs were precipitated by Ni2+-NTA agarose beads and binding of 12.5 nM [3H]MPP+ was measured at 37°C in the presence of various concentrations of unlabeled MPP+. MPP+ binding was measured and numbers of transporter monomers were determined as in Fig. 2B. Mean values ± S.D. of three experiments are indicated. A two-site binding model was fitted to the data. In the right panel, MPP+ binding and the fitted curve at low MPP+ concentrations are shown.
Evidence that Arg440 Is Involved in MPP+-Binding to a Different Low-Affinity MPP+ Binding Site than Trp218 and Asp475.
To determine whether the mutations studied in NDs formed with DMPG exhibit effects on the high-affinity MPP+ binding site, we also measured MPP+ binding after reconstitution of the mutants in NDs formed with POPC (Fig. 3, B, D, and E; Fig. 5A; Table 2). In NDs formed with POPC the mutations F160Y, W218Y, D475E, and the double mutant W218Y/D475E exerted similar effects on low-affinity MPP binding as in NDs formed with DMPG. Bmax of low-affinity MPP+ binding per monomer was reduced from about two to one in mutants W218Y, D475E, and W218Y/D475E, and the affinity for low-affinity binding was decreased in mutant F160Y. Bmax for high-affinity binding remained unaltered. The apparent KD for high-affinity binding of MPP+ was increased in mutant W218Y, suggesting an allosteric effect of this mutation on the high-affinity MPP+ binding site.

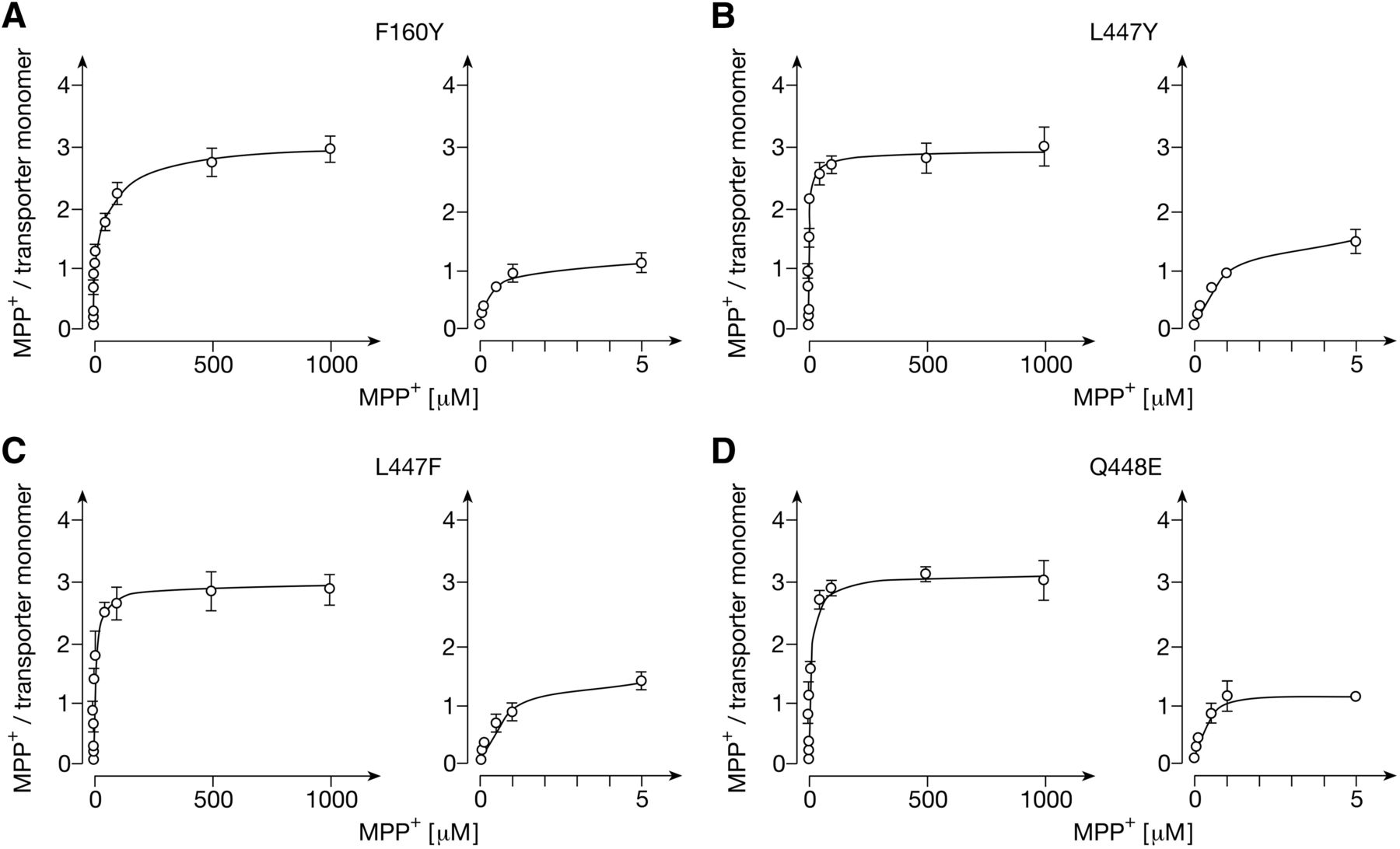
Binding of MPP+ to nanodiscs composed of POPC and MSP1E3D1 containing GFP-rOCT1 mutants with amino acid exchanges in positions 160 (A), 447 (B,C), and 448 (D). His-tagged GFP-rOCT1 mutants were reconstituted by cell-free expression in the presence of empty NDs formed from POPC and His-tagged MSP1E3D1. The NDs were precipitated by Ni2+-NTA agarose beads and binding of 12.5 nM [3H]MPP+ was measured at 37°C in the presence of various concentrations of unlabeled MPP+. MPP+ binding was measured and numbers of transporter monomers were determined as in Fig. 2B. Mean values ± S.D. of three experiments are indicated. A two-site binding model was fitted to the data. In the right panels, MPP+ binding and the fitted curves at low MPP+ concentrations are shown.
Trying to localize the second low-affinity and the high-affinity MPP+ binding site, we also analyzed mutations of Arg440, Leu447, and Gln448 which also have been localized within the outward-open cleft of our rOCT1 model (Volk et al., 2009; Gorboulev et al., 2018) (Fig. 3C; Fig. 5, B–D; Table 2). In mutants L447Y, L447F, and Q448E, maximal binding to the low- and high-affinity binding sites per monomer was not altered; however, the apparent KD values for low-affinity binding were decreased. The data indicate that Leu447 and Gln448 are not essential for low- or high-affinity binding, but that mutations in these positions may exhibit allosteric effects on MPP+ binding sites. After replacement of Arg440 by lysine, Bmax of low-affinity binding was decreased from 2.29 ± 0.05 to 1.24 ± 0.06 per monomer (Fig. 3C; Table 2). The common apparent KD value for low-affinity binding of MPP+ was doubled, whereas high-affinity binding remained unchanged. To determine whether the R440K mutation blunted MPP+ binding to a low-affinity site different from the D475E mutation, we measured MPP+ binding to double mutant R440K/D475E (Fig. 3F; Table 2). Noteworthy, in this mutant no low-affinity MPP+ binding was detectable, whereas Bmax per transporter monomer of high-affinity MPP+ binding was not altered.
Evidence That Both Low-Affinity MPP+ Binding Sites Are Involved in Transport.
To elucidate the relevance of the MPP+ binding sites for transport, we reconstituted cell-free-expressed GFP-rOCT1 wild-type and GFP-rOCT1 mutants into proteoliposomes and measured the substrate dependence of MPP+ uptake. The proteoliposomes were formed from PS, PC, and cholesterol, and MPP+ uptake was measured after a 1-second incubation at 37°C in the presence of an inside-negative potassium diffusion potential, as described for rOCT1 (Keller et al., 2008). As with rOCT1 in proteoliposomes (Keller et al., 2005, 2008) and rOCT1 expressed in oocytes or human embryonic kidney (HEK) 293 cells (Busch et al., 1996; Gorboulev et al., 2018), for GFP-rOCT1 in proteoliposomes, a hyperbolic concentration dependence was observed that could be fitted to the Michaelis-Menten equation (Fig. 6A). No indication of an additional high-affinity uptake was detectable. For GFP-rOCT1, an apparent Km value of 19 ± 3.1 μM (n = 3) was obtained that was lower than the apparent Km of 35 ± 1.5 μM determined for cell-free-expressed rOCT1 in proteoliposomes (Keller et al., 2008).
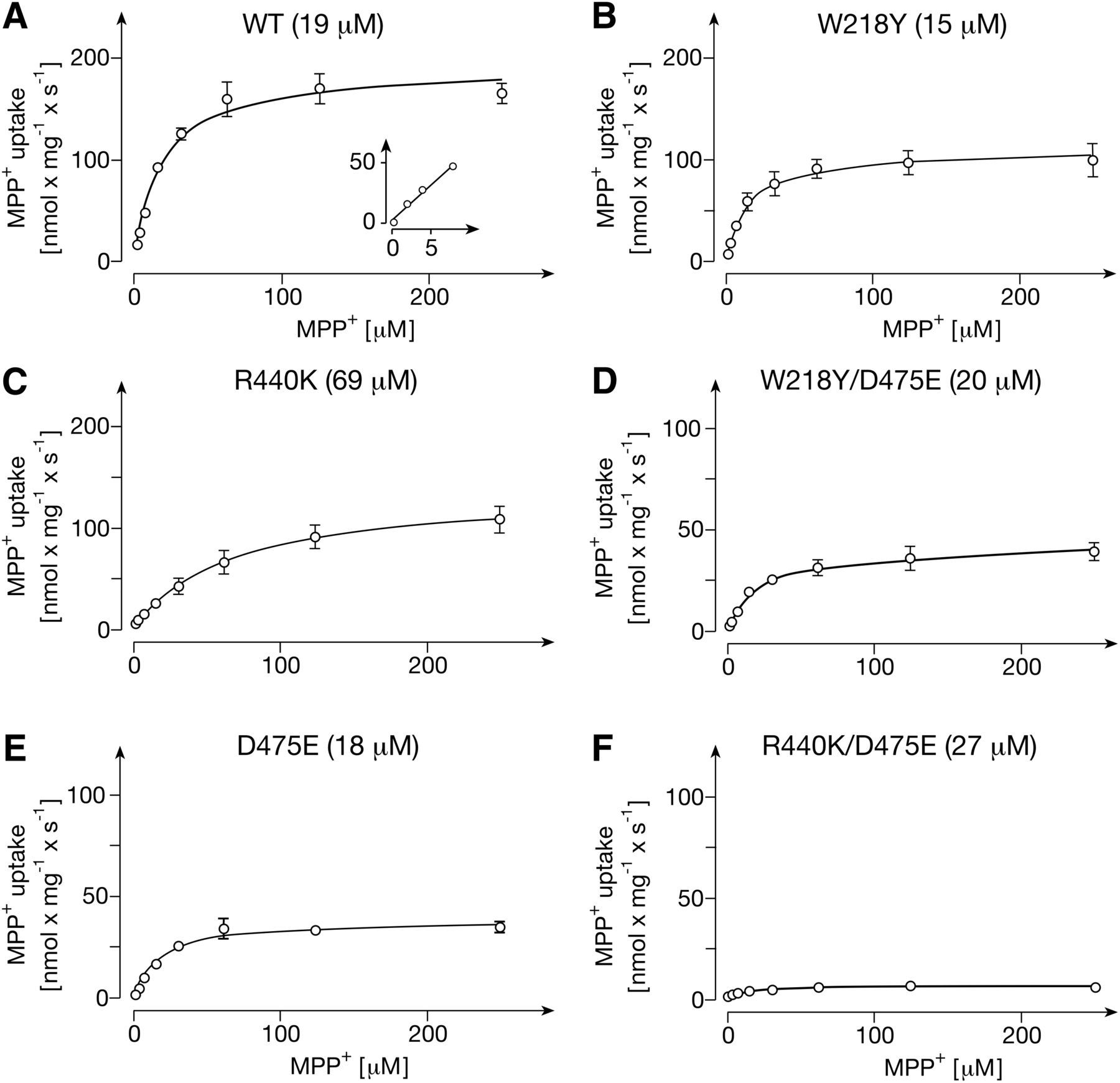
Transport of MPP+ into proteoliposomes containing GFP-rOCT1 wild-type (A) or mutants with amino acid exchanges in positions 218 (B), 440 (C), 475 (E), 218 and 475 (D), and 440 and 475 (F). His-tagged GFP-rOCT1 (WT) or GFP-rOCT1 mutants were reconstituted into proteoliposomes formed with cholesterol, PC, and PS. Uptake of 12.5 nM [3H]MPP+ into proteoliposomes was measured after a 1-second incubation at 37°C in the presence of an outward-directed K+-diffusion potential and various concentrations of unlabeled MPP+. Nonspecific uptake measured in the presence of quinine was subtracted. Mean values ± S.D. of three experiments are indicated. The Michaelis-Menten equation was fitted to the data. The apparent Km values are indicated in parenthesis.
In mutants W218Y and R440K, where binding of MPP+ to one or the other low-affinity binding site was largely impaired, and which led to 41% (W218Y) and 46% (R440K) decrease in total low-affinity binding, the Vmax of MPP+ transport was reduced by 44% (W218Y) and 29% (R440K) (Fig. 6, B and C; Fig. 7A; Table 2). When the Trp218- and Asp475-related binding site was blocked by exchange of Asp475 with glutamate, total low-affinity binding was decreased by 44%, similar to the W218Y mutation; however, a more pronounced 79% decrease of Vmax was observed (Fig. 6E; Fig. 7A; Table 2). The more pronounced effect on Vmax reflects the dual role of Asp475 for substrate affinity and transport-related conformational changes (Egenberger et al., 2012). The double mutant W218Y/D475E exhibited properties similar to the D475E mutant, showing a 54% reduction in low-affinity binding and a 78% reduction in Vmax (Fig. 6D; Fig. 7A; Table 2). In double mutant R440K/D475E, for which no low-affinity binding was detected, the Vmax was reduced by 96% (Fig. 6F; Fig. 7A; Table 2). The data indicate that MPP+ binding to the two identified low-affinity sites is critically involved in translocation.

Comparison between kinetic constants of GFP-rOCT1 wild-type and mutants with amino acid exchanges in positions 218, 440, and 475 determined for MPP+ uptake into proteoliposomes and for MPP+ binding to NDs formed with POPC. (A) Comparison between Vmax values for MPP+ uptake and Bmax values for MPP+ binding. (B) Comparison between apparent Km values for MPP+ uptake and apparent KD values for low-affinity MPP+ binding. The compiled data are presented in Figs. 3 and 6. The compared mean ± S.D. values were calculated from three individual experiments. The values are shown in Table 2. (*P < 0.05; ***P < 0.001; significance of differences between Vmax and Bmax values and between apparent Km and apparent KD (low-aff.) values of individual mutants that were determined by two-way ANOVA using Bonferroni correction.)
In mutants W218Y, D475E, and W218Y/D475E, the apparent Km value for MPP+ uptake was not altered significantly, whereas in mutant R440K, the apparent Km value was increased 3-fold (Fig. 7B). The effects on apparent Km have some similarity to the effects on the common apparent KD (low aff.) value, which was not altered in mutant D475E, decreased by 42% in mutants W218Y and W218Y/D475E, and 2.3-fold increased in mutant R440K. The small differences between the effects of the R440K and W218Y/D475E mutations on apparent Km versus apparent KD (low aff.) values (Fig. 7B) may be revealing the difference in lipid composition between the NDs and proteoliposomes and/or that the affinity for substrate binding may not exclusively determine the Km for translocation.
In mutants F160Y, L447Y, L447F, and Q448E no significant changes in the determined total number of three binding sites per transporter monomer and the Vmax values for MPP+ transport were observed (Figs. 5 and 8; Table 2). However, apparent KD (low aff.) and apparent Km values were increased in mutant F160Y and decreased in mutants L447Y and L447F. The data support the interpretation that the investigated mutations of Phe160 and Leu447 induce allosteric effects on MPP+ binding to the low-affinity sites that are critically involved in translocation.
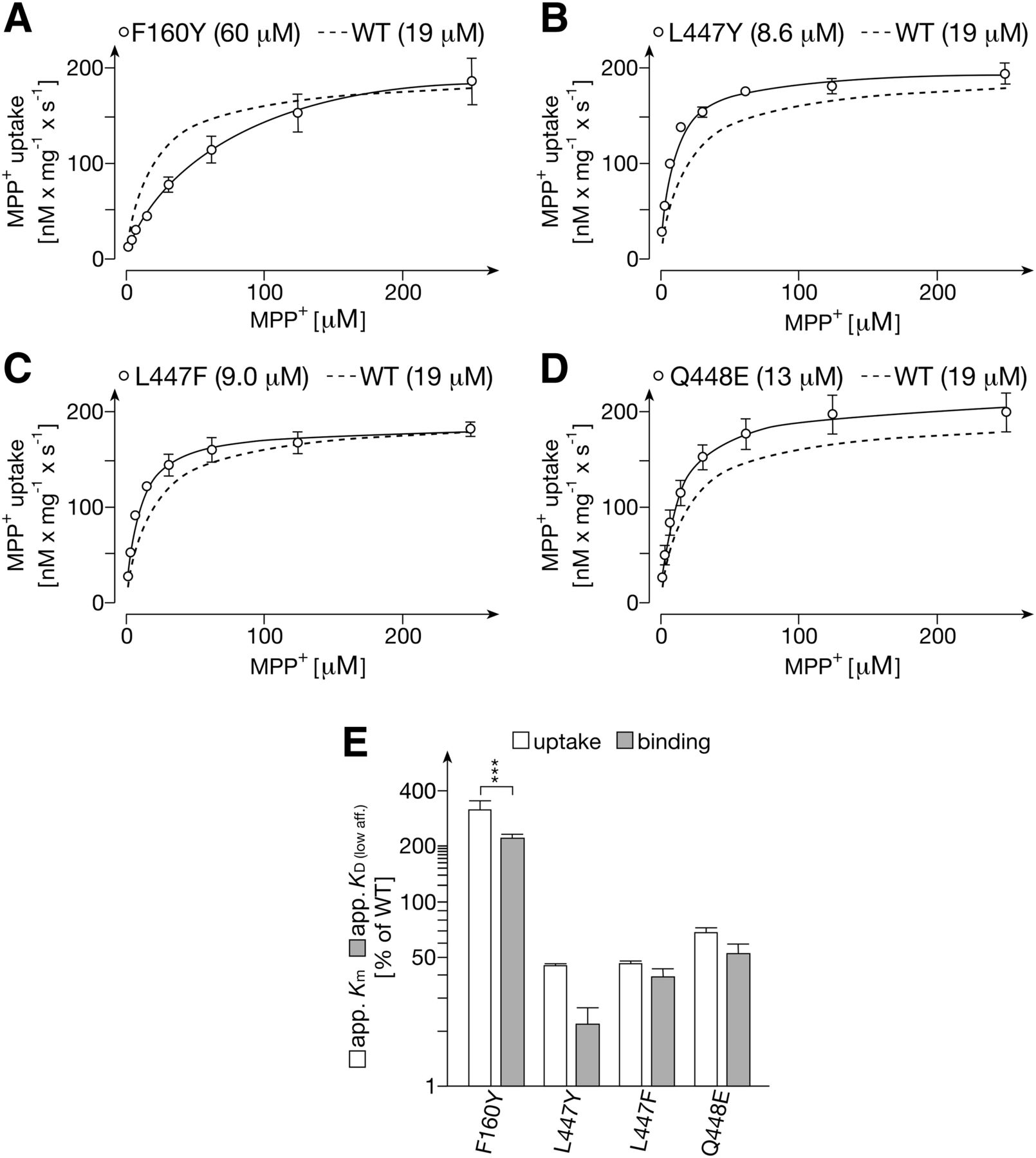
Transport of MPP+ into proteoliposomes containing GFP-rOCT1 mutants with exchanges of amino acids in positions 160, 447, and 448 (A–D) and comparison between apparent Km values for uptake and apparent KD (low aff.) values for binding (E). GFP-rOCT1 mutants were reconstituted into proteoliposomes formed with PC, PS, and cholesterol. Uptake of 12.5 nM [3H]MPP+ into proteoliposomes was measured after a 1-second incubation at 37°C in the presence of an outward-directed K+-diffusion potential and various concentrations of unlabeled MPP+. (A–D) The Michaelis-Menten equation was fitted to the compiled data sets. The curve obtained for the concentration dependence of MPP+ uptake in proteoliposomes containing GFP-rOCT1 (WT) is indicated for comparison. The apparent Km values (see Table 2) are indicated in parenthesis. (E) Comparison of apparent Km values for MPP+ uptake into proteoliposomes with apparent KD values for low affinity MPP+ binding to NDs formed with POPC. Mean values ± S.D. from three experiments are indicated. Significance of difference between apparent Km and apparent KD (low aff.) values of individual mutants were determined by two-way ANOVA with Bonferroni correction (***P < 0.001). For mutant GFP-rOCT1(L447Y) significance of a 2-fold difference between apparent Km and apparent KD (low aff.) was not indicated by ANOVA but by Student’s t test (P < 0.01).
Docking of MPP+ to Modeled Outward-Open and Inward-Open Clefts of rOCT1.
Previously, we generated a 3D homology model of the inward-open conformation of rOCT1 on the basis of the crystal structure of lactose permease from E. coli that belongs to the MFS superfamily like the OCTs (Popp et al., 2005). In addition, we also built a 3D model of rOCT1 in the outward-open conformation by assuming a rigid-body movement of the six N-terminal helices of rOCT1 with respect to the six C-terminal helices, a model similar to that generated modeled and experimentally verified for lactose permease (Holyoake and Sansom, 2007; Kaback et al., 2007; Volk et al., 2009). Using as templates the crystal structures of various transporters of the MFS superfamily, an outward-open 3D model of human OCT1 (hOCT1) was built (Dakal et al., 2017). In this hOCT1 model, the locations of Phe160, Trp218, Arg440, and Asp475 within the outward-open cleft of rOCT1 were confirmed (Gorboulev et al., 2018). In the outward-open models of rOCT1 and hOCT1 Trp218 (Trp217 in hOCT1) and Asp475 (Asp474) were in close proximity, whereas Arg440 (Arg439) was located more distantly to the two other residues. This is consistent with our mutagenesis data suggesting that Asp475 and Trp218 contribute to the same low-affinity MPP+ binding site, whereas Arg440 is important for integrity of the second low-affinity MPP+ binding site. In the inward-open cleft model of rOCT1, Asp475 is located distantly from Trp218 and Arg440 (Volk et al., 2009). This suggests that in the reconstituted NDs, MPP+ binding to the outward-open conformation has been analyzed. Trying to obtain additional insight on MPP+ binding close to Trp218, Arg440 and Asp475, we performed in silico docking simulations of MPP+ into the outward-facing and inward-facing clefts of our 3D homology models of rOCT1. To screen potential binding sites close to the above listed residues, docking was restrained to within a 7-Å radius around these residues. The docking simulations have been deposited at the Protein Model Database (see end of paragraph).* The 30 docked MPP+ molecules with the highest docking energies in both conformations were analyzed (Fig. 9). In the outward-open and inward-open rOCT1 conformations, 26 and 22 molecules were found docked within the cleft, respectively. In the outward-open cleft 18 MPP+ molecules were located close to Trp218 and Asp475, whereas in the inward-open conformation 10 MPP+ were close to both Trp218 and Asp475. Hence, the docking experiments did not allow us to decide whether low-affinity binding of two MPP+ molecules to the outward-open or inward-open cleft was determined by the binding measurements of GFP-rOCT1 in NDs. [*Protein Model database (https://bioinformatics.cineca.it/PMDB/main.php): accession no. PM0081570, MPP+ docking to the outward-open conformation of rOCT1; accession no. PM0081569, MPP+ docking to inward-open conformation of rOCT1.]

Docking of MPP+ to the modeled outward-open and inward-open binding cleft of rOCT1. Docking was performed to previously built 3D homology models of rOCT1. Docking of MPP+ molecules was restrained to occur within spheres of 7 Å around Trp218, Arg440, and Asp475. Thirty MPP+ molecules with the highest docking energies for each transporter conformation were selected. Twenty-six and 22 molecules were found to be located inside the outward-open and inward-open cleft, respectively. Owing to highly similar docking positions, not all MPP+ moieties are shown in the presentations.
Discussion
On the basis of mutagenesis experiments in rOCT1 combined with binding and transport measurements of the model cation MPP+, our study provides novel insight into recognition and transport of drugs by polyspecific organic cation transporters (OCTs) of the SLC22 transporter family. The experiments were performed with cell-free-expressed GFP-rOCT1 fusion protein that was reconstituted into NDs and proteoliposomes. Evidence is provided that each rOCT1 monomer contains two low-affinity MPP+ binding sites that are involved in MPP+ transport and one high-affinity MPP+ binding site that does not directly participate in transport but may mediate allosteric effects on the low-affinity MPP+ binding sites. It cannot be excluded that the determined numbers of MPP+ binding sites per transporter monomer are underestimated, because it has not been proven experimentally that all transporter monomers in the NDs are functional. However, functionality of all transporters in our preparation is probable, because the reconstitution of GFP-rOCT1 into NDs was performed by cotranslational insertion during cell-free expression of the transporter in the absence of detergent (Roos et al., 2013). Thus, post-translational protein modifications and protein aggregation causing protein heterogeneities were avoided. More importantly, because only about 1% of the NDs contained GFP-rOCT1, a homogeneous population of NDs containing functional transporter is expected.
The identified high-affinity MPP+ binding site was only accessible in specific lipid environments, namely in NDs formed with POPC or a mixture of cholesterol, PC, and PS, but not with DMPG or DPPC. The lipid dependence suggests that a minimal membrane fluidity (POPC vs. DPPC) and/or an optimal membrane thickness (DMPG vs. POPC) is required for an in vivo-like transporter conformation with an accessible high-affinity binding site. Phospholipid binding to a lipid binding site of rOCT1 as has been described for some transporters (Laganowsky et al., 2014), is also possible.
The involvement of individual low-affinity sites in transport can be concluded from the observed parallel decrease in Bmax of MPP+ binding and Vmax of MPP+ transport after inactivation of one or the other low-affinity MPP+ binding site in mutants W218Y and R440K, as well as the total absence of uptake after inactivation of both low-affinity binding sites in the double mutant R440K/D475E. A direct involvement of the high-affinity MPP+ binding site in transport could be excluded because Bmax of high-affinity MPP+ binding remained unchanged when both low-affinity MPP+ binding sites were blocked and MPP+ transport was abolished. In addition, neither a high-affinity MPP+ transport site nor positive cooperativity could be detected by measuring the substrate dependence of MPP+ uptake in proteoliposomes (Fig. 6A) or in those rOCT1-expressing oocytes in which high-affinity MPP+ binding sites have been identified (Busch et al., 1996; Gorbunov et al., 2008).
The properties of rOCT1 we discovered provide a molecular framework for understanding how various factors can influence the affinities of inhibitors. For example, for inhibition of MPP+ uptake into rOCT1-expressing HEK293 cells by a nontransported inhibitor, widely divergent IC50 values were obtained when different concentrations of MPP+ far below the apparent Km for MPP+ uptake were used for uptake measurements (Gorboulev et al., 2018). Under these conditions only one low-affinity MPP+ binding site is supposed to be involved in transport and a different occupation is assumed for the high-affinity MPP+ binding site. A high complexity of drug-drug interaction should then be anticipated, considering that OCTs contain both allosteric high-affinity cation binding sites (Gorbunov et al., 2008; Minuesa et al., 2009) as well as partially overlapping, transport-related, low-affinity cation binding sites, and that cationic drugs with largely different structures may interact with the high- and low-affinity sites. (Koepsell, 2013). Hence, the procedures recommended by the U.S. Food and Drug Administration (US Food and Drug Administration, 2012, 2017) and European Medicine Agencies and in the literature to determine whether NMEs interact with hOCT1 or hOCT2 by measuring inhibition of uptake of one concentration of MPP+ or metformin as test substrate are insufficient for drug development, because effects of substrate structure and functionally relevant high-affinity inhibition are missed (Koepsell, 2018). Considering the complexity of drug interactions with OCTs, more sophisticated approaches are required. Hence, we suggest employing three structurally diverse substrates including a clinically relevant compound at three different concentrations far below the respective Km values for in vitro testing of NMEs by inhibition experiments. Pharmacophore models based on the different experimental conditions may be generated and applied one after the other to achieve a predictive potential that is high enough to justify in silico exclusion of NMEs from in vitro testing. Such a line of action is mandatory for in silico prediction, because pharmacophore models so far described for hOCT1 and hOCT2 are insufficient, because they are based on uptake measurements performed at a single substrate concentration that is close to the respective Km value. By these models only 70%–82% of the interacting drugs have been identified (Koepsell, 2018).
The current understanding of the molecular mechanism by which human polyspecific drug transporters recognize compounds with different molecular structures has been limited by its reliance on modeling and on indirect experimental evidence. So far, crystal structures of mammalian polyspecific transporters and of transporter-ligand complexes have been missing, and homology models derived from the crystal structures of bacterial transporters belonging to the same superfamily and docking experiments of ligands to homology models have provided only limited information. Extensive mutagenesis studies have been performed only with a few polyspecific drug transporters to measure the effects of mutations on apparent Km values of substrates and on IC50 values for inhibition of transport. These measurements depicted secondary effects of the mutations rather than direct effects on substrate- or inhibitor binding. Recently, crystal structures of four dipeptide complexes with different conformations of the proton-dependent peptide transporter PepTStT from Streptococcus thermophiles were reported (Martinez Molledo et al., 2018). The data suggest that binding of different dipeptides to PepTST occurs at the same binding site and is enabled by movements of the interacting amino acid residues in combination with adjustments of dipeptide positions and interposition of water molecules. Remarkably, the EC50 values obtained for the interaction of the cocrystallized dipeptides exhibiting different but not completely divergent structures from solubilized PepTStT ranged between 0.56 and >50 mM. In contrast, organic cations with completely different molecular structures, such as TBuA+, metformin+, and MPP+, interacting with OCTs had EC50 values that ranged between subnanomolar and millimolar concentrations. Consistent with this different degree of polyspecificity between proton-peptide cotransporters and OCTs, we have provided evidence for the existence of different cation binding sites in rOCT1. Each of these binding sites in rOCT1—and presumably different binding sites in all OCTs—may recognize a group of structurally related compounds employing mechanisms as described for PepTStT. In total this allows the recognition of a large variety of structurally diverse compounds by OCTs.
By measuring binding of MPP+ to GFP-rOCT1 in NDs, binding to low-affinity binding sites within either the outward-open or the inward-open cleft of the transporter can be determined. On the basis of homology modeling and docking data, no distinction between binding to the outward-open and inward-open cleft can be made. Likewise, the determined apparent Km values for MPP+ uptake and the apparent KD values for low-affinity MPP+ binding do not allow a distinction between binding to the outward-open or inward-open cleft because we do not know whether rOCT1-mediated cellular influx and efflux of MPP+ are asymmetric and have different Km values for MPP+ transport and/or different KD values for low-affinity MPP+ binding. The observation that apparent Km values of 35 and 19 μM for MPP+ uptake in proteoliposomes containing cell-free expressed rOCT1 (Keller et al., 2008) or GFP-rOCT1 were higher than the apparent Km values measured for rOCT1-mediated MPP+ uptake in oocytes (4.9 μM) and HEK293 cells (3.9 μM) could indicate that rOCT1 in the proteoliposomes is oriented inside-out and the Km for MPP+ efflux is higher compared with influx. However, different properties of rOCT1 in proteoliposomes compared with rOCT1 in native plasma membranes cannot be excluded. Because a high-affinity MPP+ binding site on rOCT1 expressed in oocytes was identified after short-term extracellular application of MPP+ (Gorbunov et al., 2008), it is highly probable that the detected high-affinity MPP+ binding site is accessible extracellularly. The experiment docking MPP+ to the outward-open cleft is consistent with the interpretation that the high-affinity and the two low-affinity MPP+ binding sites are located within the outward-open cleft.
In summary, the data described in this paper provide a rationale for improving in vitro and in silico testing of NMEs for interaction with OCTs. From the demonstration of three different binding sites for one substrate in one rOCT1 monomer, in combination with the property of OCTs to translocate structurally diverse compounds, we conclude that these highly polyspecific transporters contain various binding sites that are mostly or exclusively located within large clefts that may have both extracellular or intracellular exposure. Future attempts to crystallize transporters of the SLC22 family may be successful if performed in the presence of structurally different ligands and lipids containing unsaturated fatty acids and/or cholesterol. The demonstration of one high-affinity and two low-affinity binding sites in rOCT1 and the identification of amino acids that participate in formation of the low-affinity binding sites will help to interpret the crystal structures obtained.
Acknowledgments
We thank Martin Lohse (Institute of Pharmacology and Toxicology, University Würzburg) for providing laboratory space and giving technical support and Michael Christof (Institute of Anatomy and Cell Biology, University Würzburg) for preparing the figures.
Authorship Contributions
Participated in research design: Keller, Mueller, Bernhard, Koepsell.
Conducted experiments: Keller, Gorboulev, Mueller, Bernhard.
Contributed new reagents or analytic tools: Bernhard, Dötsch.
Performed data analysis: Keller, Mueller, Koepsell.
Wrote or contributed to the writing of the manuscript: Koepsell.
Footnotes
- Received June 29, 2018.
- Accepted November 5, 2018.
This work was supported by the Deutsche Forschungsgemeinschaft (KO 862/6-1).
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- 3D
- three-dimensional
- ANOVA
- analysis of variance
- Bmax
- maximal binding
- CHAPS
- 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate
- DMPG
- 1,2-dimyristoyl-sn-[phospho-rac-(1-glycerol)]
- DPPC
- 1,2-dipalmitoyl-sn-glycero-3-phosphocholine
- EC50
- half-maximal effective concentration
- GFP
- green fluorescent protein
- HEK
- human embryonic kidney
- hOCT
- human OCT
- Km
- Michelis-Menten constant
- MFS
- major facilitator superfamily
- MPP+
- 1-methyl-4-phenylpyridinium+
- MSP
- major scaffold protein
- ND
- nanodisc
- Ni2+-NTA
- Ni2+-nitriloacetic acid
- NME
- novel molecular entity
- OCT
- organic cation transporter
- PBS
- phosphate buffered saline
- PC
- 1,2-diacyl-sn-glycero-3-phospho-L-serine
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- PS
- 1,2-diacyl-sn-glycero-3-phospho-L-serine
- rOCT
- rat organic cation transporter
- TBuA+
- tetrabutylammonium+
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}