
Abstract
The P-glycoprotein (P-gp) is a 170-kDa protein that acts as an energy dependent, transmembrane efflux pump and is encoded by the MDR1 gene. It has been shown to be responsible for multidrug resistance (MDR) in a defined subpopulation of breast cancer patients and thus represents a molecular target for circumventing MDR in this tumor indication. MDR modulators have been developed and demonstrated high selectivity for P-gp with inhibitory activities in the low nanomolar range. Although some objective responses were achieved in clinical trials, combination therapy with these MDR modulators, such as Ca2+ antagonists caused unacceptable toxicity. Targeting P-gp inhibitors to the tumor site is a mean to increase their therapeutic index, and in this context binding of tailor-made prodrugs to circulating albumin is an established technology to reduce the toxicity and enhance the efficacy of anticancer drugs. In this study, we consequently developed an acid-sensitive albumin-binding prodrug of the P-gp inhibitor zosuquidar (LY335979) in a two-step synthesis using a maleimide hydrazone linker system established in our laboratory that first introduces acetylbenzoic acid at the HO-group of zosuquidar followed by derivatization with 6-maleimidocaproyl hydrazide to form the acid-sensitive hydrazone bond. The maleimide group enables the prodrug to bind rapidly and selectively to the cysteine-34 position of endogenous albumin after intravenous administration. HPLC analysis demonstrated rapid albumin binding of the zosuquidar prodrug as well as the quantitative release of the acetylbenzoic ester derivative of zosuquidar at pH 5.0. Subsequently, its ability to circumvent MDR was tested in two doxorubicin-resistant breast carcinoma cell lines (MCF-7/ADR and MT-3/ADR). The MDR status of these cell lines can be reversed by zosuquidar which was confirmed in a rhodamine 123 assay using fluorescence microscopy and FACS analysis. Furthermore, zosuquidar as well its acid-sensitive albumin conjugate re-sensitized cells to doxorubicin as well as to an albumin-binding prodrug of doxorubicin, i.e., the 6-maleimidocaproyl hydrazone derivative of doxorubicin, achieving IC50 values in the same order of magnitude as the parental cell lines. Thus, a novel formulation of zosuquidar has been developed that could have the potential to improve the toxicity issues and tumor targeting properties of the original compound.
Similar content being viewed by others

Introduction
Intrinsic or acquired multidrug resistance (MDR) is a major problem in the treatment of breast cancer. A number of biochemical mechanisms have been described to be responsible for drug resistance, including mutations in the cellular target of the respective drug, alterations in enzymatic activation and detoxification mechanisms, defective apoptotic pathways, membrane changes as well as elimination of the drug from the tumor cell through the action of drug efflux pumps [1]. The latter represents a frequent cause of drug resistance that allows tumor cells to survive by lowering the intracellular concentrations of the drugs. The cell-membrane efflux pumps include P-glycoprotein (P-gp), multiple resistance protein (MRP), and breast cancer resistance protein (BCRP) which belong to the ATP-binding cassette (ABC) transporter family [2–4].
A well-characterized efflux pump is the P-gp (ABCB1), which is a 170-kDa energy-dependent transmembrane protein encoded by the MDR1 gene [5, 6]. Typical substrates for the P-gp are hydrophobic or amphiphilic compounds and include cytotoxic drugs, such as taxanes, vinca alkaloids, anthracyclines, epipodophyllotoxins, actinomycin D, and mitomycin C [7, 8].
The P-gp is often found over-expressed in resistant breast tumor cells. In 1997, Trock et al. [9] conducted a meta-analysis of 31 studies that evaluated tumor specimens from ~1,200 breast cancer patients. Their analyses showed that ~40% of breast tumors expressed MDR1 mRNA. There was substantial heterogeneity in the values across the individual studies, however, largely attributable to different analytical methodology. Data from nine studies comprising patients treated with cytotoxic agents or hormones showed a significant increase in the proportion of patients with tumors expressing P-gp subsequent to therapy. Consequently, patients with P-gp expressing tumors were threefold less likely to achieve an objective response after treatment compared to patients whose tumors were MDR1/P-gp negative. A study by Mechetner et al. [10] in 1998 confirmed these results: in 359 breast cancer specimens, P-gp expression was only detected in 11% of untreated patients compared to 30% of treated patients; the extent of P-gp expression correlated with in vitro resistance to doxorubicin and paclitaxel in functional assays.
A recent study assessed the RNA expression of MRP, BCRP, MDR1, and LRP (lung resistance-related protein) in 59 primary breast tumor specimens and showed that MDR1 expression was inversely related with the efficacy of first-line chemotherapy, and high expression levels were a significant predictor of poor prognosis [11]. The predictive value of the expression levels for the other efflux pumps was, however, limited in this study.
In the most recent review on the role of P-gp in breast cancer [4], the authors concluded that P-gp is detected in a significant proportion of untreated breast cancers (on average 40 by immunohistochemistry) without a clear and consistent association with cancer stage and that exposure to chemotherapy increased its expression. A stronger association of P-gp/MDR1 with response rates has been observed when expression or an increase in expression is detected immediately following chemotherapy. Correlations with prognosis appear more evident in studies using immunohistochemistry in the adjuvant and neoadjuvant setting. In summary, although other resistance mechanisms play a role in breast cancer, the P-gp is responsible for MDR in a defined subpopulation of breast cancer patients and thus represents a molecular target for circumventing MDR.
Consequently, there has been a concerted effort over the past 20 years to develop specific P-gp modulators [12–21]. P-gp inhibitors have been developed primarily using combinatorial chemistry and demonstrated high selectivity for P-gp with inhibitory activities in the low nanomolar range. Prominent examples are shown in Fig. 1.

Examples of potent P-gp inhibitors that have undergone or are currently being investigated in clinical trials
Several clinical studies have been performed and demonstrated that the pharmacokinetic profile of co-administered anticancer drugs was not altered [2, 13–21]. Although some objective responses were achieved, combination therapy with these MDR modulators caused unacceptable toxicity, e.g., severe neurotoxicity [22].
In summary, therapeutic strategies that combine a P-gp inhibitor systemically with anticancer agents have the following inherent disadvantages: (a) systemic toxicity of the MDR modulators, (b) sufficient concentrations of the MDR modulator and the anticancer drug in resistant tumor cell are not achieved simultaneously as the therapy schedule for two pharmacokinetically unrelated drugs remains unpredictable and needs to be assessed empirically, and (c) a lack of tumor targeting for the MDR modulators as well as anticancer.
Thus, we focused our efforts on developing dual acting prodrugs for the treatment of resistant breast cancer of patients with intrinsic or acquired resistance caused by an over-expression of the P-gp. Recently, we reported on the synthesis and cleavage properties of a dual prodrug AJ-077 comprising doxorubicin and the potent P-gp inhibitor ONT-093, currently in clinical trials [23], that binds to the cysteine-34 position of circulating albumin through its maleimide group [22]—see Fig. 2. Both drugs were bound to a bispecific linker through a dipeptide substrate Phe-Lys-PABC (para-aminobenzyloxycarbonyl) that is effectively cleaved by cathepsin B, a protease that is over-expressed in breast cancer [24].

Structure of the dual prodrug AJ-077 comprising doxorubicin (violet) and a β-alanine derivative of the P-gp-inhibitor ONT-093 (green) that are bound through a cathepsin substrate Phe-Lys-PABC (para-aminobenzyloxycarbonyl) (red) a bifunctional maleimide linker (blue). The maleimide acts as a thiol-binding group that binds rapidly and selectively to the cysteine-34 position of circulating albumin and after accumulation of the thus formed drug albumin conjugate in tumor tissue and cells releases doxorubicin and the β-alanine derivative of ONT-093, i.e., AJ-038 due to cleavage by cathepsin B of the Phe–Lys dipeptide followed by a 1,6-elimination of PABC [22]
The new dual prodrug AJ-077 binds rapidly and specifically through its thiol-binding maleimide group to the cysteine-34 position of endogenous albumin and exploits the passive targeting properties of albumin for solid tumors [25, 26]. Albumin is playing an increasing role as a drug carrier in the clinical applications, and albumin uptake in malignant and inflamed tissue is mediated by the pathophysiology of tumor tissue, characterized by angiogenesis, hypervasculature, a defective vascular architecture, and an impaired lymphatic drainage combined with an absent or defective lymphatic drainage system, a phenomenon coined as the enhanced permeability and retention (EPR) of macromolecules for solid tumors [27]. In situ binding of maleimide-based prodrugs to the cysteine-34 position of circulating albumin is meanwhile a preclinically and clinically validated technology of increasing the therapeutic index of anticancer agents. An acid-sensitive prodrug of doxorubicin, the (6-maleimidocaproyl)hydrazone derivative of doxorubicin (INNO-206, previously known as DOXO-EMCH) developed in our group has been studied clinically [28] and has entered phase II studies in soft tissue sarcoma (see http://www.cytrx.com).
Furthermore, a series of albumin-binding prodrugs with enzymatically cleavable linkers have been developed in the past 5 years demonstrating superior efficacy and tolerability in preclinical tumor-bearing mice models [25, 29].
Finally, Abraxane®, an albumin paclitaxel nanoparticle which acts as an albumin paclitaxel complex in vivo was approved for treating metastatic breast cancer in 2007 [25]. Thus, there is a good rationale for using albumin as a carrier for treating metastatic and resistant stages of this tumor indication.
The dual prodrug shown in Fig. 2 could have the advantage that once bound to circulating albumin transports an anticancer agent and an inhibitor of P-gp simultaneously on a molecular level to resistant cancer cells that express P-gp thus preventing the efflux of the anticancer drug with a concomitant increase in tumor cell death. The anticancer agent of choice was doxorubicin as an anthracycline-based regimen is routinely used to treat patients with breast cancer and in addition the extent of P-gp expression correlated with in vitro resistance to doxorubicin in functional assays [30]. As a potent P-gp inhibitor, we selected ONT-093 because its structure–activity relationships have been intensely studied and it possesses suitable functional groups for chemical modification [31]. ONT-093 reversed resistance to doxorubicin and paclitaxel in six different cell lines expressing P-gp in the low nanomolar range with an average EC50 value of 32 nM [32].
As the MDR modulator ONT-093 has no suitable functional groups for direct derivatization, a structural modification was required. Zhang et al. [31] reported that the replacement of the isopropyl amino groups in ONT-093 with dimethyl amino groups or other substituents has only a very minor effect on the activity of the substance. Prompted by these results, we replaced one isopropyl amino group with a primary amino group, and replaced the other isopropyl amino group with a tertiary amine to prevent any competition reaction thus obtaining the derivative of ONT-093, AJ-038 (see Fig. 2), that is also the released compound after cathepsin B cleavage of the albumin conjugate of AJ-077 [22].
Subsequently, we investigated the effect of the release product of the new albumin conjugate HSA-AJ-077, i.e., AJ-038 on doxorubicin in drug resistant P-gp over-expressing breast cancer cell lines (MCF-7/ADR, MT-3/ADR). Unexpectedly, however, the modified ONT-093 derivative, AJ038 demonstrated essentially no reversal of MDR, and its P-gp inhibitory effect was obviously lost (see “Results and discussion” section) which was not in accordance with the structure–activity relationships reported in the literature for ONT-093 derivatives [31].
We therefore decided to investigate one of the newest and most potent inhibitors of the P-gp efflux pump. For this purpose, we chose zosuquidar (LY335979) that has been investigated in clinical trials [33, 34]. Furthermore, its chemical structure allows chemical modification through its hydroxyl group (see Fig. 1).
The objective of this study was to investigate the ability of an albumin-binding prodrug of zosuquidar to circumvent the MDR phenotype in MCF-7 and MT-3 resistant cell lines in the presence of doxorubicin and the acid-sensitive albumin-binding prodrug of doxorubicin, the 6-maleimidocaproyl hydrazone derivative of doxorubicin (DOXO-EMCH, INNO-206—see Fig. 3), currently in clinical trials. We chose to synthesize an acid-sensitive albumin-binding prodrug of zosuquidar in a two-step synthesis avoiding the disadvantages related to the more complex multi-step synthesis of dual prodrugs. Furthermore, this strategy allows to readily evaluate and optimize the most efficacious ratio of the P-gp inhibitor and anticancer drug in vitro. In this study, we show that zosuquidar as well as its acid-sensitive albumin conjugate were able to reverse MDR in two P-pg expressing breast cancer cells (MCF-7ADR and MT-3/ADR).

Chemical structure of the (6-maleimidocaproyl)hydrazone derivative of doxorubicin, DOXO-EMCH
Materials and methods
General
Doxorubicin (Dox) hydrochloride was purchased from Yick-Vic Chemicals & Pharmaceuticals (HK, China). Dry solvents were from Fluka Sure Seal grade. All other chemicals and solvents were purchased from Sigma-Aldrich, Fluka, Merck and Roth (Germany) and were used without further purification. The buffers used were vacuum filtered through a 0.2-μm membrane (Sartorius, Göttingen, Germany) and thoroughly degassed with ultrasound before use. Centrifugation was carried out with a Tomy Capsule microcentrifuge model HF-120 (Peninsula Laboratories, Belmont, CA). The solvents were removed under reduced pressure at 40°C. Electron spray ionization mass spectra (ESI-MS) were obtained on a Thermo Electron LCQ Advantage with associated MAT SS 200 data.
Analytical reversed-phase HPLC was carried out on a Kontron System using a solvent delivery system (HPLC Pump 422), a variable wavelength UV–Visible detector (HPLC Detector 430) with the column symmetry 300 (250 × 4.6 mm) from Waters. For peak integration, Geminyx software (v 1.91 by Goebel Instrumentelle Analytik, Germany) was used. HPLC conditions were as follows: flow rate: 1 ml/min, λ = 254 and 220 nm; injection volume: 50 μl, gradient: 0–5 min 100% mobile phase A; 5–40 min increase to 100% mobile phase B; 40–45 min 100% mobile phase B; 45–50 min decrease to 100% mobile phase A; 50–60 min 100% mobile phase A; mobile phase A: 10 mM sodium phosphate buffer (pH 7.0)/acetonitrile 70:30; mobile phase B: 10 mM sodium phosphate buffer (pH 7.0)/acetonitrile 20:80.
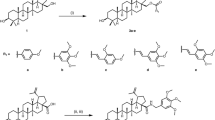
Synthesis of the prodrug 2
Zosuquidar was obtained according to the literature in a 8-step synthesis resulting in 2 g of a light yellowish compound [35]. A solution of zosuquidar (1 equiv, 4 mmol, 625 mg) and triethylamine (6 equiv, 6 mmol, 815 μl) in dry dichloromethane (50 ml) was added to a solution of 4-acetylbenzoyl chloride [36] (6 equiv, 6 mmol, 1075 mg) in dry dichloromethane (25 ml). The reaction solution was stirred at room temperature for 30 min (TLC; chloroform/methanol, 1:1), and was then treated with methanol (400 μl). The solvents were removed under reduced pressure, and the residue was purified by flash chromatography eluting with chloroform/methanol 30:1 to yield 1 as a white solid (475 mg, 72%). C41H37F2N3O4, ESI-MS: calcd [M + H]+ 674.28, found 674.00; HPLC analysis: (λ = 254 nm) > 97%—see Supporting information for MS spectrum.
A solution of ε-maleimidocaproic acid hydrazide trifluoroacetic acid (EMCH·TFA) (4 equiv, 2.32 mmol, 790 mg) [37] in dry methanol (5 ml) (Scheme 1, middle) was added to a solution of 1 (1 equiv, 0.58 mmol, 390 mg) in dry methanol (100 μl), and the reaction solution was stirred at room temperature for 1.5 h (TLC; chloroform/methanol, 1:1). The solvent was removed under reduced pressure, and the residue was purified by flash chromatography eluting with chloroform/methanol 30:1 to yield 2 as a white solid (453 mg, 89%). C51H50F2N6O6, ESI-MS: calcd [M + H]+ 881.38, found 881.20; HPLC analysis: (λ = 254 nm) >98%—see Supporting information for MS spectrum.

Synthesis of the acid-sensitive zosuquidar prodrug 2 and subsequent formation of the albumin-bound form HSA-2
Preparation of the albumin conjugate HSA-2
For conjugation experiments, a stock solution (2 mg/ml, 2.27 mM) of the prodrug 2 was prepared by dissolving the prodrug (2 mg) in Cremophor EL/ethanol (1/1) (60 μl) followed by dilution with saline (940 μl).
2 (100 μl, 2.27 mM) was subsequently incubated with fully reduced human serum albumin (HSA) containing ~90% free thiol groups (2.26 ml, 135 mg) [37] under slight stirring at 37°C at a final concentration of 100 μM and pH 7.0. Aliquots (50 μl) were taken at the time points stated in the chromatograms (see Figs. 4, 6) and analyzed by HPLC at 220 and 254 nm using the HPLC conditions previously described preciously. Samples were kept frozen at −80°C and thawed before HPLC analysis.

Chromatograms of incubation studies of the prodrug 2 with HSA at λ = 254 nm. The characteristic peak of 2 (100 μM) (retention time ~ 38 min) disappears and is ~90% bound to HSA within 2 min (green chromatogram). When the cysteine-34-position is no longer available after prior reaction with an excess of 6-maleimidocaproic acid (EMC), the thiol-binding prodrug 2 does not bind to albumin (red line) even after 15 min. See “Materials and methods” section for incubation and HPLC conditions
In an analogous experiment and to show that the thiol-reactive prodrug 2 binds selectively and rapidly to the cysteine-34 sulfhydryl group of HSA with the maleimide group of the prodrug, 2 (25 μl stock solution, 2.27 mM) was added slowly at a final concentration of 100 μM to a solution of HSA containing ~90% free thiol groups (0.18 μmol free thiol groups, 200 μl, 12 mg) that was first pre-incubated with a threefold excess of ε-maleimidocaproic acid (EMC) (0.54 mmol, 114 mg) for 5 min. Aliquots (50 μl) were taken after 15 min and analyzed by HPLC at 220 and 254 nm using the HPLC conditions described above.
Albumin-binding properties of the prodrug 2 in human plasma
Human blood plasma (EDTA-stabilized) was taken from healthy volunteers. Prodrug 2 (25 μl of the stock solution, 2.27 mM) was added to human plasma (500 μl) at a final concentration of 100 μM, and the sample was incubated at 37°C. Aliquots (50 μl) were taken at the time points stated in the chromatograms (see Fig. 5) and analyzed by HPLC at 220 and 254 nm using the HPLC conditions described above.

Chromatograms of incubation studies of the prodrug 2 with human plasma at λ = 254 nm. A chromatogram of the cleavage product 1 (see Scheme 1) is shown as a reference. The characteristic peak of 2 (100 μM) (retention time ~ 38 min) disappears in ~90% after incubation with plasma within 2 min (green chromatogram). Hydrolysis of HSA-2 to the acetylbenzoic acid ester derivative of zosuquidar 1 (retention time ~ 40 min) was not observed after 24 h and showed only small amounts of unbound 2. See “Materials and methods” section for HPLC conditions
Cleavage studies of the conjugate HSA-2 in acidic medium
300 μl of the HSA-2 stock solution (100 μM) at 37°C was brought to pH 5.0 with sodium phosphate buffer (10 mM, pH 3.0). Aliquots (50 μl) were taken at 10 and 70 min and analyzed by HPLC at 220 and 254 nm. To study the stability of HSA-2 at pH 7.0, 150 μl of the HSA-2 stock solution (pH 7.0, 100 μM) were incubated at 37°C and analyzed after 24 h by HPLC. The data are shown in Fig. 6.

Chromatograms of an incubation study of the albumin conjugate HSA-2 at pH 5.0 (10 min, 70 min) and pH 7.0 (24 h) at 37°C. Acid-sensitive hydrolysis of the hydrazone bond yields 1 (retention time ~ 40 min) as the cleavage product, the acetylbenzoic acid ester derivative of zosuquidar within an incubation time of 70 min. A chromatogram of the cleavage product 1 (see Scheme 1) is shown as a reference. The conjugate was stable at pH 7.0 and compound 1 was not detected after a 24 h incubation time and merely showed small amounts of unbound 2. See “Materials and methods” section for HPLC conditions
Cellular experiments
Cell culture and generation of luciferase expressing cell lines
MT-3, MT-3/ADR, MCF-7, and MCF-7/ADR were generous gifts from Dr. Iduna Fichtner, Max Delbrück Center for Molecular Medicine, Berlin, Germany [38, 39]. All cell lines (established from human mammary cancer) were cultured in DMEM containing 10% FBS and 1% penicillin–streptomycin in 10% CO2 at 37°C. The doxorubicin-resistant cell lines MCF-7/ADR and MT-3/ADR were maintained in media containing 0.1 μg/ml doxorubicin. Luciferase expressing cell pools were generated via transduction using a lentivirus encoding a luciferase-neomycin fusion protein and selected using 1 mg/ml G418 [40].
Rhodamine 123 efflux assay
The rhodamine efflux assay is based on the observation that rhodamine 123, which is a cell permeable green fluorescent dye, is a substrate of P-gp/ABCB1. Cells over-expressing the MDR pump do not accumulate rhodamine 123 [41]. However, treatment of these cells with a P-gp inhibitor, such as zosuquidar, blocks the P-gp-mediated efflux, resulting in an accumulation of rhodamine. The assay was carried out as described in the literature [41]. In short, 1.5 × 106 MCF-7, MCF-7/ADR, MT-3, and MT-3/ADR cells were pelleted in 15 ml Falcon tubes, washed once with 10 ml DMEM without antibiotics, and re-suspended in 1.5 ml DMEM without antibiotics. 250 μl of the cell suspension were then each distributed into Eppendorf tubes, and to two tubes were added 250 μl DMEM without antibiotics, the latter including either 1 mg/ml rhodamine 123 or latter including 1 μM zosuquidar. After 30 min at 37°C at 10% CO2, one half of the tubes were left on ice, whereas the medium of the other tubes was replaced by DMEM without antibiotics, but containing 1 μM zosuquidar. After an additional 90-min incubation period at 37°C and 10% CO2, the cells were taken up in ice-cold PBS, and analyzed by FACS, or fixed under a coverslip, and analyzed by fluorescence microscopy.
Results and discussion
As described in the “Introduction” section, we first determined the IC50 concentrations of doxorubicin in sensitive (MCF-7, MT-3) and drug resistant P-gp over-expressing breast cancer cell lines (MCF-7/ADR, MT-3/ADR) in the presence and absence of ONT-093 as well as the release product of the new albumin conjugate HSA-AJ-077, i.e., AJ-038 (see Fig. 2). Cyclosporine A, a potent P-gp inhibitor was additionally assessed as a positive control. The results of these studies are shown in Table 1.
MCF-7 and MT-3 non-resistant cells were sensitive to doxorubicin, with IC50 values of 0.3 and 1.0 μM, respectively (data not shown). As expected, however, the P-gp expressing cells were far less sensitive, with IC50 values of >50 μM for the MCF-7/ADR and ~20 μM for the MT-3/ADR cell line (Table 1). The original ONT-093 compound showed relatively low toxicity, with IC50 values ~14 and ~7 μM toward MCF-7/ADR and MT-3/ADR cells, respectively. Furthermore, the addition of 5 μM ONT-093 to the resistant cell lines in combination with a semi-logarithmic dilution series of doxorubicin resulted in a shift of the IC50 value indicating a reversal of the MDR phenotype. The interference with the P-gp was more pronounced in the MT-3 cell line, which showed doxorubicin sensitivity that was even lower than that of the wild-type cells (IC50 value = 1.0 μM vs. IC50 value = 0.28 μM, see Table 1). MCF-7/ADR cells showed a less pronounced effect, but sensitivity to doxorubicin was restored to some degree (Table 1). Unexpectedly, however, the modified ONT-093 derivative, AJ-038, demonstrated essentially no reversal of MDR and appeared to have lost its P-gp inhibitory activity (Table 1). Cyclosporine A, a competitive P-pg inhibitor included as a positive control, showed slightly increased general toxicity compared to ONT-093. It showed complete MDR reversal in the MT-3/ADR cell line (IC50 value = 0.24 μM vs. IC50 value = 0.3 μM for doxorubicin in the non-resistant cell line) and adequate reversal in the MCF-7 cell line (IC50 value = 2.9 μM vs. IC50 value = 1.0 μM for doxorubicin) activity in both cell lines (Table 1).
This surprising finding, which was not in agreement to the published reports on the structure–activity relationships of ONT-093 derivatives which had shown that replacing the isopropyl amino groups with a spectrum of substituents did not have an influence on the ability to inhibit P-gp [31], precluded further preclinical assessment of the new dual acting prodrug.
We therefore decided to investigate another potent inhibitor of the P-gp efflux pump, i.e., zosuquidar (LY335979) that has been investigated in clinical trials [33, 34]. The objective of the study was to evaluate its potential as well as of a respective acid-sensitive albumin-binding prodrug of reversing MDR in resistant breast cancer cells toward doxorubicin and the albumin-binding prodrug of doxorubicin (DOXO-EMCH).
Design of the albumin-binding prodrug of zosuquidar
Zosuquidar was synthesized according to the literature [35]. For the design of the prodrug (see Scheme 1), we chose to introduce a maleimide linker incorporating an acid-sensitive hydrazone bond as this bond is stable after binding of the maleimide group to the cysteine-34 group of HSA and is cleaved in acidic conditions present in the extracellular environment of solid tumors as well as in the endosomes/lysosomes of tumor cells [26].
For developing such a prodrug (Scheme 1), we used our established strategy of introducing a carbonyl group through reaction of the building block 4-acetylbenzoyl chloride [36] with the hydroxyl group of a pharmaceutically active compound that then allows the derivatization with a hydrazide group. We have previously used this strategy to develop acid-sensitive prodrugs of paclitaxel and respective poly(ethylene) conjugated retained the anticancer activity of paclitaxel in three human tumor cell lines (MACL MCF-7 mammary carcinoma, MEXF 514 melanoma carcinoma, and RXF 944 renal cell carcinoma) [36].
In analogy, the hydroxyl group of zosuquidar was esterified with 4-acetylbenzoyl chloride to yield 1 and subsequent reaction with 6-maleimidocaproic acid hydrazide (EMCH) in dry methanol furnished the prodrug 2 which was purified by chromatography to yield a yellowish powder. The prodrug 2 was characterized by analytical HPLC and mass spectroscopy (see Supporting information). It showed water solubility of 2 mg/ml in saline when diluted with 6% Cremophor EL/ethanol (1/1).
Binding, cleavage, and plasma stability of the albumin-binding prodrug of zosuquidar
Following the synthesis of the prodrug 2, we studied its albumin-binding properties. In analogy to our previous study [37, 42], 2 was quantitatively and selectively bound through its maleimide moiety to the cysteine-34 group of HSA within 2 min at 37°C and pH 7.0 to yield the albumin-bound prodrug HSA-2 as shown by HPLC (see Fig. 4). While the free prodrug elutes at ~38 min, the signal disappeared almost completely after a 2-min incubation period with HSA, and the peak eluting at the retention time of HSA ~16 min can be assigned to the formed conjugate. To prove that the fast binding results from a Michael addition of the sulfhydryl group of HSA with the maleimide group of the prodrug, analogous HPLC experiment was carried out in which the cysteine-34 position of HSA was blocked with an excess of ε-maleimidocaproic acid (EMC) before incubation with 2 (Fig. 4). In this case, no binding to HSA was observed after incubating HSA with 2.
The plasma stability of an albumin-bound form of a prodrug is an important criterion for the biological activity of a synthesized prodrug. It should be stable long enough to enable the transport of the active molecule to the tumor and tumor cells through passive targeting of albumin mediated by the EPR effect.
To determine the plasma stability of 2, the respective albumin conjugate HSA-2 was generated by incubation of the prodrug 2 with EDTA-stabilized human plasma at a final concentration of 100 μM at 37°C, and subsequently samples were analyzed after an incubation period of 2 min and analyzed by HPLC (Fig. 5). The signal corresponding to the free prodrug (~38 min) disappeared almost completely after a 2-min incubation period with HSA, and the peak eluting at the retention time of HSA ~16 min can be assigned to the formed conjugate. No hydrolysis to compound 1, the acetylbenzoic ester derivative of zosuquidar, was observed after 24 h. As with exogenous HSA, HPLC studies demonstrated an essentially quantitative, selective and fast binding to endogenous albumin in analogy to our previous experience with albumin-binding prodrugs (Fig. 5) [37].
The cleavage properties of HSA-2 were evaluated at pH 5.0 and 37°C. Reverse phase HPLC showed that the prodrug 2 that incorporates a carboxylic hydrazone bond releases the zosuquidar derivative 1 completely when incubated at pH 5.0 (37°C) for 70 min (Fig. 6).
The prodrug was stable at pH 7.0 for 24 h and no cleavage to the derivative 1 was detected by HPLC (Fig. 6). HSA-2 contains an acid-sensitive hydrazone linker that principally allows the acetylbenzoic acid ester derivative of zosuquidar 1 to be released either extracellularly in the slightly acidic environment often present in tumor tissue (pH 6.0–6.8) or intracellularly in acidic endosomal or lysosomal compartments after cellular uptake of the conjugate by the tumor cell (pH 6.0–4.0). Due to the pronounced cleavage at pH 5.0 and stability at pH 7.0 (see Fig. 6), we would expect that 1 is primarily released intracellularly after cellular uptake of HSA-2.
Cellular experiments
Zosuquidar prevents the efflux of rhodamine 123 in P-gp expressing resistant cells MCF-7/ADR and MT-3/ADR as shown by fluorescence microscopy and FACS analysis
Zosuquidar has been shown to fully reverse the MDR phenotype in CEM/VLB100 cells when incubated simultaneously with doxorubicin, paclitaxel, or vinblastine [15] rendering the compound an attractive target for our prodrug strategy of circumventing MDR. To confirm the MDR status of the MCF-7/ADR and MT3/ADR cells, a rhodamine 123 efflux experiment was performed. Rhodamine 123 is a substrate of P-gp and P-gp expressing tumor cells therefore do not accumulate the dye. Resistant and sensitive breast cancer cells (MT-3, MT-3/ADR, MCF-7, and MCF-7/ADR) were thus incubated with rhodamine 123 in the presence or absence of zosuquidar followed by a chase again in the presence or absence of zosuquidar. As shown in Fig. 7, whereas wild-type MT-3 and MCF-7 cells readily accumulated the dye in the absence of zosuquidar, their doxorubicin-resistant counterparts efficiently pumped the fluorophore out (Fig. 7a, b).

a, b Rhodamine 123 efflux assay using MT-3 and MCF-7 wild-type as well as the P-gp expressing resistant cell lines MCF-7/ADR and MT-3/ADR. After a 30-min exposure of cells to rhodamine 123 at 37°C, cells were kept either on ice or were incubated in medium with or without 1 μM zosuquidar for an additional 90 min at 37°C and analyzed either by FACS (a) or by fluorescence microscopy (b)
Addition of zosuquidar, however, efficiently blocked the efflux of the dye leading to an accumulation of the fluorescent label in the cells (Fig. 7a, b).
Cytotoxic activity of doxorubicin as well the albumin conjugates of DOXO-EMCH in the presence of zosuquidar and the albumin conjugate of zosuquidar, HSA-2
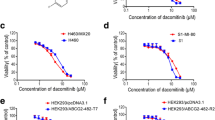
In the next step, we studied the effects of doxorubicin as well of HSA–DOXO-EMCH, the albumin conjugate of DOXO-EMCH, on wild-type and resistant MT-3, MT-3/ADR, MCF-7, and MCF-7/ADR cells. Furthermore, the ability of zosuquidar and its albumin conjugate (HSA-2) to reverse the MDR phenotype were tested. The prodrugs were measured in their albumin-bound forms as formed in situ after intravenous administration and binding to the cysteine-34 group of albumin. Cytotoxicity was determined using the luciferase assay.
While the MDR cell lines showed the expected robust resistance toward doxorubicin with IC50 values > 50 μM, the IC50 values of the sensitive cell lines were 0.29 μM (MCF-7) and 1.27 μM (MT-3). The IC50 value of zosuquidar was ~10 μM in both cell lines (Table 2).
When combining the low-molecular weight P-gp inhibitor with the cytotoxic drug, we found no effect on the doxorubicin sensitivity of wild-type MCF-7 cells, but a slight sensitization of the wild-type MT-3 cells (Table 2). In contrast, the P-gp inhibitor zosuquidar—as expected—reversed the MDR in the resistant cell lines. Exposure of resistant cells to 1 μM zosuquidar, i.e., a concentration well below the IC50 value of the P-gp inhibitor, resulted in a shift of the doxorubicin IC50 value from >50 to ~1 μM in the MCF-7/ADR cells, and 0.3 μM in the MT-3/ADR cells (Table 2).
In a next step, the albumin-bound form of zosuquidar (HSA-2) was tested instead of the free P-gp inhibitor. The IC50 value of HSA-2 was found to be >10 μM in all cell lines (Table 2).
The chemical modifications introduced at the hydroxyl group of the P-gp inhibitor zosuquidar to form the prodrug may have affected the IC50 value of native zosuquidar in its ability to restore doxorubicin sensitivity in the MDR cell lines. To examine whether this was the case, we kept the doxorubicin concentration constant at 5 μM, and determined the concentrations of zosuquidar or HSA-2 that were required to induce cell killing by inhibiting P-gp and circumventing doxorubicin resistance. Indeed, the amount of P-gp inhibitor that was necessary to revert MDR shifted from single digit nM concentrations for the free drug to 40 to 80-fold higher values for the albumin drug conjugate HSA-2 (Table 2). However, with effective concentrations between 30 and 250 nM, the albumin-binding prodrug is still a very active compound. Thus, when evaluated for its potential to reverse MDR, 5 μM HSA-2 included in the assay, shifted the IC50 of doxorubicin in MCF-7/ADR as well as MT-3/ADR cell lines to a value comparable to that of the parental cell lines (Table 2).
Last but not least, the ability of zosuquidar as well as its albumin conjugate HSA-2 to reverse resistance against HSA–DOXO-EMCH was tested. The albumin-bound form of DOXO-EMCH is not as active in cell culture experiments as the free drug, due to a slower uptake by adsorptive endocytosis compared to the free diffusion of the latter. Thus, confirming previous results, the IC50 values against the parental MCF-7 and MT-3 cells were two- to threefold higher than those of the free drug (0.29 vs. 0.65 μM in MCF-7 cells and 1.27 vs. 4.14 μM in MT-3 cells) [37].
As expected, the doxorubicin-resistant cell lines MCF-7/ADR and MT-3/ADR were insensitive against HSA–DOXO-EMCH. The presence of zosuquidar or HSA-2 sensitized the MT-3/ADR cells resulting, as for the free doxorubicin above, in lower IC50 values than those in the parental cells. MCF-7/ADR cells were sensitized as well, but kept a level of resistance with IC50 values ten times higher than their wild-type counterparts (Table 2).
Ultimately, a combination of two albumin-binding prodrugs might represent a promising strategy as the biodistribution of the bound drugs is determined by the pharmacokinetic property of HSA, rather than that of the individual drug allowing for a fine tuned time-scheduled delivery of the two compounds at an optimal ratio to tumor cells in vivo.
Conclusions
In conclusion, we have demonstrated that zosuquidar and an acetyl benzoic ester derivative of zosuquidar (1) as well as its acid-sensitive albumin conjugate are able to reverse the doxorubicin resistance of two breast cancer cell lines MCF-7/ADR and MT-3/ADR when additionally exposed to either doxorubicin or an albumin conjugate of the acid-sensitive albumin prodrug of doxorubicin i.e., the 6-maleimidocaproyl hydrazone derivative of doxorubicin (DOXO-EMCH) an albumin-binding prodrug, renamed INNO-206 that is undergoing clinical trials. These results form a good basis for evaluating the antitumor efficacy of doxorubicin as well as DOXO-EMCH together with the administration of zosuquidar as well with the albumin-binding prodrug 2 that releases the active benzoic ester derivative of zosuquidar 1. Although other resistance mechanisms play a role in solid tumors due to the heterogeneity and genomic instability of the tumor cell population, it is feasible that P-gp is responsible for MDR in a defined subpopulation of breast cancer patients and thus represents a molecular target for circumventing MDR. In addition, if a preclinical proof of principle can be demonstrated for combining albumin-binding prodrugs that transport a potent P-gp inhibitor as well as doxorubicin to resistant breast cancer cells, then such prodrugs would be attractive candidates for the treatment of mammary carcinoma patients who over-express the P-gp. Using new radioscintigraphy with 99Tc-sestamibi or 99mTc-methoxyisobutylisonitrile (MIBI) as P-gp substrates, the diagnostic tools are meanwhile available to select eligible patients with P-gp over-expression [43–45].
References
Lehnert M (1998) Chemotherapy resistance in breast cancer. Anticancer Res 18(3C):2225–2226
Thomas H, Coley HM (2003) Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control 10(2):159–165
Wang RB, Kuo CL, Lien LL, Lien EJ (2003) Structure–activity relationship: analyses of p-glycoprotein substrates and inhibitors. J Clin Pharm Ther 28(3):203–228
Leonessa F, Clarke R (2003) ATP binding cassette transporters and drug resistance in breast cancer. Endocr Relat Cancer 10(1):43–73
Sikic BI (1999) Modulation of multidrug resistance: a paradigm for translational clinical research. Oncology (Williston Park) 13(5A):183–187
Sikic BI, Fisher GA, Lum BL, Halsey J, Beketic-Oreskovic L, Chen G (1997) Modulation and prevention of multidrug resistance by inhibitors of P-glycoprotein. Cancer Chemother Pharmacol 40(Suppl):S13–S19
Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM (1999) Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol 39:361–398
Lu L, Leonessa F, Clarke R, Wainer IW (2001) Competitive and allosteric interactions in ligand binding to P-glycoprotein as observed on an immobilized P-glycoprotein liquid chromatographic stationary phase. Mol Pharmacol 59(1):62–68
Trock BJ, Leonessa F, Clarke R (1997) Multidrug resistance in breast cancer: a meta-analysis of MDR1/gp170 expression and its possible functional significance. J Natl Cancer Inst 89(13):917–931
Mechetner E, Kyshtoobayeva A, Zonis S, Kim H, Stroup R, Garcia R, Parker RJ, Fruehauf JP (1998) Levels of multidrug resistance (MDR1) P-glycoprotein expression by human breast cancer correlate with in vitro resistance to taxol and doxorubicin. Clin Cancer Res 4(2):389–398
Burger H, Foekens JA, Look MP, Meijer-van Gelder ME, Klijn JG, Wiemer EA, Stoter G, Nooter K (2003) RNA expression of breast cancer resistance protein, lung resistance-related protein, multidrug resistance-associated proteins 1 and 2, and multidrug resistance gene 1 in breast cancer: correlation with chemotherapeutic response. Clin Cancer Res 9(2):827–836
Chan HS, DeBoer G, Thiessen JJ, Budning A, Kingston JE, O’Brien JM, Koren G, Giesbrecht E, Haddad G, Verjee Z et al (1996) Combining cyclosporin with chemotherapy controls intraocular retinoblastoma without requiring radiation. Clin Cancer Res 2(9):1499–1508
Fischer V, Rodriguez-Gascon A, Heitz F, Tynes R, Hauck C, Cohen D, Vickers AE (1998) The multidrug resistance modulator valspodar (PSC 833) is metabolized by human cytochrome P450 3A. Implications for drug–drug interactions and pharmacological activity of the main metabolite. Drug Metab Dispos 26(8):802–811
Gruber A, Bjorkholm M, Brinch L, Evensen S, Gustavsson B, Hedenus M, Juliusson G, Lofvenberg E, Nesthus I, Simonsson B et al (2003) A phase I/II study of the MDR modulator Valspodar (PSC 833) combined with daunorubicin and cytarabine in patients with relapsed and primary refractory acute myeloid leukemia. Leuk Res 27(4):323–328
Dantzig AH, Shepard RL, Cao J, Law KL, Ehlhardt WJ, Baughman TM, Bumol TF, Starling JJ (1996) Reversal of P-glycoprotein-mediated multidrug resistance by a potent cyclopropyldibenzosuberane modulator, LY335979. Cancer Res 56(18):4171–4179
Toppmeyer D, Seidman AD, Pollak M, Russell C, Tkaczuk K, Verma S, Overmoyer B, Garg V, Ette E, Harding MW et al (2002) Safety and efficacy of the multidrug resistance inhibitor Incel (biricodar; VX-710) in combination with paclitaxel for advanced breast cancer refractory to paclitaxel. Clin Cancer Res 8(3):670–678
Starling JJ, Shepard RL, Cao J, Law KL, Norman BH, Kroin JS, Ehlhardt WJ, Baughman TM, Winter MA, Bell MG et al (1997) Pharmacological characterization of LY335979: a potent cyclopropyldibenzosuberane modulator of P-glycoprotein. Adv Enzyme Regul 37:335–347
Stewart A, Steiner J, Mellows G, Laguda B, Norris D, Bevan P (2000) Phase I trial of XR9576 in healthy volunteers demonstrates modulation of P-glycoprotein in CD56+ lymphocytes after oral and intravenous administration. Clin Cancer Res 6(11):4186–4191
Shepard RL, Cao J, Starling JJ, Dantzig AH (2003) Modulation of P-glycoprotein but not MRP1- or BCRP-mediated drug resistance by LY335979. Int J Cancer 103(1):121–125
Rubin EH, de Alwis DP, Pouliquen I, Green L, Marder P, Lin Y, Musanti R, Grospe SL, Smith SL, Toppmeyer DL et al (2002) A phase I trial of a potent P-glycoprotein inhibitor, zosuquidar·3HCl trihydrochloride (LY335979), administered orally in combination with doxorubicin in patients with advanced malignancies. Clin Cancer Res 8(12):3710–3717
Oza AM (2002) Clinical development of P glycoprotein modulators in oncology. Novartis Found Symp 243:103–115 discussion 115–118, 180–185
Abu Ajaj K, Kratz F (2009) Development of dual-acting prodrugs for circumventing multidrug resistance. Bioorg Med Chem Lett 19(3):955–1000
Velingkar VS, Dandekar VD (2010) Modulation of P-glycoprotein (PGP) mediated multidrug resistance (MDR) in cancer using chemosensitizers. Int J Pharm Sci Res 1(2):104–111
Sloane BF, Yan S, Podgorski I, Linebaugh BE, Cher ML, Mai J, Cavallo-Medved D, Sameni M, Dosescu J, Moin K (2005) Cathepsin B and tumor proteolysis: contribution of the tumor microenvironment. Semin Cancer Biol 15(2):149–157
Kratz F (2008) Albumin as a drug carrier: design of prodrugs, drug conjugates and nanoparticles. J Control Release 132(3):171–183
Kratz F, Beyer U, Schütte MT (1999) Drug-polymer conjugates containing acid-cleavable bonds. Crit Rev Ther Drug Carrier Syst 16(3):245–288
Maeda H, Wu J, Sawa T, Matsumura Y, Hori K (2000) Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release 65(1–2):271–284
Kratz F (2007) DOXO-EMCH (INNO-206): the first albumin-binding prodrug of doxorubicin to enter clinical trials. Expert Opin Investig Drugs 16(6):855–866
Kratz F, Muller IA, Ryppa C, Warnecke A (2008) Prodrug strategies in anticancer chemotherapy. ChemMedChem 3(1):20–53
Kaklamani VG, Gradishar WJ (2003) Epirubicin versus doxorubicin: which is the anthracycline of choice for the treatment of breast cancer? Clin Breast Cancer 4(Suppl 1):S26–S33
Zhang C, Sarshar S, Moran EJ, Krane S, Rodarte JC, Benbatoul KD, Dixon R, Mjalli AM (2000) 2,4,5-Trisubstituted imidazoles: novel nontoxic modulators of P-glycoprotein mediated multidrug resistance. Part 2. Bioorg Med Chem Lett 10(23):2603–2605
Newman MJ, Rodarte JC, Benbatoul KD, Romano SJ, Zhang C, Krane S, Moran EJ, Uyeda RT, Dixon R, Guns ES et al (2000) Discovery and characterization of OC144–093, a novel inhibitor of P-glycoprotein-mediated multidrug resistance. Cancer Res 60(11):2964–2972
Ruff P, Vorobiof DA, Jordaan JP, Demetriou GS, Moodley SD, Nosworthy AL, Werner ID, Raats J, Burgess LJ (2009) A randomized, placebo-controlled, double-blind phase 2 study of docetaxel compared to docetaxel plus zosuquidar (LY335979) in women with metastatic or locally recurrent breast cancer who have received one prior chemotherapy regimen. Cancer Chemother Pharmacol 64(4):763–768
Morschhauser F, Zinzani PL, Burgess M, Sloots L, Bouafia F, Dumontet C (2007) Phase I/II trial of a P-glycoprotein inhibitor, zosuquidar·3HCl trihydrochloride (LY335979), given orally in combination with the CHOP regimen in patients with non-Hodgkin’s lymphoma. Leuk Lymphoma 48(4):708–715
Barnett CJ, Huff B, Kobierski ME, Letourneau M, Wilson TM (2004) Stereochemistry of C-6 nucleophilic displacements on 1,1-difluorocyclopropyldibenzosuberanyl substrates. An improved synthesis of multidrug resistance modulator LY335979 trihydrochloride. J Org Chem 69(22):7653–7660
Rodrigues PCA, Scheuermann K, Stockmar C, Maier G, Fiebig HH, Unger C, Mülhaupt R, Kratz F (2003) Synthesis and in vitro efficacy of acid-sensitive poly(ethylene glycol) paclitaxel conjugates. Bioorg Med Chem Lett 13(3):355–360
Kratz F, Warnecke A, Scheuermann K, Stockmar C, Schwab J, Lazar P, Drückes P, Esser N, Drevs J, Rognan D et al (2002) Probing the cysteine-34 position of endogenous serum albumin with thiol-binding doxorubicin derivatives: improved efficacy of an acid-sensitive doxorubicin derivative with specific albumin-binding properties compared to that of the parent compound. J Med Chem 45:5523–5533
Naundorf H, Rewasowa EC, Fichtner I, Buttner B, Becker M, Gorlich M (1992) Characterization of two human mammary carcinomas, MT-1 and MT-3, suitable for in vivo testing of ether lipids and their derivatives. Breast Cancer Res Treat 23(1–2):87–95
Stein U, Walther W, Lemm M, Naundorf H, Fichtner I (1997) Development and characterisation of novel human multidrug resistant mammary carcinoma lines in vitro and in vivo. Int J Cancer 72(5):885–891
Elsadek B, Graeser R, Esser N, Schafer-Obodozie C, Tsurumi C, Abu Ajaj K, Warnecke A, Unger C, Saleem T, Kratz F (2011) In vivo evaluation of a novel albumin-binding prodrug of doxorubicin in an orthotopic mouse model of prostate cancer (LNCaP). Prostate Cancer Prostatic Dis 14(1):14–21
Lee JS, Paull K, Alvarez M, Hose C, Monks A, Grever M, Fojo AT, Bates SE (1994) Rhodamine efflux patterns predict P-glycoprotein substrates in the National Cancer Institute drug screen. Mol Pharmacol 46(4):627–638
Kratz F, Mueller-Driver R, Hofmann I, Drevs J, Unger C (2000) A novel macromolecular prodrug concept exploiting endogenous serum albumin as a drug carrier for cancer chemotherapy. J Med Chem 43(7):1253–1256
Del Vecchio S, Salvatore M (2004) 99mTc-MIBI in the evaluation of breast cancer biology. Eur J Nucl Med Mol Imaging 31(Suppl 1):S88–S96
Del Vecchio S, Zannetti A, Aloj L, Salvatore M (2003) MIBI as prognostic factor in breast cancer. Q J Nucl Med 47(1):46–50
Yoon JH, Bom HS, Song HC, Lee JH, Jaegal YJ (1999) Double-phase Tc-99m sestamibi scintimammography to assess angiogenesis and P-glycoprotein expression in patients with untreated breast cancer. Clin Nucl Med 24(5):314–318
Acknowledgments
We gratefully acknowledge the support of the Fördergesellschaft an der Klinik für Tumorbiologie an der Albert-Ludwigs-Universität Freiburg e.V.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Khalid Abu Ajaj and Ralph Graeser have contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10549_2011_1937_MOESM1_ESM.doc
1. Supporting information.doc: This file contains as a supporting information the mass spectrum of the final product. (DOC 49 kb)
10549_2011_1937_MOESM2_ESM.pdf
2. BMCL-2009.pdf: This paper describes the recently developed dual acting prodrug comprising doxorubicin and a derivative of the p-glycoprotein inhibitor ONT-093. It is attached for the benefit of the editor and the benefit of the referees. (PDF 381 kb)
Rights and permissions
About this article
Cite this article
Abu Ajaj, K., Graeser, R. & Kratz, F. Zosuquidar and an albumin-binding prodrug of zosuquidar reverse multidrug resistance in breast cancer cells of doxorubicin and an albumin-binding prodrug of doxorubicin. Breast Cancer Res Treat 134, 117–129 (2012). https://doi.org/10.1007/s10549-011-1937-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-011-1937-9