


Abstract
The α7 nicotinic acetylcholine receptor (nAChR) is a potential drug target for the treatment of a number of neurologic and inflammatory disorders. Silent agonists are an emerging class of drugs that bind to the receptor but do not open the channel. Instead they shift the receptor to a desensitized state. Silent agonists may be able to target a subset of α7 nAChR–mediated signaling processes. Here we use noncanonical amino acid mutagenesis to characterize the binding to α7 by the silent agonist 1,4-diazabicyclo[3.2.2]nonan-4-yl(5-(3-(trifluoromethyl)phenyl)furan-2-yl)methanone (NS6740). We find that, like α7 agonists, NS6740 forms a cation-π interaction with Y115 (TyrA). We also showed that NS6740 makes a novel hydrogen bond to TyrA. This interaction is necessary for the silent agonist activity of NS6740; when the hydrogen bond is blocked, silent agonist NS6740 converts to a conventional partial agonist and appreciably opens the channel in the absence of a positive allosteric modulator (EC50 150 nM).
Significance Statement Noncanonical amino acids were used to show that a hydrogen bond to tyrosine (Y115) is required for silent agonist activity of NS6740 at the α7 nicotinic acetylcholine receptor.
Introduction
The homopentameric α7 nicotinic acetylcholine receptor (nAChR) is a ligand gated ion channel characterized by low opening probability due to rapid, agonist concentration-dependent desensitization (Williams et al., 2011, 2012; Papke, 2014; Corradi and Bouzat, 2016). The α7 nAChR is the second most abundant nAChR in the brain and is expressed both synaptically and nonsynaptically, as well as in non-neuronal cells (Corradi and Bouzat, 2016). It has been implicated as a drug target for the treatment of schizophrenia, pain, and inflammation, among other neurologic disorders (Dineley et al., 2015; Egea et al., 2015).
Classically, α7 nAChR-mediated signaling occurs because ligand binding triggers the channel to open, allowing ion influx and generating an electric current across the membrane. Recent studies suggest that α7 may exhibit ligand-mediated metabotropic signal transduction, independent of ion channel opening (King et al., 2015; Kabbani and Nichols, 2018). One way to potentially study metabotropic signaling occurring from a desensitized state is by using silent agonists.
Silent agonists are a novel class of drugs that, upon binding, do not open the channel but rather induce a conformational change stabilizing a desensitized state (Fig. 1). This can be confirmed by the coapplication of a type 2 positive allosteric modulator (PAM) such as 1-(5-chloro-2,4-dimethoxy-phenyl)-3-(5-methyl-isoxazol-3-yl)-urea (PNU-120596), which shifts the equilibrium toward a conducting open state, presumably by destabilizing the desensitized state and/or stabilizing the open state. It has been proposed that the desensitized and open states associated with a silent agonist are different from those associated with a conventional agonist (Papke et al., 2014, 2018). Silent agonists should not be thought of as competitive antagonists. Competitive antagonists simply block agonist binding and do not induce a conformational change that is sensitive to binding of a type 2 PAM.
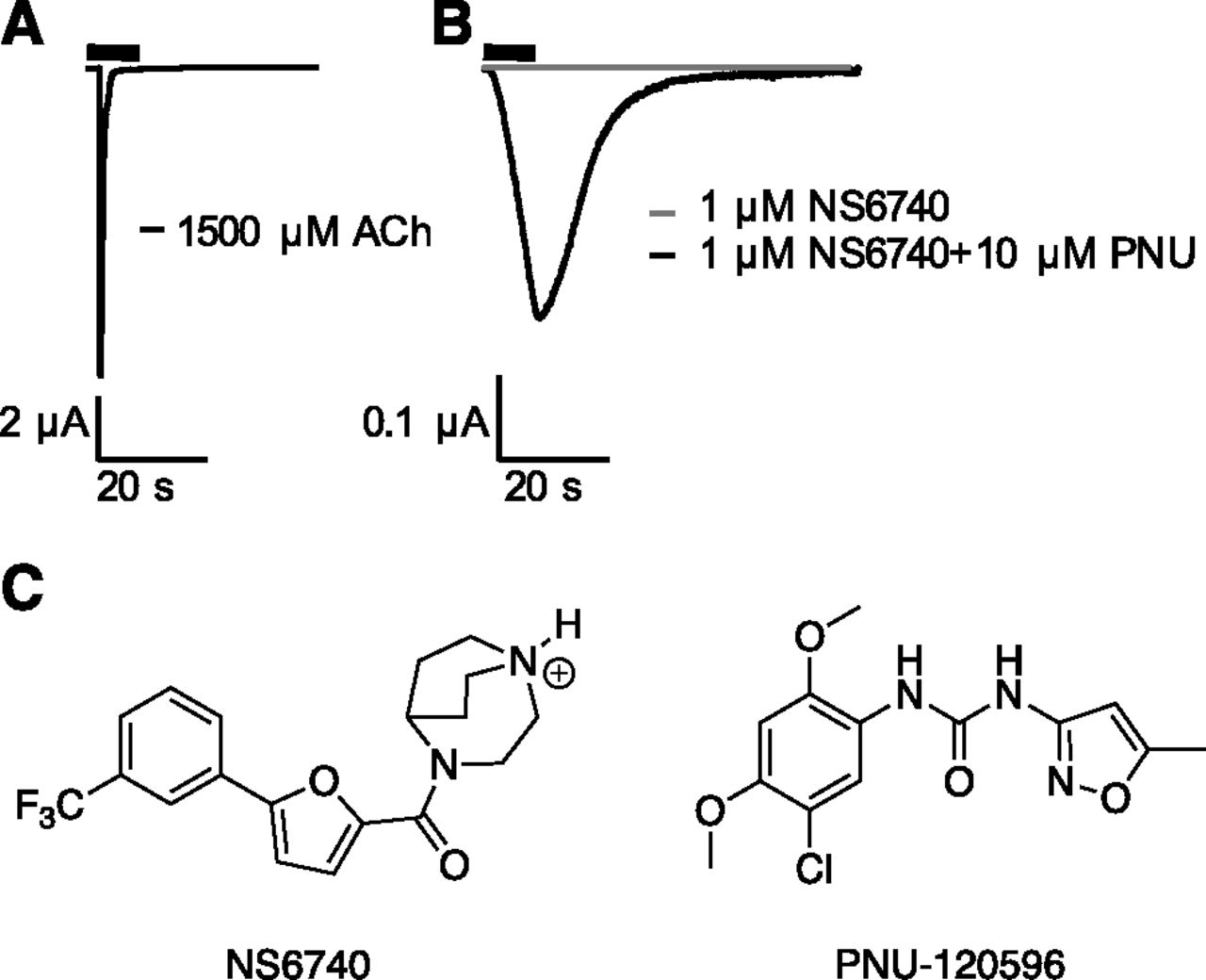
Silent agonist activity. (A) Representative current trace for the application of acetylcholine (ACh) on α7. (B) Representative current trace shapes for application of the silent agonist, NS6740, on α7 and the coapplication of NS6740 and the type 2 positive allosteric modulator (PAM) PNU-120596. (C) Structures of NS6740 and PNU-120596.
The first silent agonist to be characterized, 1,4-diazabicyclo[3.2.2]nonan-4-yl(5-(3-(trifluoromethyl)phenyl)furan-2-yl)methanone (NS6740) (Briggs et al., 2009), has been shown in vitro to modulate the inflammatory response of microglia cells as part of the cholinergic anti-inflammatory pathway (Thomsen and Mikkelsen, 2012) and to reduce the symptoms of neuropathic pain in mouse models (Papke et al., 2015). Another silent agonist, PMP-072 [(R)-N-(4-methoxyphenyl)-2-((pyridin-3-yloxy)methyl)piperazine-1-carboxamide], was shown in mouse models to have antiarthritic effects (van Maanen et al., 2015). The fact that these drugs are able to induce a physiologic response despite not being able to open the channel supports the proposed metabotropic signaling pathway for α7. If true, silent agonists represent novel therapeutic agents for the selective targeting of a specific subset of α7-meditated signaling pathways, such as those related to chronic inflammation and neuropathic pain (Horenstein and Papke, 2017).
Little is known about the core pharmacophore of silent agonists, and a better understanding of the binding of silent agonists could guide the design of new silent agonists. So far all characterized silent agonists at α7 nAChR have a cationic nitrogen, either a protonatable amine or a quaternary ammonium, and are believed to bind at the orthosteric site (Papke et al., 2015). Initial studies on alkyl ammonium ligands showed that increasing bulk around the cation reduced agonist activity while maintaining desensitization activity (Papke et al., 2014; Quadri et al., 2016). NS6740 has a bulky bicyclo[3.2.2] ring system, but derivatives of NS6740 lacking the bulky bicyclic ring system still exhibit silent agonist activity (Chojnacka et al., 2013).
There is currently no atomic scale structural information for the full α7 receptor, let alone in the open, closed, and desensitized states. The α7-chimeric acetylcholine binding protein (AChBP) crystal structures give some insight into the orthosteric binding site (Li et al., 2011; Nemecz and Taylor, 2011). However, it is unclear if these represent an open or desensitized state, and there is no crystal structure of a silent agonist bound. High-resolution electrophysiology can be used to gain vital structure-function information on ion channels. However, silent agonists require a PAM to generate an electrophysiological signal, complicating matters.
This study uses noncanonical amino acid mutagenesis (Fig. 2) to probe the binding of the silent agonist NS6740 at the orthosteric site of the α7 nAChR (Dougherty, 2013, Van Arnam and Dougherty, 2014) and compares the results to previous work from our laboratory on the binding of typical agonists such as acetylcholine (ACh) and nicotine (Marotta et al., 2015). We find that the silent agonist NS6740 makes a cation-π interaction to TyrA and a never before characterized, functionally significant hydrogen bond with TyrA.

Structures of the side chains of noncanonical amino acids used in this work. (A) Tyrosine derivatives. (B) Tryptophan derivatives. (C) Backbone amide to ester mutation strategy for perturbing a hydrogen bond.
Materials and Methods
Molecular Biology.
The rat nAChR α7 subunit (pAMV vector), Ric3 (pAMV vector), and NACHO (pAMV vector), were used as the bases for all constructs. Residue numbering was based on the full-length protein containing the signaling sequence as found on the UniProt database (Q05941). Site-directed mutagenesis was performed by polymerase chain reaction using the Stratagene QuikChange protocol with primers from Integrated DNA Technologies (Coralville, IA). The circular cDNA plasmids were linearized with NotI-HF (New England BioLabs, Ipswich, MA) and purified (Qiagen, Valencia, CA). DNA was then transcribed in vitro using the T7 mMessage Machine kit (Ambion, Austin, TX), and the mRNA was isolated using the RNeasy purification kit (Qiagen). The Amber (UAG) stop codon was used for incorporating noncanonical amino acids in the α7 subunit. The 74-nucleotide THG73 transfer RNA (tRNA) and 76-nucleotide THG73 tRNA were in vitro transcribed using the MEGAshortscript T7 (Ambion) kit and isolated using CHROMA SPIN DEPC-H2O columns (Clontech, Mountain View, CA). The final concentrations were determined by UV-visible.
Oocyte Preparation and Injection.
Xenopus laevis oocytes (stage IV and V) were retrieved as described elsewhere (Nowak et al., 1998). For conventional mutagenesis experiments, α7 and Ric3 mRNA were mixed 1:1 by weight, and the oocytes were injected with 50 nl of solution containing 20 ng mRNA. Cells were incubated at 18°C for 24 hours in ND96 solution (96 mM NaCl, 1.8 mM CaCl2, 2 mM KCl, 1 mM MgCl2, 5 mM HEPES at pH 7.5) enriched with theophylline (6.7 mM), sodium pyruvate (2.5 mM), and kanamycin (0.1 mg/ml). Kanamycin was used because other antibiotics have been shown to reduce α7 expression levels (Amici et al., 2005).
Noncanonical Amino Acid Incorporation.
The NVOC protected, cyanomethylester form of the noncanonical amino acid was coupled to the dinucleotide dCA and enzymatically ligated to the UAG-suppressor 74-mer THG73 tRNACUA. The product was verified via matrix-assisted laser desorption/ionization (MALDI) time of flight mass spectrometry using a 3-hydroxypicolinic acid matrix (Nowak et al., 1998). The noncanonical amino acid-coupled tRNA was deprotected with a M365LP1 365 nm 1150 mW LED lamp (Thorlabs, Newton, NJ) immediately before coinjection with mRNA containing a UAG mutation at the site of interest. Then mRNA and tRNA were injected in a 1:1 or 2:1 volume ratio in a total volume of 50 or 75 nl, respectively, so that cells were injected with a 40 ng 1:1 mixture of α7-UAG and Ric3 mRNA.
For noncanonical amino acids that showed little or no response after 24 hours of incubation, oocytes were subjected to a second round of injection and incubation as before. If this procedure was not effective, oocytes were double injected with a 75 nl of solution containing 40 ng mRNA of α7-UAG, Ric3 and NACHO 1:1:1 mixture. A read-through/reaminoacylation test served as a negative control. Full-length unacylated 76-mer tRNA was coinjected with mRNA. Lack of current showed no detectable reaminoacylation at the suppression site.
Drug Preparation.
Acetylcholine chloride (Sigma-Aldrich, St. Louis, MO) was dissolved in ND96 Buffer to make a 1 M stock solution. NS6740 was synthesized as described elsewhere (Papke et al., 2015) and dissolved in DMSO to form a 20 mM stock solution. PNU-120596 (Selleckchem, Houston, TX) was dissolved in DMSO to 150 mM stock. Further dilutions were made for experimentation using ND96 buffer.
Electrophysiology.
Agonist-induced currents were recorded using an OpusXpress 6000A (Axon Instruments/Molecular Devices, Union City, CA) in two-electrode voltage clamp mode at a voltage clamped holding potential of −60 mV. Voltage and current electrodes were filled with 3 M KCl. Oocytes were perfused with ND96 medium at a rate of 3 ml/min. Drug applications consisted of coapplication of a 1-ml dose of the drug solution over 8 seconds followed by a 15-second pause to allow the response to reach maximum before being washed out for 300 seconds at a rate of 3 ml/min (Supplemental Fig. 1; Supplemental Table 1). Concentrations of NS6740 were varied over several orders of magnitude to generate a concentration–response curve. For efficacy experiments, 1 ml of EC100 dose of drug was applied over 8 seconds followed by a 1-minute incubation followed by 10-minute washout.
Data Analysis.
Data were sampled at 50 Hz and then low-pass filtered at 5 Hz. For NS6740 coapplied with PNU-120596, EC50 measurements were recorded as peak height. Data for concentration–response curves were baselined, and normalized on a per cell basis, and then averaged on a per-concentration basis; the Hill equation was fit to the data using Prism 6 (GraphPad Software, San Diego, CA). EC50 and Hill coefficient errors are presented as S.E.
For relative efficacy experiments, ACh doses were normalized to an initial ACh dose, and all other drug applications were normalized to an immediately preceding acetylcholine dose and reported as Rmax = Imax(drug)/Imax(ACh). In data tables, N refers to the total number of oocytes analyzed. Cells from different frogs on at least two different days were used for each point; if N < 10, then cells from a third frog on a 3rd day were injected. Cation-π binding energies were calculated, ab initio (6-311G**), for a gas phase Na+ ion binding to each noncanonical amino acid side chain as described by Duffy (2014).
Results
Measuring Binding Interactions in the Presence of a PAM.
Our metric for measuring the effect of noncanonical amino acid incorporation is the half-maximal effective concentration (EC50). This is a composite measure of several equilibria, including agonist binding and channel gating events. There is some ambiguity about how a given mutation in a protein shifts EC50, affecting either gating or binding. Single channel measurements can provide more detailed kinetic analysis on which equilibrium is being perturbed. However, single channel studies are not feasible in this study, given the number of mutations probed and protein expression limitations for α7.
Given that noncanonical amino acid mutagenesis allows for very subtle and precise modifications of the protein and given our knowledge of the structure of the binding site, it is clear we are probing interactions within the binding site, and that shifts in EC50 are a result of attenuated binding interactions. The ambiguity concerns which of the multiple equilibria is most perturbed by the mutation, but it is clear we are perturbing binding interactions between the drug and the protein.
A complicating factor in this project is the need to coapply a PAM with the silent agonist of interest for a signal to be observed. In ion channels, allosteric modulators can induce a long-range conformational change to the orthosteric binding site, altering the binding affinity of the agonist, or they can modulate the gating transition of the receptor. If the former mechanism were operative, it would complicate analysis of a silent agonist. However, we have previously shown that in α7 the type 2 PAM PNU-120596 does not alter the orthosteric binding site, as determined by detailed interactions with the native agonist ACh (Marotta et al., 2015). This was done by comparing the impact of a mutation on receptor function with or without the coapplication of the PAM. Given that PNU-120596 does not alter the agonist binding site, we can assume that any change from mutations made to the binding site are from an interaction with NS6740 not PNU-120596. As such, studies of NS6740 were performed in the presence of 10 μM PNU-120596, producing robust signals in our Xenopus oocyte assay.
Binding Interaction of TyrA with NS6740.
The cation-π interaction plays a prominent role in binding of natural and synthetic agonists to nAChRs (and other Cys-loop receptors), but α7 is unique in the family in that ACh makes a cation-π interaction with TyrA (Y115, so named because it lies on loop A of the agonist binding site), rather than TrpB (W171), the more commonly used residue (Puskar et al., 2011; Van Arnam and Dougherty, 2014). It is possible to identify a cation-π interaction by progressively fluorinating the aromatic group, reducing the π electron density of the aromatic ring surface, and thus weakening the interaction. When plotted against calculated cation-π binding energies, this results in a “fluorination” plot. This approach also applies to other deactivating substituents on the aromatic side chain, including cyano and bromo functional groups. However, fluorination affects the pKa of the hydroxyl functional group of tyrosine as well as the strength of the cation-π interaction. Therefore, to study tyrosine residues such as TyrA it is necessary to either methylate the hydroxyl group, producing TyrOMe, or remove the hydroxyl group entirely and use Phe derivatives. We employed both strategies here.
The fluorination plot shows a linear relationship indicating that NS6740 forms a cation-π interaction with TyrA (Fig. 3; Table 1). The slope of the fluorination plot (−0.16 ± 0.03) is similar to that of acetylcholine (−0.15 ± 0.02) at TyrA for α7 (Marotta et al., 2015). This suggests a similar strength of interaction, despite the increased bulkiness of the ligand. Using acetylcholine, we have previously shown that a sterically significant substituent is needed at the four-position of the A-site residue (Puskar et al., 2011; Marotta et al., 2015). This effect is less pronounced for NS6740 but still noticeable, with Phe and 4-F1Phe not falling on the line.

Cation-π plots for TyrA and TyrC2 in the α7 nicotine acetylcholine receptor (nAChR). (A) NS6740 forms a cation-π interaction at TyrA in the presence of PNU-120596 with a similar strength to acetylcholine (ACh) at TyrA, as indicated by the slopes of linear regression fit to the Log10[EC50(mut)/EC50(wt)] (NS6740, −0.16 ± 0.03 and ACh −0.15 ± 0.02). (B) TyrC2 shows no cation-π interaction with NS6740 or acetylcholine. Acetylcholine data are from Marotta et al. (2015).
EC50 values of aromatic box residues
A novel observation is that TyrA forms a functionally relevant hydrogen bond for activation of NS6740. A large gain of function was observed when Tyr was replaced by TyrOMe (Table 1). The hydroxyl group of TyrA can act as both a hydrogen bond donor and acceptor. Methylation of the hydroxyl group removes the hydrogen bond donating ability of the residue but not its hydrogen bond accepting ability. Note that activation by ACh is unaffected by the Tyr-to-TyrOMe mutation, both in the presence or absence of PNU-120596 (Marotta et al., 2015). While we have seen a loss of function for this mutation before (Nowak et al., 1995; Lummis et al., 2005; Daeffler et al., 2014), most notably in muscle type nAChR, this is the first time we have ever seen gain of function for TyrOMe.
The shift in EC50 was accompanied by an increase in relative efficacy for the NS6740/PNU-120596 combination, being 1.9 ± 0.32 for the TyrOMe mutant versus 0.027 ± 0.004 for the wild type, relative to activation by ACh (Table 2). A comparable effect was not seen when judging the impact of PNU-120596 on ACh activation—wild type and the TyrOMe mutant show only a small difference.
Relative efficacy to Imax acetylcholine (S.E.M.)
The increase in efficacy for the TyrOMe mutant was so great that we hypothesized that the PAM PNU-120596 may no longer be needed for NS6740 to open the channel (Fig. 4; Table 3). This proved to be true, as NS6740 acts as partial agonist on the mutant channel with an EC50 of 150 nM. The shape of the signal changed in the absence of PNU-120596 to resemble a much more traditional α7 agonist trace, which is characterized by its sharp peak and fast desensitization (Fig. 4). This is understandable, given that PNU-120596 acts by disrupting the desensitized state, resulting in broader signals when coapplied with an agonist.

Effect of the α7 TyrA TyrOMe mutation on NS6740 activity. (A) EC50 curve for application of NS6740 on α7 TyrA TyrOMe without any type 2 PAM. (B) Representative current traces for NS6740 on α7 TyrA TyrOMe. (C) Representative current traces for NS6740 on α7 wild type receptors.
EC50 values at TyrA:TyrOMe
During efficacy experiments, no suppression of signal for applications of ACh after application of NS6740 was observed for wild type receptors in contrast to previous work, which indicated NS6740 had a slow binding-off rate (Papke et al., 2018). This is most likely due to variations in the experimental setup. However, for the TyrOMe mutation a significant decrease in the efficacy of ACh after application of NS6740 was observed. This indicates that TyrOMe may lower the binding-off rate for NS6740.
Characterization of Other Binding Interactions of NS6740 in the Orthosteric Binding Site: Other Potential Cation-π Sites.
Previous work has shown that the high-affinity agonist epibatidine makes a cation-π interaction with TyrC2 (Y217) at α7 in addition to the cation-π interaction at TyrA (Puskar et al., 2011). Low currents (Table 1) prevented a complete study of TyrC2; no signal was detected for more strongly perturbing residues such as 4-CNPhe or 2,3,5,6-F4TyrOMe. However, it is still clear that NS6740 does not make a strong cation-π interaction (Fig. 3); the most perturbing residue measured, 4-BrPhe, was essentially wild type.
Acetylcholine also does not form a cation-π interaction at TyrC2, but two trends were noted. First, bulk is required at the four position for ACh binding (Marotta et al., 2015). This is not true for NS6740—Phe and TyrOMe have similar fold shits, 0.96 and 0.74, respectively. This may be because NS6740 is bulkier and compensates for the loss of bulk at the 4 position. Second, TyrC2 is sensitive to 3- or 5- substitutions on the ring system for ACh binding (Marotta et al., 2015). This is also true for NS6740; the largest losses of function at TyrC2 were for 3-F1TyrOMe and 3,5-F2TyrOMe. However, the magnitude of the effect was much smaller (approximately 2.5-fold for NS6740 compared with greater than 10-fold for ACh). Again, the bulk of the silent agonist may explain this. Overall, interaction of NS6740 with TyrC2 does not appear to play an important functional role.
We have also probed other members of the aromatic box, and we find that NS6740 does not make meaningful interactions with any of the other residues. Insertion of the highly perturbing residue F3Trp at TrpB resulted in a modest 5.5-fold shift (Table 1), indicating, at best, a very weak interaction. Substitution by F3Trp at TrpD (W77) showed a slight gain of function, consistent with observations for acetylcholine binding to α7 (Table 1). TrpD has never been implicated in a cation-π interaction with an agonist in any nAChR. TyrC1 (Y210), the final aromatic residue that composes the classic binding box, was not probed because historically any perturbation at this site has resulted in very large losses of function (Puskar et al., 2011) and it has never been implicated in a cation-π interaction in any Cys-loop receptor (Van Arnam and Dougherty, 2014).
Backbone Hydrogen Bonding at the Orthosteric Binding Site.
Next we looked at important backbone hydrogen-bonding interactions within the orthosteric binding site. Incorporation of α-hydroxy analogs at appropriate locations eliminates the hydrogen bond donating backbone NH and weakens the hydrogen-bonding ability of the carbonyl (i-1) by converting it to an ester (Fig. 2). This assay allows for the study of functionally relevant hydrogen bonds only: crystal structures may show the presence of a hydrogen bond, but EC50 will only show a shift if that hydrogen bond is functionally relevant.
Two agonist-backbone hydrogen bonds historically have been found to be functionally relevant to agonist binding in nAChRs. The carbonyl of TrpB forms a hydrogen bond with nonquaternary amine cation agonists. This is true for NS6740 as well (Table 1, S172). Second, L141 acts as a hydrogen bond donor for many agonists of nAChRs, and at α7 it forms a functionally relevant hydrogen bond for epibatidine but not acetylcholine or varenicline (Van Arnam et al., 2013). NS6740 does not make a functionally relevant hydrogen bond at this site (Table 1).
Discussion
This work focuses on the binding interactions of the silent agonist NS6740 at the α7 nAChR. Noncanonical amino acids were used to probe the orthosteric site, and the results for NS6740—in the presence of the type 2 PAM PNU-120596—were compared with the endogenous agonist ACh. This resulted in three major findings: first, we confirmed that NS6740 binds at the orthosteric site; second, TyrA forms a hydrogen bond that discourages activation in the presence of NS6740, and this hydrogen bond is a requirement for silent agonist activity; and third, NS6740 forms a cation-π interaction with TyrA, consistent with several agonists of α7.
It was predicted that NS6740 binds at least partially in the orthosteric binding site, given the cationic nitrogen at physiologic pH and competitive inhibition of the native agonist (Papke et al., 2015). Using noncanonical amino acids to probe binding interactions of the orthosteric binding site, we showed that NS6740 does indeed bind at the orthosteric binding site. This is evidenced by the cation-π interaction to TyrA and hydrogen bonds to TyrA and to the backbone carbonyl of TrpB. This is the most direct evidence so far concerning the binding site of NS6740. Furthermore, we observed subtle differences in those interactions that give insight into the nature of silent agonist binding.
An intriguing finding of the present work is the key role of a hydrogen bond involving the OH of TyrA as the donor. This was revealed by the strong impact of the TyrOMe mutation, an effect not seen with this substitution in other studies. This relatively subtle mutation profoundly impacts receptor response to NS6740, turning the silent agonist—which produces no signal on its own—to a conventional partial agonist. In principle, the hydrogen bond acceptor could be another side chain residue, the protein backbone, or the silent agonist itself. We propose that the most likely hydrogen bonding partner is the amide carbonyl of NS6740. Related compounds lacking the amide carbonyl, but maintaining a similar overall structure, have been reported in the literature as partial agonists of α7 (Briggs et al., 2009), supporting the importance of the carbonyl of NS6740 to its silent agonist activity. Second, the distance between the cationic nitrogen and carbonyl of NS6740 is ∼5.9 Å. Recall that the N+H of NS6740 makes a hydrogen bond to the backbone carbonyl of TrpB. While there is some variability, a typical distance observed in X-ray crystal structures between the backbone carbonyl of TrpB and the TyrA OH is 5.8 Å, as seen the in apo structure of the α7-AChBP chimera crystal structures (Li et al., 2011). This suggests that the receptor could easily adapt to form an arrangement such as shown in Fig. 5. We propose that the hydrogen bond between the carbonyl of NS6740 and the OH of TyrA locks the receptor closed or in an alternative desensitized state (D*). Removal of the TyrA OH, by making TyrOMe, removes this interaction and turns NS6740 into a partial agonist. Additionally, removal of the Tyr OH appears to lower the binding off rate for NS6740, which further indicates an altered binding site.

Scheme of purposed binding interactions of NS6740 at the orthosteric binding site of α7 nAChR, showing a hydrogen bond and a cation-π interaction to TyrA and a hydrogen bond to TrpB.
Figure 6 presents a simplified model to describe the results reported here. ACh stabilizes the open state (O), enabling channel activation. In the α7 nAChR, the desensitized state D is readily reached via either the open or the closed ACh-bound state (the latter path is not shown in Fig. 6). A silent agonist such as NS6740 is thought to stabilize an alternative desensitized state, D*, which cannot gate (Papke et al., 2018). Addition of a type 2 PAM such as PNU-120596 destabilizes D* and/or stabilizes an alternative open state, O*, allowing channel gating. The TyrA to TyrOMe mutation results in a sharp peak (Fig. 4B), typical of normal agonist activity and fast desensitization, indicating that removal of the hydrogen bond results in NS6740 stabilizing a more typical open state and acting on its own as a partial agonist. Figure 6 shows the mutation acting in a manner similar to that of ACh, but other mechanisms are possible.

Schematic summarizing our results. The scheme is based in part on previously published models of Papke et al. (2018). Shown are the conventional closed (C), open (O), and desensitized (D) states, as well as alternative desensitized and open states (D* and O*) that are accessed by a silent agonist.
The final major finding is that, like all agonists tested at α7, NS6740 forms a cation π interaction to TyrA. A typical dependence of EC50 on the cation-π binding ability of the residue at the TyrA position is seen (Fig. 3). A caveat is that, when probing for a cation-π interaction at TyrA, we have altered the OH of the Tyr so that the hydrogen bond of Fig. 5 does not form. It is conceivable that this change turns on a cation-π interaction that is absent in the wild type receptor. We consider this unlikely because no other aromatic residue shows a cation-π interaction, and a cation-π interaction to TyrA is a universal feature of compounds that activate the α7 nAChR.
We have used noncanonical amino acids and nonsense suppression to probe the binding of NS6740, a silent agonist. This is the first structure–function study of a silent agonist, and we showed that a specific hydrogen bond inhibits agonist activity and is important for suppressing channel opening for silent agonists. Given these results, in the future it may be possible to probe the desensitized state using nonsense suppression and silent agonists.
Acknowledgments
The authors thank Dr. Chris Marotta for providing constructs.
Authorship Contributions
Participated in research design: Blunt, Dougherty.
Conducted experiments: Blunt.
Performed data analysis: Blunt.
Wrote or contributed to the writing of the manuscript: Blunt, Dougherty.
Footnotes
- Received February 21, 2019.
- Accepted May 28, 2019.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- ACh
- acetylcholine
- AChBP
- acetylcholine binding protein
- nAChR
- nicotinic acetylcholine receptor
- NS6740
- 1,4-diazabicyclo[3.2.2]nonan-4-yl(5-(3-(trifluoromethyl)phenyl)furan-2-yl)methanone
- PAM
- positive allosteric modulator
- PMP-072
- (R)-N-(4-methoxyphenyl)-2-((pyridin-3-yloxy)methyl)piperazine-1-carboxamide
- PNU-120596
- 1-(5-chloro-2,4-dimethoxy-phenyl)-3-(5-methyl-isoxazol-3-yl)-urea
- tRNA
- transfer RNA
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}