


Abstract
Phosphatidylinositol (3,4,5) trisphosphate (PIP3)-dependent Rac exchanger 1 (P-Rex1) is a Rho guanine-nucleotide exchange factor that was originally discovered in neutrophils and is regulated by G protein βγ subunits and the lipid PIP3 in response to chemoattractants. P-Rex1 has also become increasingly recognized for its role in promoting metastasis of breast cancer, prostate cancer, and melanoma. Recent structural, biochemical, and biologic work has shown that binding of PIP3 to the pleckstrin homology (PH) domain of P-Rex1 is required for its activation in cells. Here, differential scanning fluorimetry was used in a medium-throughput screen to identify six small molecules that interact with the P-Rex1 PH domain and block binding of and activation by PIP3. Three of these compounds inhibit N-formylmethionyl-leucyl-phenylalanine induced spreading of human neutrophils as well as activation of the GTPase Rac2, both of which are downstream effects of P-Rex1 activity. Furthermore, one of these compounds reduces neutrophil velocity and inhibits neutrophil recruitment in response to inflammation in a zebrafish model. These results suggest that the PH domain of P-Rex1 is a tractable drug target and that these compounds might be useful for inhibiting P-Rex1 in other experimental contexts.
SIGNIFICANCE STATEMENT A set of small molecules identified in a thermal shift screen directed against the phosphatidylinositol (3,4,5) trisphosphate–dependent Rac exchanger 1 (P-Rex1) pleckstrin homology domain has effects consistent with P-Rex1 inhibition in neutrophils.
Introduction
Phosphatidylinositol (3,4,5) trisphosphate (PIP3)-dependent Rac exchanger 1 (P-Rex1), a signaling protein that is highly expressed in neutrophils and the brain, is a regulator of Rac-dependent neutrophil function (Welch et al., 2002, 2005; Dong et al., 2005; Lawson et al., 2011; Herter et al., 2013). In neutrophils, P-Rex1, along with Vav family guanine-nucleotide exchange factors, mediates N-formylmethionyl-leucyl-phenylalanine (fMLP)-induced neutrophil responses (Lawson et al., 2011) by catalyzing nucleotide exchange on small GTPases such as Rac (Welch, 2015). Activation of G protein–coupled receptors, such as the fMLP receptor, leads to the liberation of heterotrimeric G protein βγ subunits and subsequent production of PIP3 through activation of phosphoinositide 3-kinase γ (Stephens et al., 1994, 1997). Both G protein βγ and PIP3, in turn, bind to and activate P-Rex1. P-Rex1–deficient mice have neutrophils that exhibit reduced recruitment in a peritonitis model and impaired C5a- and fMLP-induced generation of reactive oxygen species, events that are downstream of Rac activation (Welch et al., 2005).
Inhibition of P-Rex1 is an attractive means for the treatment of atherosclerosis, which is characterized by endothelial dysfunction and subsequent transendothelial migration of immune cells and lipids from circulation to the arterial wall, where they accumulate and form plaques (Soehnlein, 2012). A major contributor to this is inflammation, wherein neutrophils infiltrate the plaque, resulting in blockage of the blood vessel (Wolf et al., 2014). Although current therapies are mostly focused on treating hyperlipidemia and preventing blood clots, targeting the inflammatory processes involved is an emerging perspective (Gomes Quinderé et al., 2014). Indeed, depletion of neutrophils has been shown to be antiatherogenic (Zernecke et al., 2008). Thus, inhibition of P-Rex1 may be an effective method to restrain neutrophil infiltration in the treatment of atherosclerosis. Furthermore, P-Rex1 mediates tumor necrosis factor-α–induced increases in vascular permeability and neutrophil infiltration into lung tissue. Ablation of P-Rex1 in this model significantly reduced neutrophil transendothelial migration (Naikawadi et al., 2012).
In addition to its role in neutrophils, P-Rex1 is upregulated in prostate, breast, and skin cancers, where it contributes to metastasis (Qin et al., 2009; Lindsay et al., 2011; Montero et al., 2011). This upregulation typically involves epigenetic mechanisms that result in P-Rex1 becoming highly expressed in tissues in which it is usually very low or nonexistent (Wong et al., 2011). In models of metastatic cancer in which P-Rex1 is overexpressed, knockdown of P-Rex1 leads to a reversal of the metastatic phenotype. Thus, P-Rex1 is a validated therapeutic target (Lindsay et al., 2011). In support of targeting P-Rex1, mice lacking P-Rex1 have only minor developmental phenotypes, such as mild neutrophilia, indicating that P-Rex1 is a nonessential protein (Welch et al., 2005).
The pleckstrin homology (PH) domain is the primary site for binding and activation of P-Rex1 by PIP3 (Fig. 1) (Cash et al., 2016). Crystal structures of the independent P-Rex1 PH domain bound to the soluble headgroup of PIP3, d-myo-inositol-1,3,4,5-tetraphosphate (IP4), and to other molecules that were present in the crystallization conditions showed that the PIP3-binding site can accommodate a variety of different molecules (Cash et al., 2016). These data and other biochemical and biologic experiments enable modeling of P-Rex1 at the cell membrane (Fig. 1C) and, moreover, have shown that PIP3 binding to the PH domain is absolutely required for P-Rex1 activation in model cell lines (Cash et al., 2016). Thus, we hypothesized that compounds competitive for IP4 at the PIP3-binding site could serve as therapeutic leads for treating P-Rex1–associated disease states (Welch, 2015; Srijakotre et al., 2017). Successful attempts to target the PIP-binding PH domains of other pharmacologically important proteins with small molecules have been reported by others (Meuillet et al., 2003; Jo et al., 2011; Joh et al., 2012). Some of these compounds exhibit specificity for Protein Kinase B and/or PH domains that bind to a particular type of PIP, such as PIP3 over PIP2 (Mahadevan et al., 2008; Miao et al., 2010). Some mimic the inositol phosphate headgroup of PIPs, whereas others are chemically distinct. The affinities of molecules targeting the PH domain of Protein Kinase B generally span from the high nanomolar to the low micromolar range (Meuillet, 2011).

Crystal structure of the P-Rex1 PH domain bound to IP4, which shows residues involved in the interaction with PIP3 (Protein Data Bank: 5D3Y). (A and B) IP4 (stick model with gray carbon, red oxygen, and orange phosphorous atoms) binds in a pocket wherein basic residues and a tyrosine side chain engage the 3- and 4-position phosphates of the inositol ring. For the protein side chains, carbon atoms are shown in purple, nitrogen in cyan, and oxygen in red. (C) A model of how the PH domain would be oriented at the cell membrane upon binding PIP3 in an activated P-Rex1 signaling molecule.
Here, we report a chemical library screen utilizing differential scanning fluorimetry (DSF) that identified six small molecules that stabilize the purified PH domain of P-Rex1. Three of these compounds were tested in human neutrophils and in breast cancer cells for their abilities to affect P-Rex1–dependent functions and activation of signaling downstream of P-Rex1. One compound was tested in vivo in a zebrafish model wherein it had a profound effect on neutrophil motility and recruitment to sites of inflammation.
Materials and Methods
Chemicals.
Compound 1 is methyl 4-{2-[(2-fluorobenzyl)oxy]phenyl}-2-methyl-5-oxo-4,5-dihydro-1H-indeno[1,2-b]pyridine-3-carboxylate (ChemDiv, Inc.), compound 2 is dimethyl 2-(1-acetyl-2,2,6-trimethyl-3-thioxo-2,3-dihydro-4(1H)-quinolinylidene)-1,3-dithiole-4,5-dicarboxylate (Vitas-M Laboratory, Ltd.), compound 3 is dimethyl 2-(4,4,9-trimethyl-1,2-dioxo-5-thioxo-1,2,4,5-tetrahydro-6H-pyrrolo[3,2,1-ij]quinolin-6-ylidene)-1,3-dithiole-4,5-dicarboxylate (Vitas-M Laboratory, Ltd.), compound 4 is 2-(4-methoxyphenyl)-3-[4-(2-pyridinyl)-1-piperazinyl]-1H-inden-1-one (ChemDiv, Inc.), compound 5 is 6-[(E)-2-(5-nitro-2-furyl)ethenyl]-6,7-dihydro[1,2,4]triazino[5,6-day][3,1]benzoxazepin-3-yl propyl sulfide (ChemDiv, Inc.), and compound 6 is 3-(3,5-dimethoxyanilino)-2-phenyl-1H-inden-1-one (ChemDiv, Inc).
Protein Production and Purification.
Human P-Rex1 cDNA was a gift from Dr. James Garrison (University of Virginia). DNA encoding the PH domain (residues 245–408) was cloned into a modified pMAL expression vector (pMALc2H10T) (Kristelly et al., 2004). Site-directed mutations were created using QuikChange (Qiagen) and confirmed by DNA sequencing. Rosetta (DE3) pLysS Escherichia coli cells (Novagen) were used to overexpress P-Rex1 PH constructs as N-terminally His10-tagged maltose binding protein (MBP)-fusion proteins. Cells were grown in Terrific Broth plus carbenicillin to an optical density of a sample measured at a wavelength of 600 nm (OD600) of 0.8, and then protein expression was induced with 0.1 mM isopropylthiogalactopyranoside at 20°C for 18 hours. Cells were harvested and lysed, and then recombinant protein was extracted using histidine affinity (Ni-nitrilotriacetic acid (NTA)) resin chromatography. Cells were lysed into a buffer containing 20 mM HEPES pH 8, 100 mM NaCl, 2 mM dithiothreitol, and protease inhibitors (0.1 mM EDTA, 0.001 mM leupeptin, 1 mM lima bean trypsin inhibitor, and 0.1 mM phenylmethylsulfonylfluoride) and eluted from the Ni-NTA resin with lysis buffer but with 200 mM NaCl and 200 mM imidazole pH 8. Proteins were then simultaneously dialyzed into 100 mM NaCl buffer and treated with 5% (w/w) tobacco etch virus protease to remove the N-terminal MBP tag. The cleaved MBP tag (which contains the His10 tag) was then removed by repassage over Ni-NTA resin. Final purity was achieved by processing over an S75 size exclusion column (GE Healthcare) in dialysis buffer.
Differential Scanning Fluorimetry.
Thermal denaturation assays were performed using a ThermoFluor plate reader (Johnson & Johnson) in black 384-well polypropylene polymerase chain reaction plates (AB-1384/K; Thermo Scientific). For medium-throughput screening, liquid dispensing was performed using a Thermo Microdrop dispenser. Melting temperatures were determined by monitoring the fluorescence change of 8-anilinonaphthalene-1-sulfonic acid (ANS) as it binds to unfolded protein. ANS and protein were used at 80 μM and 0.1 mg/ml final concentrations, respectively. These components were protected from light prior to analysis. The reaction was prepared by first adding 5 μl buffer (20 mM HEPES pH 8, 200 mM NaCl, 2 mM dithiothreitol) to each well, and then compounds were added from 5 mM DMSO stocks using a Perkin Elmer Sciclone ALH3000 liquid handler with a 50 nl pintool (V&P Scientific). Five microliters of a 2× P-Rex1 PH plus ANS stock was added to columns 1–22 of a 384-well plate. As a positive control, 0.5 mM Ins(1,3,4,5)P4 (IP4) was used (Cayman Chemical). A positive control stock of 2× P-Rex1 PH/ANS/IP4 was added to wells in column 23 at 5 μl per well. The reactions were overlaid with 1.8 μl silicone oil (10 cSt viscosity) and centrifuged at 2000g for 2 minutes. A Thermo Multidrop 384 was used for bulk dispensing of all reagents except the silicone oil, for which a Matrix Equalizer Pipettor was used. For the dose-response tier and analysis of P-Rex1 PH variants, screening was performed without robotics, and samples were measured in triplicate. Melting curves were analyzed by fitting to a Boltzmann model using ThermoFluor Acquire 3.0 software.
X-Ray Crystallography.
Initially, previously described conditions used to crystallize the P-Rex1 PH domain in complex with IP4 were used to try to cocrystallize the PH domain in complex with compounds (Cash et al., 2016). PH domain at 5.6 mg/ml was incubated with 500 μM compound for 2–4 hours, and then 0.22 μm was filtered prior to setting hanging drop crystallization trays. Crystals formed were orange or red in color and were shown to be compound alone. Furthermore, 96-well sparse matrix screening led to the identification of 0.1 M BisTris pH 5.5, 200 mM lithium sulfate, and 25% polyethylene glycol 3350 as a crystallization condition. Variant PH domain K280A (Cash et al., 2016) was crystallized in this condition in hanging drop trays, producing microcrystals. These crystals were crushed and used to seed empty drops in the same condition, producing large, well diffracting (sub 2 Å) crystals that were used for further soaking experiments. In these experiments, crystals were slowly transferred from the initial mother liquor to an intermediate solution containing mother liquor reduced to 100 mM sulfate plus 5% DMSO and then to a final solution containing mother liquor without sulfate plus 10% DMSO and 500–1000 μM compound. Each soak lasted 1–12 hours, with the final soak lasting 1 hour before crystal harvesting and freezing. Diffraction data were collected at 110 K on a CCD detector at beamline 21-ID-D or 21-ID-G at the Advanced Photon Source. Data were integrated and scaled using HKL2000 (Otwinowski and Minor), and initial phases were provided by molecular replacement using Phaser (Mccoy et al., 2007; Winn et al., 2011). Alternating rounds of model building and maximum-likelihood refinement were performed in Coot (Emsley et al., 2010) and Refmac5 (Murshudov et al., 1997).
Cell Culture Reagents.
Eight-well chamber slides were purchased from Ibidi (cat. no. 80826). Fibronectin was obtained from Sigma. Polycarbonate filters with a 3-µm pore size were from Neuroprobe. One-step polymorphs were obtained from Accurate Chemical and Scientific Corporation (cat. no. AN221725).
Human Neutrophil Isolation.
Human neutrophils were isolated from peripheral blood of healthy donors. All the steps were performed on ice with ice-cold buffers. The cells were isolated as previously described (Nuzzi et al., 2007). Briefly, 4.5 ml of blood was carefully layered on top of one-step polymorphs, and tubes were centrifuged (1000g, 45 minutes, 20°C). The top two layers of plasma and monocytic layer were aspirated off carefully, and the buffy coat of neutrophils (third layer) was transferred to the fresh, ice-cold tubes. Tubes were centrifuged (400g, 10 minutes, 4°C) to remove residual polymorphs solution. The supernatant was aspirated off, and red blood cells were lysed using 4.5 ml of 0.1× PBS (hypotonic solution) for 30 seconds, and 1.5 ml of 4× PBS was immediately added to the tubes. The tubes were centrifuged (400g, 10 minutes, 4°C), the pellet containing neutrophils was resuspended in modified HBSS (mHBSS), and the cells were counted.
Neutrophil Cell Spreading.
Each well of an eight-well chamber slide was coated with 5 µg of fibronectin overnight at 4°C. Freshly isolated human neutrophils were preincubated with vehicle or indicated P-Rex1 inhibitors for 15 minutes before plating on fibronectin-coated glass slides. Cells were allowed to adhere to the fibronectin-coated wells for 30 minutes at 37°C, 5% CO2, and unadhered cells were washed off with mHBSS. Three random fields were imaged at 40× magnification by differential interference contrast microscopy before and after 100 nM fMLP stimulation. Images from three independent experiments were analyzed by counting the number of spread cells and total cells in a blinded manner to determine the spreading of the cells. The number of cells spread after fMLP treatment was counted as 100%.
G-LISA Assay.
The effects of compounds on Rac activation mediated by fMLP were assessed using an absorbance-based G-LISA Rac activation assay kit (cat. no. BK125; Cytoskeleton, Inc.) in dPLB985 (PLB) cells. PLB cells were grown in RPMI 1640 (RPMI + GlutaMAX) medium with 10% FBS and 1% penicillin-streptomycin. PLBs were differentiated to neutrophil-like cells (dPLBs) at a density of 0.4 × 106 cells per milliliter for 5 days with 1.3% DMSO. On the sixth day, dPLBs were serum starved for 24 hours in 1.3% DMSO containing RPMI medium. On the seventh day, cells were resuspended in NaCl buffer as described previously (Lehmann et al., 2008) at a density of 3 × 106 cells per milliliter, and protein concentration was measured using Precision Red advanced protein assay reagent provided with the kit. The cells were diluted to a final protein concentration of 0.3 mg/ml per well. Diluted cells were treated with 30 µM compounds or vehicle for 15 minutes before stimulation with 100 nM fMLP for 20 seconds. Immediately after stimulation, cells were pelleted (10,000g, 2 minutes, 4°C) and washed twice with ice-cold PBS. The cells were then lysed using the lysis buffer provided with the kit containing protease inhibitors and 20 mM NaF for 10 minutes on ice. The cell lysate was centrifuged (10,000g, 5 minutes, 4°C) to remove the cell debris, and the supernatant was further processed per the manufacturer’s protocol. The effect of compound 1 on Rac activation stimulated by epidermal growth factor (EGF) in MCF-7 breast cancer cells was assessed using the same G-LISA assay. MCF-7 cells were grown in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin. Cells were plated in a six-well plate at 0.35 × 106 cells per well and allowed to adhere overnight, followed by serum starvation for 24 hours. Cells were then pretreated with either 30 μM compound 1 or vehicle for 15 minutes before stimulation with 100 nM EGF for 1 minute (Sosa et al., 2010; Montero et al., 2011). Immediately afterward, cells were washed twice with ice-cold PBS and lysed in 300 μl of lysis buffer. The lysate was then centrifuged (10,000g, 5 minutes, 4°C) to remove the cell debris, and the supernatant was further processed per the manufacturer’s protocol.
Transwell Chemotactic Assay.
Human neutrophils at a density 3 × 106 cells per milliliter in mHBSS were pretreated with vehicle or P-Rex1 inhibitors for 15 minutes. DMSO or 100 nM fMLP was added in the lower chamber, and a 3-μm polyvinylpyrrolidone-free polycarbonate membrane filter was placed on top of the lower chamber. Fifty microliters of treated cells was added to the top chamber and allowed to migrate for 1 hour at 37°C and 5% CO2. The filter facing the top side of the chambers was scraped off to remove unmigrated cells, and the other side of the filter was fixed and stained. Three random fields were imaged at 20× using a light microscope, and cells were counted using ImageJ. The data are shown as the chemotaxis index, which is the ratio of the number of cells (treated or untreated) that migrated in response to fMLP versus migrated toward DMSO.
Live Imaging of Zebrafish Larvae.
Imaging was performed as previously described (Hsu et al., 2019). Briefly, larvae at 3 days postfertilization were settled on a glass-bottom dish, and imaging was performed at 28°C. Time-lapse fluorescence images of the head mesenchyme were acquired with a laser-scanning confocal microscope (LSM710; Zeiss) with a Plan-Apochromat 20×/0.8 M27 objective at 1-minute intervals for 30 minutes. The green channel was acquired with ∼0.1%–0.3% power of the 488-nm laser with a 200-μm pinhole at a speed of 1.27 microseconds per pixel and averaged. The fluorescent stacks were flattened using the maximum intensity projection and overlaid with a single slice of the bright-field image. Neutrophil speed and directionality were quantified using the ImageJ plug-in MTrackJ.
Inflammation Assays in Zebrafish.
Zebrafish wounding and infection were performed as described (Hsu et al., 2017). Briefly, larvae at 3 days postfertilization were treated with DMSO or compound 1 at indicated doses for 1 hour and amputated posterior to the notochord. The larvae were fixed in 4% paraformaldehyde at 1 hour postwounding. Neutrophils were stained with Sudan Black, and the numbers at the indicated regions were quantified.
Statistical Analyses.
For experiments with human neutrophils, cells were counted in a blinded manner. The data are means ± S.D. of three different fields for each condition from three independent biologic experiments performed in technical duplicates and analyzed by one-way ANOVA using Tukey’s multiple comparison test in GraphPad Prism version 7. ****P < 0.0001, ***P < 0.001, **P < 0.01. Not significant indicates P > 0.05. For experiments in PLB cells, the data are means ± S.D. of four independent experiments. For zebrafish larvae experiments, results are representative of three biologic repeats, each containing 20 fish per group. P values were calculated using unpaired Student’s t test: ****P < 0.0001, **P < 0.01.
Results
Medium-Throughput Screen to Identify Compounds That Bind the P-Rex1 PH Domain.
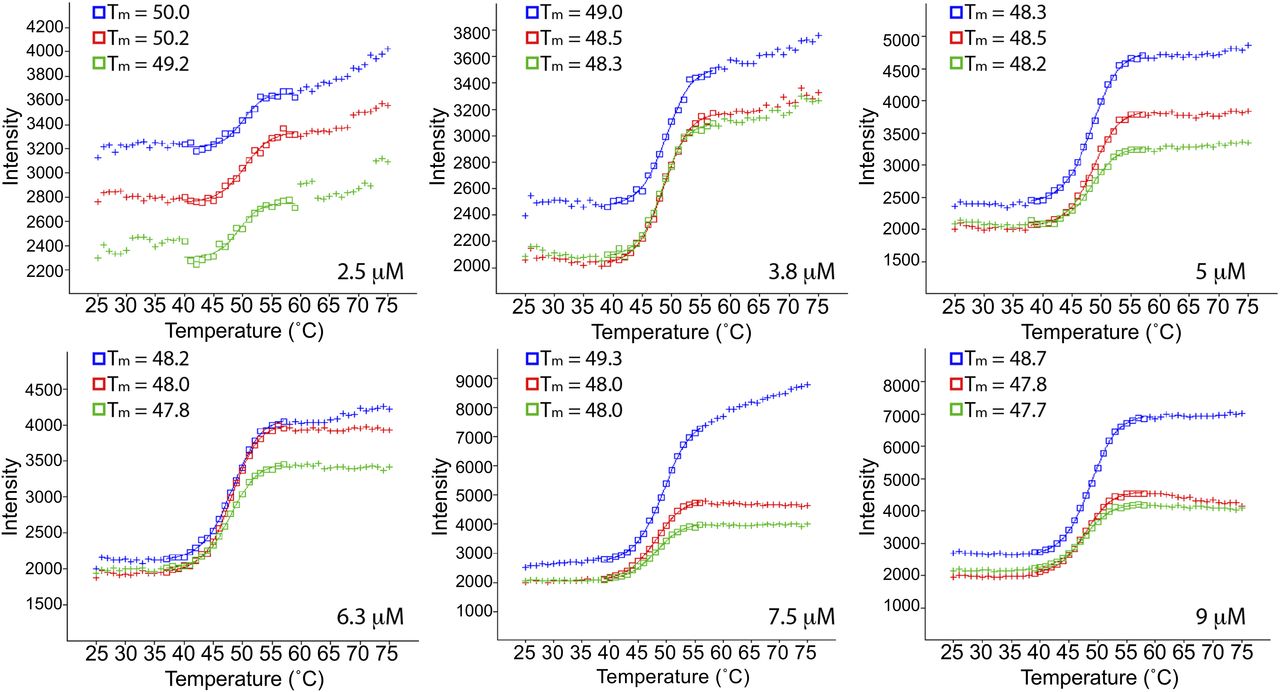
To discover small molecules that bind to the human P-Rex1 PH domain—in particular, those that block binding of the PH domain to PIP3—we optimized a DSF assay that detects binding of small molecules to the isolated PH domain. We first screened a range of protein concentrations from 2.5 to 9 μM to find the concentration that generated a strong signal in the assay while using the least amount of protein to minimize protein concentration and the concentration of small molecule needed to observe an effect, settling on 5 μM PH domain (Fig. 2). A robotic liquid handling system was then used to pipette reagents into 384-well plates for the screen. PH domain alone and PH domain plus 0.5 mM IP4 were used as negative and positive controls, respectively. Small molecules were multiplexed at four compounds per well for the primary screen and included those from the Chemical Diversity 100K, Prestwick & LOPAC, and Navigator Fragments libraries provided by the University of Michigan Center for Chemical Genomics (Fig. 3). The assays exhibited an average plate Z score of 0.91. The hits from the first tier of screening were then deconvoluted, testing one compound per well, resulting in 47 confirmed hits. This list of compounds was triaged primarily for commercial availability and promiscuity, as observed in other screens performed by the CCG, resulting in 20 compounds that were moved on to dose-response analysis.
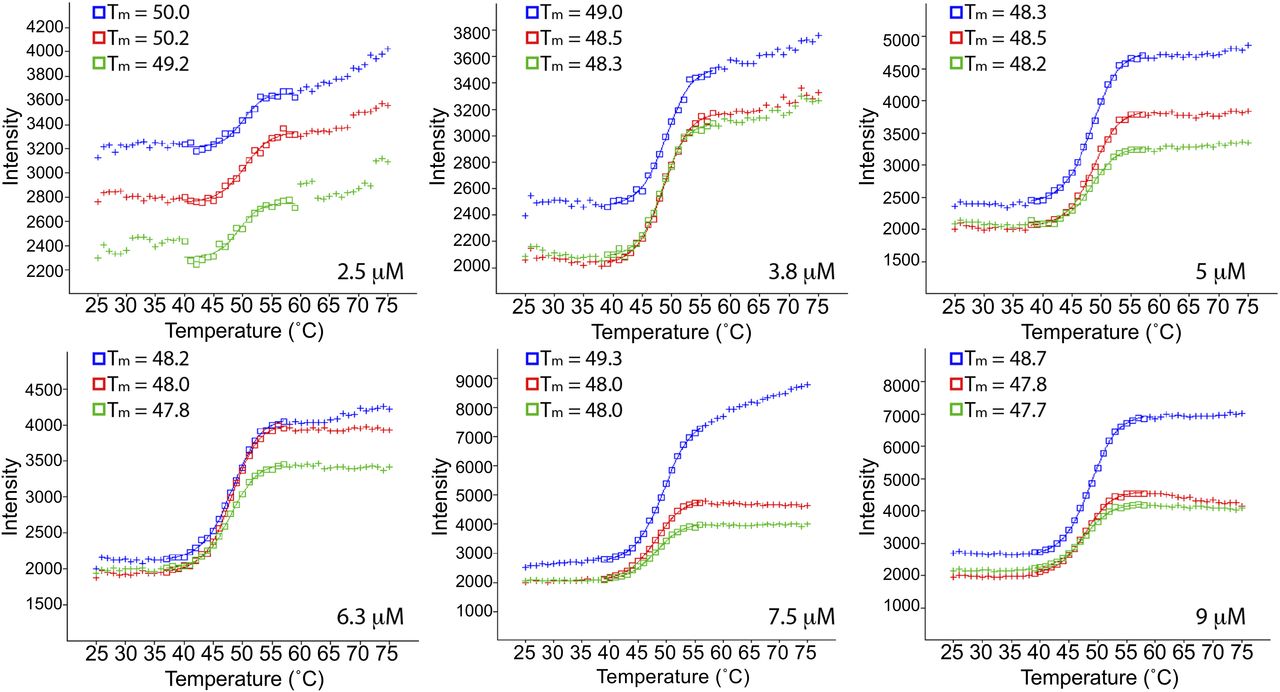
DSF of the independent P-Rex1 PH domain was optimized for use in a screen to discover small molecules that bind to this domain. PH domain concentration was screened, and 5 μM was chosen for use in the assay. Each curve represents one sample, and the melting temperature (Tm) for each is shown.

Flowchart for the stages and outcomes of the DSF screen. Screening led to six confirmed hits, and three of these compounds (1–3) were moved forward into further assays.
Dose-Response Analysis.
The remaining compounds were screened at concentrations of 5, 20, and 100 μM in a 384-well plate pipetted by hand. Of these, six increased the melting temperature of the PH domain at one or more concentration of compound (Fig. 4). Some of the compounds had very low solubility in aqueous solution and could not be tested at higher concentrations. Compound 1 exhibited a dose response (Fig. 4). Interestingly, compounds 2 and 3 share chemical similarity. Compound 3 showed the strongest thermal stabilization of the PH domain at the lowest concentration of compound tested but was also the least soluble of the group. Compounds 1–3 were chosen as the top three hits and were assessed further.

Confirmed hits from the final tier of compound screening. (A) Chemical structures are shown along with their corresponding number used in the main text and figures. Compounds are numbered according to appearance in the text. The identifier for each listed in the Center for Chemical Genomics library is 1: 114319, 2: 107713, 3: 114256, 4: 129917, 5: 105072, and 6: 129984. (B) The DSF dose-response assays confirmed that these compounds are stabilizers of the P-Rex1 PH domain. Each concentration was analyzed in triplicate, and error bars represent S.D. of the mean. At concentrations in which data are not shown, the compounds were not soluble. (C) DSF curves for compound 1 are shown as an example. Each curve represents one experiment, and corresponding calculated melting temperatures (Tm) are shown.
Cocrystallization of Compounds with the P-Rex1 PH Domain.
We attempted to determine co-crystal structures of the PH domain bound to the compounds. We began by using crystallization conditions previously used to crystallize the P-Rex1 PH domain (Cash et al., 2016) but instead used PH domain preincubated with compound. However, the resulting crystals were composed of compound alone. A new crystallization condition was identified using sparse matrix screening, and microseeding experiments using this condition produced large, well diffracting crystals. Unfortunately, the precipitant contained sulfate, which competitively binds the PIP3-binding site and interacts with basic residues in the pocket. To alleviate this effect, we mutated one of these residues, Lys280, to alanine and crystallized this variant. We then performed soaking experiments to slowly transfer these crystals into solutions with reduced sulfate concentration and increased DMSO concentration plus compound, which was needed to solubilize compound. The goal in combining the K280A mutation with the soaking experiments was to reduce the affinity of and remove the sulfate from the binding site and replace it with compound. However, out of ∼30 data sets analyzed, structures never revealed more than what is likely partial occupancy of the PIP3-binding site. We attribute this to the low solubility of the compounds in aqueous solution.
Thermal Stabilization of PIP3-Binding Deficient Variants of the P-Rex1 PH Domain.
We next tested the effects of PIP3-binding site mutations, including K280A, R289A, and K368A, which greatly reduce binding of IP4 (Cash et al., 2016), on the ability of the top three compounds to thermally stabilize the PH domain (Fig. 5; Table 1). As predicted, the K280A, R289A, and K368A mutations eliminated thermal stabilization by IP4. However, these mutations did not reduce thermal stabilization by the screen compounds and, in some cases, enhanced it. For example, mutation K368A increased thermal stabilization by compounds 1, 2, and 3, and Y300F enhanced thermal stabilization by both compounds 2 and 3. Variant K280A exhibited increased thermal stabilization by compound 2, but only at the 20 µM concentration. Because the compounds are hydrophobic, it is possible that these mutations allow more favorable binding by reducing the overall charge of the pocket. However, the compounds may also simply bind outside the PIP3-binding site and affect the ability of this domain to bind its native ligand allosterically.

Binding of compounds to P-Rex1 PH domain PIP3-binding site variants. To test whether disruption of the PIP3-binding site would alter binding of the top three compounds to the PH domain, single-point mutations of residues shown in Fig. 1 were made in the binding pocket, and these variants were tested in DSF for stabilization by the compounds. Shown are melting temperatures (Tm) for each variant, and error bars represent 95% confidence intervals. Shown as controls are Tm values in the absence of ligand and in the presence of IP4. See also Table 1.
Effects of point mutations in the P-Rex1 PH domain PIP3-binding site on thermal stabilization by compounds 1, 2, and 3
Shown are ΔTm values representing the change in melting temperature of each protein in the presence of compound.
Neutrophil Adhesion Assay.
To examine the ability of the compounds to inhibit endogenous P-Rex1 activity in cells, we tested the effects of the compounds on neutrophil functions downstream of P-Rex1 as stimulated by formyl peptide receptor 1 (Welch et al., 2005). Human neutrophils were pretreated with compounds derived from the screen for 15 minutes and then seeded onto slides coated with fibronectin followed by washing to remove cells that had not adhered. DMSO-treated cells had round, nonpolarized morphology (Fig. 6A, left panel), and addition of the formyl peptide receptor 1 agonist fMLP caused the cells to spread on the surface and polarize (Fig. 6A, middle panel). All of the tested compounds strongly suppressed the fMLP-stimulated morphologic transition at 30 μM (Fig. 6B). Compound 1 inhibited fMLP-stimulated cell spreading and polarization with an IC50 of 3 μM (Fig. 6, A and C). Next, we tested the effects of compounds 1–3 on fMLP-stimulated migration of human neutrophils in a transwell chemotaxis assay. Deletion of P-Rex1 in mouse neutrophils does not significantly affect chemotaxis in this assay (Lawson et al., 2011). Consistent with these results, the three compounds tested at the limit of their solubility had no effect on fMLP-stimulated transwell migration (Fig. 6D). This demonstrated a lack of toxicity and the specificity of these compounds for a subset of fMLP responses.
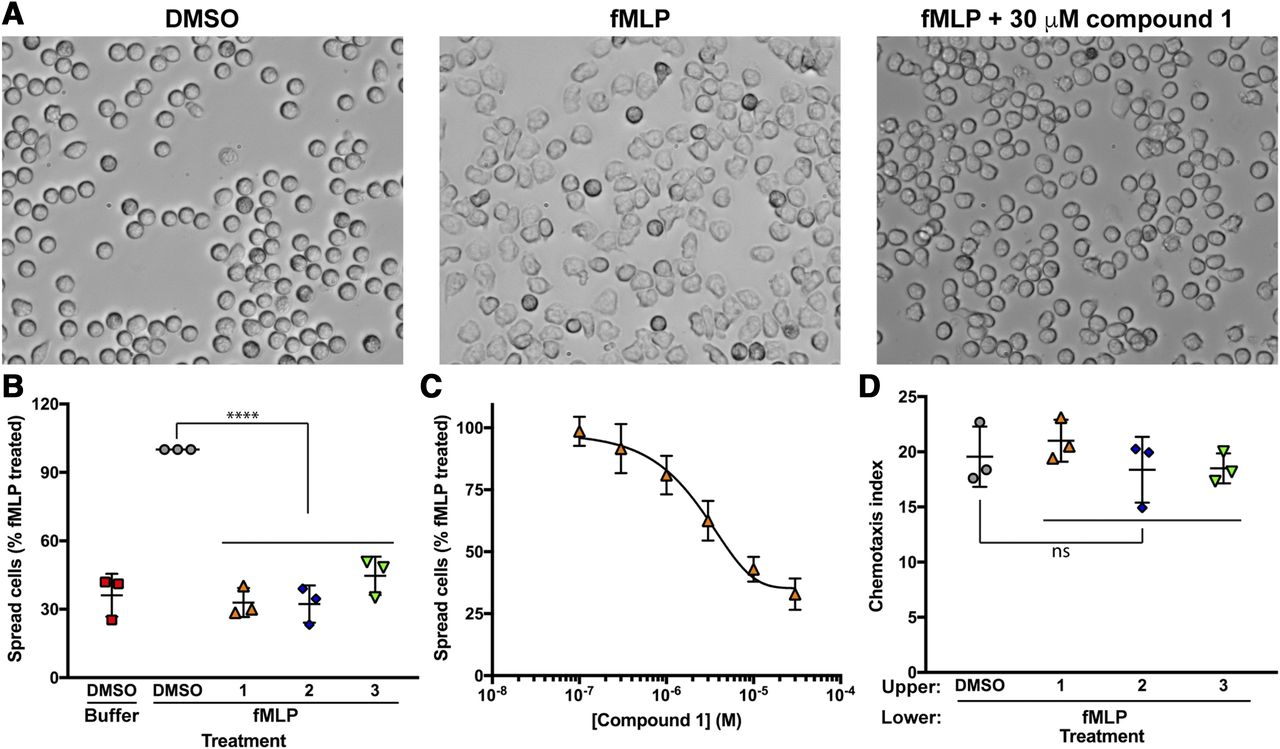
Treatment of human neutrophils with the top three compounds from the screen prevents fMLP-induced cell spreading but does not affect chemotaxis toward fMLP. Human neutrophils were preincubated with vehicle or indicated P-Rex1 inhibitors before plating on fibronectin-coated glass slides. Cells were washed and imaged by differential interference contrast microscopy at 40× magnification before and after 100 nM fMLP stimulation. (A) Representative images of human neutrophils depicting the cell spreading before and after treatment with fMLP with or without pretreatment with 30 μM compound 1. (B) Images from three independent experiments were analyzed by counting the number of spread cells and total cells in a blinded manner to determine the spreading of the cells. The data are represented as the percentage of cells spread compared with fMLP in the same experiment. The data are means ± S.D. of three different fields for each condition from three independent experiments analyzed by one-way ANOVA. ****P < 0.0001. (C) Pretreatment with compound 1 results in a concentration-dependent decrease in fMLP-induced cell spreading. Human neutrophils were treated with the indicated concentrations of compound 1 and analyzed as in panel B. Data are pooled from three independent experiments and fit with a dose-response inhibition equation. (D) Human neutrophils were preincubated with vehicle or indicated P-Rex1 inhibitors (30 μM) and allowed to migrate for 1 hour at 37°C toward 100 nM fMLP in the lower chamber. Chemotaxis index is the number of cells that migrated in the presence of fMLP divided by the number of cells that migrated in the absence of fMLP. Data are means ± S.D. from three independent experiments performed in duplicate analyzed by one-way ANOVA. ns (not significant) indicates P > 0.05.
Rac Activation Assay.
Rac2 regulation of actin polymerization is responsible for fMLP-dependent changes in neutrophil cell spreading (Lawson et al., 2011). Because P-Rex1 is highly expressed in neutrophils and is a guanine nucleotide exchange factor for Rac2, we next assessed the activation of Rac upon treatment of cells with compounds 1–3. For these assays, we used a human neutrophil-like cell line that is relatively easy to grow compared to primary human neutrophils. Stimulation of dPLB cells with fMLP caused significant Rac activation at 2 minutes, and pretreatment with each compound at 10 μM significantly inhibited Rac activation (Fig. 7A). Compound 1 inhibited fMLP-dependent Rac activation at concentrations as low as 3 μM (Fig. 7B), consistent with the neutrophil adhesion assay.

Compound 1 inhibits Rac activation downstream of P-Rex1 in cells. Treatment of differentiated PLB cells with compounds 1–3 prevents fMLP-induced Rac activation. (A) PLB cells were serum starved overnight and treated with 30 μM inhibitor for 15 minutes before stimulation with fMLP for 20 seconds. Rac activation was measured with a Rac G-LISA kit. The data are represented by means ± S.D. of four independent experiments analyzed by one-way ANOVA. ***P < 0.001. (B) Treatment of PLB cells with compound 1 results in concentration-dependent inhibition of fMLP-induced Rac activation. The data are pooled from four independent experiments. **P < 0.01. (C) The effect of compound 1 on Rac activation stimulated by EGF in MCF-7 breast cancer cells was assessed using the same G-LISA assay. Cells were plated and allowed to adhere overnight, followed by serum starvation for 24 hour. Cells were then pretreated with either 30 μM compound 1 or vehicle for 15 minutes before stimulation with 100 nM EGF for 1 minute. OD, optical density.
To examine whether compound 1 can inhibit P-Rex1 in other biological settings, we examined Rac activation downstream of the EGF receptor in MCF-7 breast cancer cells, which is also dependent on P-Rex1 (Sosa et al., 2010; Montero et al., 2011). Stimulation of MCF-7 cells with 100 nM EGF caused approximately 3-fold activation of Rac after 1 minute (Fig. 7C). Pretreatment of cells with 30 μM compound 1 significantly inhibited this effect and nearly eliminated EGF-stimulated Rac activation.
In Vivo Neutrophil Migration Assay.
P-Rex1 is important for neutrophil recruitment in response to inflammation in mouse models (Pan et al., 2015). To examine the ability of compound 1 to inhibit endogenous P-Rex1 activity in vivo, we used a transgenic zebrafish model with GFP-labeled neutrophils and examined neutrophil motility under normal physiological and inflammatory conditions. Similar to their mammalian counterparts, neutrophil motility and wound response in zebrafish is dependent on phosphoinositide 3-kinase (Yoo et al., 2010). Zebrafish larvae were pretreated with DMSO or 100 μM compound 1 for 1 hour to allow compound penetration. Overnight treatment with compound 1 at this concentration did not cause any morphologic changes in the larvae, indicating that the compound is not toxic. The velocity, but not directionality, of neutrophil migration was inhibited by compound 1 (Fig. 8, A–C; Supplemental Video 1). Consistently, acute neutrophil recruitment to a tailfin amputation site was also inhibited in a dose-dependent manner (Fig. 8, D and E). Together, these results suggest that compound 1 can penetrate tissue and inhibit P-Rex1 in a vertebrate model organism.
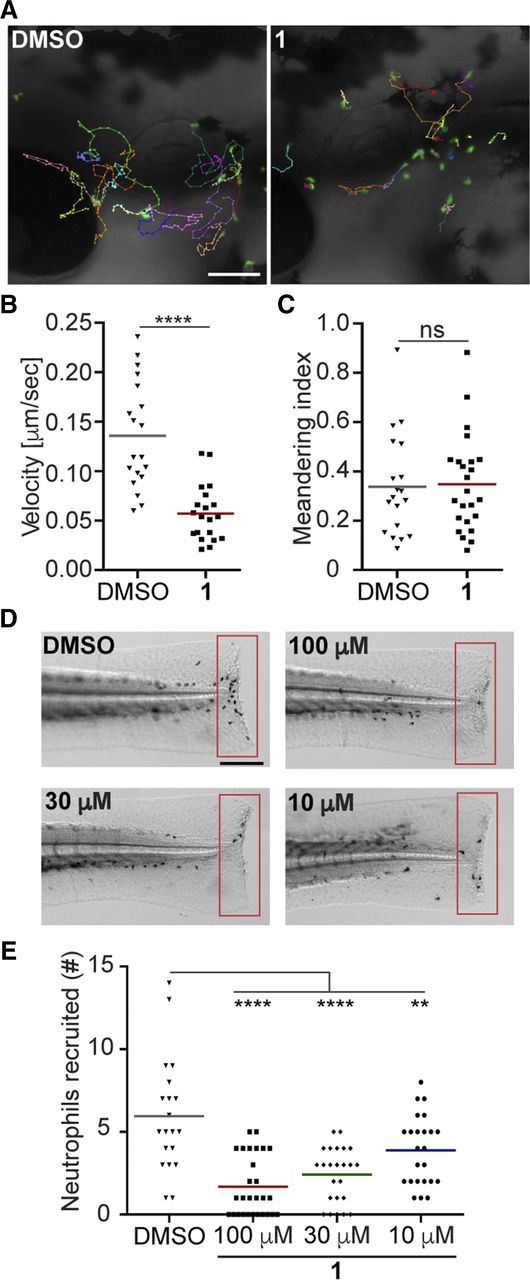
Treatment of zebrafish larvae with compound 1 inhibits neutrophil motility and recruitment to injury sites. (A) Representative images, (B) velocity, and (C) meandering index of neutrophil motility in zebrafish larvae treated with DMSO or compound 1. Scale bar, 100 μm. Three embryos each from three biological repeats were imaged, and quantification of neutrophils from one representative video is shown. (D) Representative images and (E) quantification of neutrophils recruited to tailfin transection sites in zebrafish larvae treated with DMSO or compound 1. Scale bar, 200 μm. Results are representative of three biologic repeats, each containing 20 fish per group. P values were calculated using unpaired Student’s t test: **P < 0.01, ****P < 0.0001. ns, not significant.
Discussion
Herein, we describe the development of a DSF-based screen to identify compounds that bind to the P-Rex1 PH domain, and we identify that some of these have effects in human neutrophils consistent with P-Rex1 inhibition. The fact that our three lead compounds (1–3) inhibit specific processes controlled by Rac2 activation in response to fMLP in primary human neutrophils and a human neutrophil-like cell line and in response to EGF in a breast cancer cell line strongly supports that these compounds inhibit P-Rex1 activation in cells. The results parallel those observed in neutrophils obtained from P-Rex1−/− mice (Lawson et al., 2011), but there are significant differences. Consistent with the mouse studies, deletion of P-Rex1 had no effect on chemoattractant (C5a)-dependent migration in a transwell assay. In contrast is the observation that blockade of P-Rex1 inhibits cell spreading. In mouse neutrophils, deletion of a second Rac exchange factor, Vav, in conjunction with P-Rex1 was required to inhibit cell spreading in response to fMLP (Lawson et al., 2011). Rac2 deletion also inhibited neutrophil spreading. There are some possible explanations for these differences. One is that human neutrophils have a network of signaling proteins different from those in mouse neutrophils. For example, Rac2 is the predominant isoform in human neutrophils (Heyworth et al., 1994), whereas mouse neutrophils have equal expression of Rac1 and Rac2 (Li et al., 2002). P-Rex1, the predominant exchange factor in neutrophils, preferentially activates Rac2 relative to Rac1 (Welch et al., 2002; Donald et al., 2004). Thus, it is possible that in human neutrophils, Rac activation downstream of fMLP is predominantly driven by P-Rex1 rather than Vav. An alternative explanation is that although the compounds were selected for binding to the P-Rex1 PH domain, they can also bind and inhibit Vav. Nonetheless, the data are consistent with these compounds targeting the PH domain of P-Rex1 to inhibit P-Rex1 activation in human cells. Furthermore, they demonstrate the tractability of targeting the P-Rex1 PH domain for treatment of maladaptive neutrophil function. Zebrafish are a well established vertebrate animal model for toxicity (Dai et al., 2014) and innate immunity, especially neutrophil biology (Henry et al., 2013). Similar to human neutrophils, zebrafish neutrophils predominantly express Rac2 compared with Rac1, at least at the mRNA level (Hsu et al., 2019). The fact that compound 1 inhibits neutrophil migration in tissue without causing gross developmental defects suggests that this compound has high potential for therapeutic use. Future directions will be to better define the compound binding site and work with medicinal chemists to improve potency, selectivity, tissue penetration, and in vivo efficacy.
Acknowledgments
The authors thank the University of Michigan Center for Structural Biology and the High-Throughput Protein Production Facility for use of their ThermoFluor instrument.
Authorship Contributions
Participated in research design: Cash, Chandan, Deng, Smrcka, Tesmer.
Conducted experiments: Cash, Chandan, Hsu, Sharma.
Performed data analysis: Cash, Chandan, Hsu, Smrcka.
Wrote or contributed to the writing of the manuscript: Cash, Chandan, Deng, Smrcka, Tesmer.
Note Added in Proof—American Cancer Society – Michigan Cancer Research Fund Postdoctoral Fellowship PF-14-224-01-DMC (J.N.C.) was accidentally not included in the Fast Forward version published January 3, 2020. The funding footnote has now been corrected.
Footnotes
- Received June 11, 2019.
- Accepted December 19, 2019.
This work was supported by the National Institutes of Health (NIH) National Cancer Institute and National Heart, Lung, and Blood Institute [Grants R01CA221289, R01HL122416, R01HL071818], a University of Michigan Center for the Discovery of New Medicines FY15 Round 1 grant, and the Walther Cancer Foundation (J.J.G.T.), the NIH National Institue of General Medical Sciences [Grant R35GM127303] (A.V.S.), a Michigan Pharmacology Centennial Fund Predoctoral Fellowship and a Benedict and Diana Lucchesi Fellowship (N.R.C.), the NIH National Institue of General Medical Sciences [Grant R35GM119787] (Q.D.), and a Purdue Cagiantas fellowship (A.Y.H). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract number DE-AC02-06CH11357. Use of the Life Sciences Collaborative Access Team Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). The authors would also like to acknowledge the contribution of American Cancer Society – Michigan Cancer Research Fund Postdoctoral Fellowship PF-14-224-01-DMC (J.N.C).
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- ANS
- 8-anilinonaphthalene-1-sulfonic acid
- DSF
- differential scanning fluorimetry
- EGF
- epidermal growth factor
- fMLP
- N-formylmethionyl-leucyl-phenylalanine
- FPR1
- formyl peptide receptor 1
- IP4
- D-myo-Inositol-1,3,4,5-tetraphosphate
- MBP
- maltose binding protein
- mHBSS
- modified HBSS
- NTA
- nitrilotriacetic acid
- PH
- pleckstrin homology
- PIP3
- phosphatidylinositol (3,4,5) trisphosphate
- P-Rex1
- PIP3-dependent Rac exchanger 1
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}