
Abstract
The Rho subgroup of the Rho GTPases consisting of RhoA, RhoB and RhoC induces a specific type of actin cytoskeleton and carry out a variety of functions in the cell. mDia and ROCK are downstream effectors of Rho mediating Rho action on the actin cytoskeleton; mDia produces actin filaments by nucleation and polymerization and ROCK activate myosin to cross-link them for induction of actomyosin bundles and contractility. mDia is potentially linked to Rac activation and membrane ruffle formation through c-Src-induced phosphorylation of focal adhesion proteins, and ROCK antagonizes this mDia action. Thus, cell morphogenesis, adhesion, and motility can be determined by the balance between mDia and ROCK activities. Though they are not oncogenes by themselves, overexpression of RhoA and RhoC are often found in clinical cancers, and RhoC has been repeatedly identified as a gene associated with metastasis. The Rho-ROCK pathway is implicated in Ras-mediated transformation, the amoeboid movement of tumor cells in the three-dimensional matrix, and transmigration of tumor cells through the mesothelial monolayer. On the other hand, the Rho-mDia1 pathway is implicated in Src-mediated remodeling of focal adhesions and migration of tumor cells. There is also an indication that the Rho pathway other than ROCK is involved in Src-mediated induction of podosome and regulation of matrix metalloproteases. Thus, Rho mediates various phenotypes of malignant transformation by Ras and Src through its effectors, ROCK and mDia.
Similar content being viewed by others


1 Rho signaling; ROCK and mDia1
The Rho family of GTPases has now expanded, consisting of more than 20 members [1, 2]. By Rho we refer here to prototypical members of Rho in a narrower sense, namely, RhoA, RhoB and RhoC, and review implication of their signaling in cancer, because these three Rho GTPases share the same group of effectors and are supposed to have similar mode of actions. Rho in this definition functions as a molecular switch in cellular processes such as cell morphogenesis, adhesion, migration and cell cycle progression including cytokinesis [1, 2]. Their conversion from the GDP-bound inactive form to the GTP-bound active form is catalyzed by the Dbl family of Rho GTPase-specific guanine nucleotide exchange factors (Rho GEFs) [3] and conversion from the GTP-bound form to the GDP-bound form is carried out by intrinsic GTPase activity stimulated by Rho-specific GTPase activating proteins (Rho GAPs) [4]. The primary action of Rho is to induce a specific type of actin cytoskeleton in the cell. Rho also modulates local dynamics of microtubules (MTs) in a stimulus-dependent manner, stabilizing a subset of microtubules. Typical actin cytoskeletons induced by Rho are stress fibers running in an interphase cell, and the contractile ring formed in a mitotic cell (Fig. 1a). Both stress fibers and the contractile ring are actomyosin bundles composed of anti-parallel actin filaments cross-liked by myosin. It is therefore reasoned that Rho induces production of actin filaments and activation of myosin, and locates thereby formed actomyosin bundles at different sites of a cell dependent on a phase of cell cycle (Fig. 1b). These actions of Rho are elicited by effectors that are activated downstream of Rho (Fig. 2a). There are two major effectors for Rho; one is ROCK (Rho-associated coiled-coil forming kinase) (Rho kinase/ROK) [5–7] and the other is mDia (mammalian homolog of Drosophila diaphanous) [8]. mDia is a formin molecule that catalyzes actin nucleation and polymerization and produces long, straight actin filaments [9], whereas ROCK is a serine/threonine kinase that can phosphorylate a variety of substrates [10]. One major substrate for ROCK is the myosin-binding subunit of myosin phosphatase, and ROCK inactivates it by phosphorylation [11, 12]. ROCK can also directly phosphorylate myosin light chain [13]. These two actions of ROCK, as a consequence, increase the myosin light chain phosphorylation, stimulate cross-linking of actin by myosin and enhance actomyosin contractility. ROCK also phosphorylates and activates LIM-kinase, which in turn phosphorylates and inactivates actin-depolymerizing and severing factor, cofilin [14]. This latter action of ROCK results in stabilization of existing actin filaments and increase in their content. It is presumed that these actions of ROCK and mDia on actin and myosin are combined downstream of Rho to induce actomyosin bundles in the cell (Fig. 2b). Indeed, expression of an active form of mDia induces stress fibers in cultured cells, and treatment of these cells with a specific ROCK inhibitor, Y-27632 [15], causes dissolution of the bundles, leaving the cells with diffusely distributed actin filaments [16]. Requirement of ROCK and mDia in the contractile ring formation and function was also reported [17, 18].

(a) Stress fibers and the contractile ring, two typical actin cytoskeletons induced by Rho. Both structures are composed of anti-parallel actin filaments cross-linked by myosin II. (b) Presumed Rho-regulated steps in assembly of actomyosin bundles. Rho is supposed to catalyze actin nucleation and polymerization to form actin filaments and activate myosin to cross-link them

(a) Rho effectors. Effector molecules for Rho are categorized by similarity of their domain structure. RBD, Rho-binding domain; PH, pleckstrin homology domain; FH, formin homology domain. ROCK has two isoforms, ROCK-I and ROCK—II, and mDia has three isoforms, mDia1, mDia2 and mDia3. PKN, Rhophilin and Rhotekin contain the homologous Rho-binding domain of about 90 amino acid stretch. (b) Site and mechanism of actions of mDia1 and ROCK in Rho-induced assembly of actomyosin bundles. mDia1 catalyzes actin nucleation and polymerization to form actin filaments. ROCK activates myosin to cross-link them. ROCK also inactivates cofilin through LIM-kinase and inhibits actin severing and depolymerization
Thus, cooperation of mDia and ROCK is required for assembly of actomyosin bundles such as stress fibers and the contractile ring. However, expression of constitutive active forms of ROCK alone results in disorganized actomyosin bundles by random bundling of actin filaments and cell contraction [19]. Co-expression of active mDia1 can correct this aberrant contraction by active ROCK and aligns actomyosin bundles as seen in stress fibers [16]. These results indicate that mDia may transmit a signal to modulate the ROCK action. Tsuji et al. [20] examined this issue by treating serum-starved Swiss3T3 cells either with botulinum C3 exoenzyme that blocks total Rho signaling [21] or with Y-27632 that inhibits the ROCK branch of Rho signaling and leaves the mDia branch intact. They compared the lysophosphatidic acid (LPA) -induced morphology of these cells, and found that treatment with Y-27632 results in formation of membrane ruffles. Analyzing the signaling mechanism therein, they found that the Rho-mDia1 signaling leads to activation of Rac through Src activation and the phosphorylation-dependent formation of Cas/Crk/DOCK180 complex, and that this pathway is suppressed by the ROCK activity. Thus, ROCK and mDia antagonize in Rho-dependent Rac activation, and the balance between the two pathways appears to determine the cell shape and pattern of stress fibers (Fig. 3). This mechanism appears to operate not only in fibroblasts but also in other types of cells. Opposing actions of mDia and ROCK were also reported in epithelial cells, though signaling pathways therein were not defined. There, mDia1 facilitates and ROCK disrupts cell-cell adhesion [22]. More consistent with the Tsuji’s finding, Arakawa et al. [23] used cultured cerebellar granule neurons, and examined involvement of mDia in SDF1α-induced neurite extension. They found that mDia1 can potentially activates Rac and facilitates axonal elongation but this action is tonically suppressed by the action of ROCK. Titration of intact Rho molecules with varying doses of C3 exoenzyme exhibited the biphasic response, that is, elongation at lower concentrations and suppression at higher concentrations. Given that activation of ROCK causes neurite retraction, these results indicates that a high level of active Rho activates both ROCK and mDia1 to induce retraction, whereas a lower level of Rho preferentially activates mDia1 to induce neurite elongation (Fig. 4). This view is consistent with the reported Kd values of the Rho-binding domains of ROCK and mDia for the GTP-bound form of Rho, 130 and 6 nM, respectively [24, 25], and can be useful in interpreting Rho actions in various settings and those exerted spatiotemporally at different locations in a single cell (see below).

Potential antagonism of the Rho-ROCK and the Rho-mDia1 pathways. mDia1 can potentially activate Rac through mobilization of c-Src and the Cas-Crk-DOCK180 complex formation. ROCK inhibits the mDia1-Rac pathway, whereas Rac can antagonize ROCK action.

Diversion of Rho signaling is dependent on the local level of Rho-GTP. Arakawa et al. (23) found in neuronal cells that the high level of Rho-GTP induces ROCK activation and actomyosin contractility, whereas the low level of Rho-GTP preferentially activates mDia1 and induces Rac activation, resulting in neurite retraction and elongation, respectively. Similar mechanism appears to operate in other types of cells and in different contexts (see text)
2 ROCK and mDia in cell migration
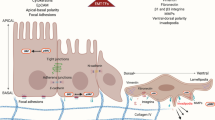
Earlier studies examining the role of Rho GTPases in cell migration used migration in two-dimensional culture for analysis such as the in vitro wound-healing assay or the assay using the Dunn chemotactic chamber [26, 27]. In these assays, cells migrate to the wound or to the chemotactic stimuli by polarizing to the direction of migration with extending protrusions at the front and the retracting tail at the rear [28] (Fig. 5a). Migrating cells then make adhesions to extracellular matrix (ECM) to stabilize the forward protrusion. Adhesions to ECM are used as sites to pull the cell body forward and are subsequently disassembled as the cell moves over them. This cycle of events enables cells to migrate to their destination. The actin cytoskeleton and microtubules (MTs) work critically in these events. Actin polymerization at the leading edge drives membrane protrusion, the association of the actin cytoskeleton with integrins regulate their binding to ECM, and the actin bundles within the body generate tension to pull the cell body forward and retract the tail. MTs are also polarized in migrating cells and are essential for the directed migration of many cell types, possibly by delivery of signaling molecules and membrane components [28]. Previous studies analyzing the effects of dominant active and negative mutants of Rho GTPases on this type of migration demonstrated that Cdc42 regulates cell polarity and Rac functions for membrane protrusion [26, 27]. One well-characterized action of Cdc42 in cell polarity is to orient the MT organizing center (MTOC) as well as the Golgi apparatus in front of the nucleus toward the leading edge [26], Subsequent studies showed that Cdc42 is indeed active at the cell front [29], and that Cdc42 activates the Par6-atypical protein kinase C (aPKC) complex there, which leads to MTOC orientation through local inhibition of GSK3β and accumulation of adenomatous polyposis coli protein (Apc) at the ends of microtubules in the leading edge [30, 31]. More recently Gomes et al. [32] suggest that the Cdc42/Par6/aPKC pathway described above maintains MTOC and Golgi at cell centroid through the microtubule-dynein interaction, while Cdc42 also induces actomyosin-dependent rearward pulling of the nuclei, and that the net action of the two forces results in MTOC orientation. As for membrane protrusion, Rac is shown to be active in the front of migrating cells [29, 33] and is thought to induce membrane protrusion by stimulating actin polymerization through activation of the WAVE-Arp2/3 complex pathway (see for review, [34]). Comparing with these Cdc42 and Rac actions, little is known concerning the role of Rho in cell migration [26, 28]. This is because inactivation of Rho abolishes cell adhesion almost completely and makes further analysis difficult [26]. Availability of selective ROCK inhibitor such as Y-27632 [12, 15] or introduction of RNAi in mammalian cells [23] has enabled us to manipulate selectively each signaling downstream of Rho and to define respective roles. One of the earliest studies along this approach revealed that the Rho-ROCK signaling is involved in tail retraction. Worthylake et al. [35] studied requirement of Rho-ROCK signaling in transendothelial migration of monocytes and found that this signaling is dispensable for attachment, spreading and crawling of monocytes on and through the endothelial layer but is required for tail retraction for completion of diapedesis. They reported that the ROCK signaling is necessary to negatively regulate integrin adhesions in the tail. The action of Rho signaling in tail retraction was also demonstrated in a simpler system. Yoshinaga-Ohara et al. [36] loaded neutrophils with C3 exoenzyme, and studied the chemotactic behavior of C3-treated neutrophils to FMLP. They found that Rho inhibition by C3 treatment did not interfere with development of polarity and protrusion of pseudopodia but impaired uropod detachment. Nonetheless, the movement of the front and cell body continued, and, as a result, C3-treated neutrophils exhibited abnormally elongated cell bodies (Fig. 5b). Worthylake and Burridge [37] further examined the mechanism of ROCK-mediated tail retraction and found that tail detachment by ROCK is not caused by myosin-based contractility, but due to attenuation of integrin-mediated adhesion. Given the fact that ROCK mediate clustering of integrin and induces large focal adhesions in fibroblasts (see fro example, 19), the mechanism suggested by their study appears paradoxical. They suggested, however, that leukocytes does not possess mature focal adhesions as seen in fibroblasts, but form small focal complexes of tethering and signaling molecules surrounding ligand-engaged integrins, and that ROCK is required to suppress them. Their findings that inhibition of ROCK in leukocytes increases the number of focal complexes and at the same time induces extensive ruffles around the perimeter of a cell may suggest that ROCK suppresses Rac-induced focal complexes and ruffle formation spatiotemporally (see below).

(a) Cell migration in two-dimensional culture. Cells migrating in two-dimensional culture polarize to the direction of migration with membrane protrusions at the front and retraction at the tail. Microtubules extending to the front are stabilized and the microtubule organizing center (MTOC) is positioned in front of the nucleus. (b) Impaired tail retraction by inhibition of the Rho-ROCK signaling. Neutrophils treated with C3 exoenzyme or Y-27632 show marked elongation of the cell body due to impaired tail retraction (Reprinted with modification from Fig. 1 of reference 36 with permission from Elsevier and courtesy of Naoko Yoshinaga-Ohara and Masataka Sasada of Kyoto University)
Compared to ROCK, study on the role of mDia in cell migration was slow due to the lack of appropriate experimental tool, but has recently been accelerated by introduction of RNAi. Grosse and collaborators [38] analyzed migration of mouse embryonic fibroblasts deficient in Gα12 and 13 in the wound-healing assay. G α12 and 13 are known to couple to Rho activation via a group of RGS-containing Rho GEFs including p115Rho-GEF and LARG (see for review, [39]). They found that MEF cells deficient in both G proteins exhibited impaired Rho activation during migration, and that the loss of Rho activation results in migration defects and impaired stabilization of microtubules directed to the front. They further observed that Rho co-localizes with mDia1 in the front edge of migrating cells and that depletion of mDia1 by RNAi also interfered with cell migration and MT stabilization. These results thus suggested requirement of Rho for cell migration in addition to that of Rac and Cdc42 shown in previous studies. Yamana et al. [40] also used RNAi for mDia1 in rat C6 glioma cells, and showed requirement of the Rho-mDia1 pathway in cell migration not only in the wound-healing assay but also in the Matrigel transwell assay. Their analysis in the Dunn chamber further showed that both directionality and locomotion were impaired in mDia1-depleted cells. Consistently, they found that mDia1 depletion interfered with not only MT stabilization as Grosse and collaborators found, but also cell polarization and focal adhesion turnover. By analyzing the mechanism underlying the polarization defect, they found that accumulation of Cdc42 and Apc at the front was impaired, and, by analyzing the mechanism for the defect in adhesion turnover, they found that c-Src accumulation and subsequent protein tyrosine phosphorylation in focal adhesions were impaired, in mDia1-depleted cells. c-Src was previously shown to induce focal adhesion disassembly [41]. On the basis of these findings, they suggested that, in migrating cells, the Rho-mDia1 pathway is activated in the front and facilitates migration, on one hand, by MT-dependent recruitment of Cdc42 and Apc in the front for cell polarization, and, on the other hand, by actin-dependent translocation of c-Src to focal adhesions to stimulate adhesion turnover (Fig. 6).

Role of the Rho-mDia1 pathway in cell migration. Yamana et al. (40) found that mDia1 localizes at the front of migrating cells, acts on both actin and microtubules, and induces adhesion turnover and cell polarization in rat C6 glioma cells
The works by the Grosse’s group and Yamana et al. thus clearly demonstrated the importance of Rho and its effector, mDia1, in cell migration, and suggested that this signaling collaborates with Rac and Cdc42 in this process. However, this view challenged the previous dogma that, among Rho GTPases, only Rac and Cdc42 are important and Rho is dispensable in cell migration. Furthermore, Arthur and Burridge demonstrated that p190Rho-GAP is activated by c-Src-dependent phosphorylation and the phosphorylated form is abundant in migrating cells and functions to down-regulate Rho [42, 43], further negating the role of Rho for migration. However, the role of Rho for cell migration is supported by a recent imaging study analyzing the localization of active Rho in migrating fibroblasts. Hahn and collaborators [44] developed a FRET biosensor for active Rho by conjugating a YFP-RhoA and CFP-Rho binding domain of Rhotekin, expressed in MEF cells, and examined localization of active Rho in cells during random migration or migration in the wound healing assay. Consistent with the previous findings [43], the Rho activity was minimal in the cell body. However, in addition to the high Rho activity in the tail of robust retraction, they found a sharp band of markedly higher Rho activity immediately adjacent to the front edge of cells with extending protrusion. This is consistent with the role of mDia1 proposed by the studies of the Grosse’s group and Yamana et al., and strengthens a significant role Rho plays for cell migration.
3 Rho signaling in metastasis and invasion
Given the roles of Rho GTPases in cell adhesion and migration, it is likely that they also play a role in tumor metastasis and invasion. Survey of genes over-expressed in clinical cancers and tumor cell lines showed frequent over-expression of RhoA and RhoC (see for review, [45]). Of the two, expression of RhoC was correlated with invasive phenotype of clinical cancers. Earlier, Suwa et al. [46] examined RhoA, RhoB and RhoC expression in 33 cases of pancreatic ductal adenocarcinoma, and found that the expression level of RhoC was higher in tumors than in non-malignant tissues, higher in metastatic lesions than in primary tumors, and correlated with perineural invasion and lymph node metastasis and poorer prognosis, whereas expression of either RhoA or RhoB did not show correlation with these clinicopathological findings. The RhoC gene was also identified experimentally as the gene involved in metastasis by genomic analysis of highly metastatic melanoma cells. Clark et al. [47] injected A375P human melanoma cells or B16F0 mouse melanoma cells intravenously into nude mice, dissected metastases occurring in the lung, expanded cells of the metastatic colonies in tissue culture and re-introduced into host mice. Repeating this procedure three times, they isolated cell populations with high metastatic potential from each cell line. By comparing gene expression between these populations and the parental populations with microarray analysis, they detected three genes that were highly expressed in all the metastatic tissues selected from both cell lines; they were fibronectin, RhoC and thymosin β4. The authors confirmed these genes derived from tumor cells and not surrounding lung tissues, and verified potential of RhoC as a metastasis gene by expressing exogenous RhoC in melanoma cells and examining lung metastasis. The selected metastatic cell population and the cells over-expressing exogenous RhoC did not show enhanced proliferation, but were more migratory and more invasive and exhibited elongated morphology, the properties suppressed by expressing dominant negative Rho mutant. This work thus confirmed experimentally the importance of RhoC in metastasis. More recently, analysis of microRNAs (miRNAs) expressed in breast cancer also identified RhoC as a metastasis-associated gene. Ma et al. [48] first detected 29 miRNAs that are differentially expressed between primary breast carcinomas and normal mammary tissues, then examined their expression in breast cancer cell lines with metastatic potential, and identified miR-10b as a candidate miRNA associated with metastasis. They then analyzed functions of this miRNA, and found that miR-10b regulates cell migration and invasion in vitro, and initiates tumor invasion and distant metastasis in vivo. Analyzing the mechanism by which miR-10b induces tumor invasion, the authors found that miR-10b directly inhibits translation of HOXD10, which results in release of HOXD10-mediated inhibition of expression of genes involved in cell migration including RhoC.
Thus, there is substantial amount of evidence for involvement of RhoC in tumor metastasis. However, little information is available how RhoC mediates such an action. It remains unclear whether the metastatic potential of RhoC is due to specific localization or specific upstream or downstream signaling. Among RhoA, B and C, RhoB is known to localize to endosomes, while no distinct localization has been reported for RhoA and C [49]. It was reported that RhoC expression leads to induction of angiogenic factors in breast epithelial cells [50]. It was also reported that RhoC interacts with ROCK more effectively than RhoA and facilitates disruption of adherens junctions of epithelial cells [20]. These mechanisms, particularly preferential binding to ROCK, may partly explain the mechanism of RhoC-mediated tumor invasion in vivo, given ROCK-mediated disruption of cell-cell junction as described above and ROCK-dependent transmigration and amoeboid movement of tumor cells as described below, though the morphology of RhoC-overexpressing cells is not consistent with the rounded morphology of ROCK-activated cells,
The earliest indication for involvement of ROCK in tumor invasion was obtained by transmigration experiment of tumor cells. In order for tumor cells to establish metastasis at sites distant from its origin and invade into tissues, they have to transmigrate through host cell layers such as the endothelial cell layer covering the blood vessels and the mesothelial cell layer covering the peritoneum. Starting with the finding that cultured rat MM1 hepatoma cells required serum stimulation and intact Rho activity to migrate through the mesothelial layer and establish tumor foci beneath the monolayer in vitro [51], Itoh et al. [52] examined involvement of ROCK in this process. They found that transfection of dominant active mutants of ROCK conferred MM1 cells the invasive activity independent of Rho and serum, whereas expression of a dominant negative ROCK mutant or treatment with a ROCK inhibitor, Y-27632, substantially attenuated invasiveness in vitro. Furthermore, continuous local infusion in vivo of Y-27632 markedly reduced dissemination and tumor nodule formation of MM1 cells injected into the peritoneal cavity of syngeneic rats. Thus, the work by Itoh et al. demonstrates that ROCK action is required not only for in vitro models of tumor invasion but also for tumor invasion in vivo. Given the work by Worthylake et al. on the role of ROCK in tail retraction discussed above, one obvious candidate of ROCK actions in this process is to retract tail while the cell body creeps beneath the monolayer, although there may be other ROCK actions that facilitate tumor invasion in vivo, one being ROCK-mediated tumor cell migration in the matrix (see below).
While many molecular mechanisms including Rho signaling underlying tumor invasion have been thought out based on the results of in vitro motility studies in the two dimensional culture, tumor cells actually invade into the three dimensional (3D) space where extensive fibrillar network of extracellular matrix (ECM) proteins such as collagen restricts their movement. This in vivo situation led to the proposal that the invasion of tumor cells requires coordination of cell adhesion/motility and proteolytic degradation of ECM substrates, a concept supported by many in vitro and in vivo model studies [53]. However, application of inhibitors targeted to ECM-degrading proteases, particularly matrix metalloproteases (MMPs), provided only weak beneficial effects in tumor models in vivo in intact animals as well as in clinical trials in humans, raising a possibility of alternative mode of cell invasion in the face of MMP inhibition. Friedl and collaborators [54] examined this issue by studying the behavior of HT1080 fibrosarcoma cells over-expressing MT1-MMP (HT1080/MT1 cells) invading the 3D-collagen matrix in vitro. They observed that, without protease inhibition, the HT1080/MT1 cells adhere the collagen matrix in an integrin-dependent manner, produce tube-like defects in the matrix by proteolysis and migrate in the formed tubes. This is consistent with the classic motility-proteolysis coordination concept. They, however, found that, when proteolytic activity was inhibited, this proteolysis-dependent mesenchymal movement was converted to the amoeboid movement, in which cells adapt spherical round shape and pass through the fibrillar network by changing the shape of their bodies by propulsive squeezing along preformed fiber strands. They suggested that this mesenchymal-amoeboid transition is a supramolecular plasticity tumor cells can adopt in tissue invasion and escape from abrogation of proteolysis. Sahai and Marshall [55] also examined the behavior of several tumor cell lines in the 3D matrix and found that some tumor cell lines migrate through the 3D matrix in the rounded form, i.e. by contraction of their bodies. The round form of migration Sahai and Marshall found apparently corresponds to the amoeboid movement described by the Friedl group. Sahai and Marshall further analyzed signal transduction therein and found that the Rho-ROCK pathway is a major driving force for this mode of migration, and suggested that inhibition of both proteases and ROCK may be beneficial for inhibition of tumor invasion. Sahai and collaborators then extended this work by showing that cells with rounded morphology pushed away the collagen in front of them for invasion and this deformation was dependent on myosin phosphorylation and ROCK activity [56]. They found that the actomyosin bundles are formed in a ROCK-dependent manner in the cell cortex perpendicular to the direction of migration just behind the invading edge, and suggest that the contraction of the cell cortex by these actomyosin bundles causes the cell body to move forward by pushing the collagen matrix away. A more recent report from this group demonstrated that localization of ROCK in the above actomyosin bundles and their contraction is dependent on PDK1 but not on its kinase activity [57], thus raising an interesting possibility for interaction of PI-3-kinase pathway and the Rho-ROCK pathway in tumor invasion.
Thus, the current understanding of tumor invasion is that there are two modes of tumor cell movement in invasion; one is the proteolysis-guided mesenchymal movement and the other is the actomyosin-driven amoeboid movement, and the inhibition of proteases, particularly matrix metallo-proteases (MMPs), can convert the mode of migration from the former to the latter, and inhibition of ROCK may convert the mode from the latter to the former, or preferentially select the fraction of tumor cells with the former mode (Fig. 7). While the latter mode is clearly Rho/ROCK-dependent, there is an indication that Rho signaling is also implicated in the former process. MMPs, either membrane bound or secreted, are localized to specialized structures at the cell-substrate boundary named podosomes/invadopodia [58]. Podosomes/invadopodia are dot-shaped actin-enriched contacts encircled by columns of integrin-adhesion protein complex that form plasma membrane extensions to ECM, and in which activated MMPs accumulate and actively degrade ECM fibers. While the actin filament assembly for podosome formation is catalyzed by the Cdc42-Arp2/3-N-WASP system [58], there are several lines of evidence that Rho signaling is also somehow involved. For example, inhibition of Rho induced podosome disruption in human and mouse dendritic cells and in mouse osteoclast-like cells [59–61], and constitutively active Rho mutant, V14-RhoA stimulated podosome assembly in osteoclasts [62]. Since Rho is not directly involved in podosome assembly, Rho signaling may be involved in induction of podosome. Podosomes are induced by activation of Src kinases, either oncogenic v-Src and a protooncogene, c-Src. Martin and collaborators [63] examined the podosome formation in Src-transformed cells, and found that inactivation of Rho by either overexpression of dominant negative N19-RhoA or botulinum C3 exoenzyme disrupted the podosome structure in these cells and strongly inhibits Src-induced proteolytic degradation of ECM proteins. They also showed that active GTP-bound form of Rho also accumulate in the podosome. Interestingly, this inhibitory activity of Rho inactivation on podosome is not mimicked by inhibition of ROCK with Y-27632, indicating that the Rho signaling other than ROCK is important in this process, and functions in situ at the podosome to maintain these structures.

Two modes of cell migration in the three-dimensional matrix. Tumor cells exhibit two modes of migration in three-dimensional matrix, the actomyosin-driven amoeboid movement and the proteolysis-guided mesenchymal movement. The Rho-ROCK pathway is involved in the former, whereas Cdc42 and possibly Rho with other effector are involved in the latter mode.
4 Rho signaling and malignant transformation
Malignant transformation of cells induces, in vitro in cell culture, morphological changes, reduced serum-dependence of proliferation, loss of contact inhibition shown by foci formation and anchorage-independent growth examined by colony formation in soft agar, and tumor formation when implanted in vivo. While the prototype of Rho GEFs, Dbl, was isolated as an oncogene, and over-expression of many Dbl family protein mutants can induce malignant transformation [64], over-expression of Rho GTPases such as RhoA, Rac1 and Cdc42 either in wild type forms or in activated mutants exhibit little or only weak transforming activity (see for example, 65). Consistently, no active mutant of Rho GTPases analogous to that for Ras was isolated as an oncogene in clinical cancers. On the other hand, however, it was shown that each of Rho GTPases is required for Ras-induced transformation [65–69]. For example, Qiu et al. [67] expressed constitutively active V14-RhoA or dominant negative N19-RhoA mutant either alone or together with active Ras mutant, V12-Ras, or active Raf mutant, RafCAAX, and found co-expression of N19-RhoA dose-dependently suppressed focus formation as well as colony formation in soft agar induced either V12 H-Ras or RafCAAX, and reversed the morphology of Ras-V12 transformed cells, whereas expression of V14-RhoA, though alone cannot induce transformation, synergizes with RafCAAX to facilitate transformation. Similar findings were also reported by Der and collaborators [65, 68]. These results indicate that Ras mobilizes not only Raf-mediated kinase cascade but also other signaling pathway(s) for efficient transformation, and that Rho functions in the latter pathway(s) to facilitate its process or Rho may exert permissive effects on these pathways. Interestingly, although Rho is required for Ras-mediated transformation, the Ras transformants typically lack stress fibers, a hallmark of Rho action. This paradox indicates that this Rho-facilitated signaling pathway contains a mechanism to cause stress fiber dissolution. More recently, Ras utilizes all three of its downstream signaling pathways, the Raf-MEK-ERK cascade, the PI-3-kinase signaling and RalGDS pathway, to induce full transformation [70]. It is possible that Rho signaling concerts with either or both of the latter two signaling pathways in Ras-induced transformation (Fig. 8a).

(a) Rho signaling in Ras-induced transformation. Rho may facilitate pathways other than the Raf pathway under Ras or promote transformation in collaboration with these pathways. A part of Rho action is mediated by ROCK (see text). (b) Involvement of Rho in translocation of Src. The Rho-mDia/ROCK pathway mediates Src translocation to focal adhesions and facilitates adhesion turnover. Whether this pathway also function in elicitation of transformation and tumorigenicity awaits clarification
Following these initial observations, Sahai and Treisman [71] then examined downstream signaling of Rho involved in transformation. They first used several point mutants in the effector loop of Val14RhoA and examined their synergism with active ΔNRaf in focus formation. Comparing the results obtained in this experiment with specificity of each mutant in binding to Rho effectors including ROCK, PKN, mDia2, citron and Rhophilin and activity of each mutant in other Rho actions such as stress fiber formation and serum response factor activation, they concluded that ROCK is involved in this process. They then used the ROCK inhibitor, Y-27632, and found that Y-27632 indeed inhibits transformation induced by activated Ras, H-Ras-R12, and co-expresison of ΔNRaf and V14RhoA [72]. On the other hand, although co-expression of active ROCK mutant with ΔNRaf induced significant enhancement of transformation induced by ΔNRaf alone, the extent of enhancement was much lower than that achieved by co-expression of V14-RhoA and ΔNRaf. These results indicate that ROCK is required for Rho-mediated enhancement of Ras-induced transformation but that it is not the sole Rho effector in this process. The Treisman’s group further examined possible involvement of ezrin, a ROCK substrate, in this process, and found that expression of T567A ezrin mutant insensitive to ROCK phosphorylaitn interfered with transformation induced by the active Ras mutant [73]. Sahai et al. [74] further examined intracellular localization of ROCK in Ras-transformed cells and found that the majority of ROCK is sequestered in an inactive pool by sustained ERK-MAP kinase activity under active Ras. They suggested that this may be one of the mechanisms for dissolution of stress fibers seen in Ras transformants. However, they presented no explanation how requirement for transformation and down-regulation for dissolution of stress fibers of ROCK can occur at the same time.
In addition to Ras, Rho signaling may also be implicated in transformation by Src. v-Src is the oldest oncogene, and its proto-oncogene, c-Src, is amplified in a variety of clinical cancers and its activity often correlates with their invasive potency [75]. Src kinases, both v-Src and c-Src, are inactive in a soluble non-myristylated form, indicating that they must be directed to a specific subcellular structure(s) in order to induce transformation. Src exhibits a variety of discrete subcellular distribution including plasma membrane, adhesion plaques, cell-cell contact and perinuclear membranes. Earlier, Hamaguchi and Hanafusa [76] used various Src mutants, and found correlation between cytoskeletal association and transforming activity. Liebl and Martin [77] prepared chimera molecules in which v-Src was conjugated with motifs targeting to a specific subcellular site, and found that v-Src and not c-Src targeted to adhesion plaques could induce transformation phenotype, though the malignant phenotype by this chimera was not exactly the same as that induced by wild type v-Src. Given their own findings that translocation of v-Src to the cell periphery is important for its transformation activity, Frame and collaborators examined mechanism of peripheral translocation using temperature-sensitive mutant of v-Src [78]. They found that v-Src accumulated in the perinuclear region at the restrictive temperature, and moves to the periphery upon the shift to the permissive temperature. In Swiss3T3 fibroblasts, this translocation of v-Src from the perinuclear pool to the periphery required serum stimulation. They found that, on serum addition, v-Src associates with serum-induced (Rho-mediated) actin stress fibers and accumulates in focal adhesions, and that intact actin filaments are required, while microtubules are dispensable, for this translocation. They then examined structural and catalytic requirement of Src for this translocation and found that the intact SH3 domain is essential for this translocation, while the myristylation and kinase activity are dispensable [79, 80]. They further found that stimulation of Swiss3T3 cells expressing GFP-Src construct with LPA, platelet-derived growth factor (PDGF) and bradykinin, stimuli known to activate Rho, Rac and Cdc42, respectively, translocated Src-GFP to focal adhesions, membrane ruffles and filopodia, respectively [81]. This translocation was mimicked by co-expression of dominant active Rho GTPases, V14-RhoA, V12-Rac1 and V12-Cdc42, and inhibited by expression of dominant negative mutants of each GTPases. Intriguingly, localization of Src-GFP to lamellipodia and filopdia was suppressed by inhibition with N17-Rac1 and N17-Cdc42, but in both cases Src-GFP then accumulated in focal adhesions. Moreover, treatment with a specific ROCK inhibitor, Y-27632, not only suppressed of Rho-mediated accumulation of Src-GFP in focal adhesions but also that in lamellipodia and filopodia induced by PDGF and bradykinin, respectively. These results indicate that Src is originally recruited to focal adhesions in a Rho and ROCK-dependent manner, and then moves to focal complexes in lamellipodis or filopodia upon remodeling of focal adhesions to focal complexes induced by Rac or Cdc42. The Frame’s group [82] more recently reported that Src in the prinuclear region and during the transit to the periphery associates with endosomes, as originally observed by Kaplan et al. [83]. The Src-containing endosomes partly overlap with those containing RhoB that resides in the endosome, and the peripheral translocation of Src is impaired in MEF cells prepared from Rho-B-/- mice, the defect rescued by re-expression of RhoB in the knockout cells. They also reported that, when cellular F-actin abolished by treatment with cytochalasin D reappear with the washout of the drug, clouds of F-actin become associated with the RhoB/Src-containing endosomes, and suggested that such actin structure may function to propel the endosomes in the cell. The Frame’s group [84] also examined translocation of Src related kinase, Yes and Fyn, and found that their translocation to the periphery also requires intact actin filaments. Interestingly, Fyn is localized to RhoD and not RhoB-containing endosomes, and this selective localization to the RhoD endosomes is dependent on palmitoylation of the N-terminal region of Fyn. Thus, the Frames group has carried out extensive study on the translocation mechanism of Src and Src-related kinases, and has found an important link between Src, actin and Rho GTPases (Fig. 8b). Curiously, however, they have discussed their results mainly in relation to Src-induced disassembly of focal adhesions, and not addressed how critical the pathway they defined is in Src-induced cell transformation. They have not examined, either, effector mechanism for actin filament assembly required for translocation of Src and Src-related kinases. Given the requirement of Rho in this process, a strong candidate is the mDia family of proteins. As discussed above, Yamana et al. [40] already reported that depletion of mDia1 resulted in impaired accumulation of c-Src in focal adhesions of migrating C6 rat glioma cells. Whether the mDia1-mediated mechanism also operates for elicitation of Src-induced malignant transformation should be explored in future studies.
5 Perspectives
As we review in this article, Rho signaling consisting of Rho, mDia and ROCK is apparently involved in elicitation of various phenotypes of tumor cells, transformation, motility, transmigration and invasion in vivo, and circumferential evidence has accumulated for strong relation of this signaling to oncogenic actions of Ras and Src. Naturally, such information raises many questions. They include; how does the Rho-ROCK pathway contribute to Ras-induiced transformation? Does it facilitate signaling pathway(s) other than the Raf-Erk pathway under Ras? Is there any Rho-mediated pathway other than ROCK functioning in Ras-induced transformation? Is the action of Rho signaling in transformation independent of its action in invasion or do they represent different aspects of the same action? Does Rho signaling function not only in Src-mediated motility and invasion but also in transformation induced by this oncogene? Are the actions of Rho signaling for Ras and Src separate and independent or are they intimately connected in elicitation of transformed phenotype by each oncogene? Finally, how much does Rho signaling contribute to tumorigenesis in intact animals and in clinical cancers? We are now in a stage where we can answer some or all of these questions. Clarifying these questions is hoped to provide an insight into how tumor cells integrate various signaling pathways including Ras, Src and Rho for expression of their malignancy.
References
Jaffe, A. B., & Hall, A. (2005). Rho GTPases: biochemistry and biology. Annual Review of Cell and Developmental Biology, 21, 247–269.
Burridge, K., & Wennerberg, K. (2004). Rho and Rac take center stage. Cell, 116, 167–179.
Zheng, Y. (2001). Dbl family guanine nucleotide exchange factors. Trends in Biochemical Sciences, 26, 724–732.
Moon, S. Y., & Zheng, Y. (2003). Rho GTPase-activating proteins in cell regulation. Trends in Cell Biology, 13, 13–22.
Leung, T., Manser, E., Tan, L., & Lim, L. (1995). A novel serine/threonine kinase binding the Ras-related RhoA GTPase which translocates the kinase to peripheral membranes. Journal of Biological Chemistry, 270, 29051–29054.
Ishizaki, T., Maekawa, M., Fujisawa, K., Okawa, K., Iwamatsu, A., Fujita, A., et al. (1996). The small GTP-binding protein Rho binds to and activates a 160 kDa Ser/Thr protein kinase homologous to myotonic dystrophy kinase. EMBO Journal, 15, 1885–1893.
Matsui, T., Amano, M., Yamamoto, T., Chihara, K., Nakafuku, M., Ito, M., Nakano, T., et al. (1996). Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. EMBO Journal, 15, 2208–2216.
Watanabe, N., Madaule, P., Reid, T., Ishizaki, T., Watanabe, G., Kakizuka, A., et al. (1997). p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO Journal, 16, 3044–3056.
Goode, B. L., & Eck, M. J. (2007). Mechanism and function of formins in the control of actin assembly. Annual Reviews of Biochemical, 76, 593–627.
Riento, K., & Ridley, A. J. (2003). Rocks: multifunctional kinases in cell behaviour. Nature Reviews Molecular Cell Biology, 4, 446–456.
Kimura, K., Ito, M., Amano, M., Chihara, K., Fukata, Y., Nakafuku, M., et al. (1996). Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science, 273, 245–248.
Uehata, M., Ishizaki, T., Satoh, H., Ono, T., Kawahara, T., Morishita, T., et al. (1997). Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature, 389, 990–994.
Amano, M., Ito, M., Kimura, K., Fukata, Y., Chihara, K., Nakano, T., et al. (1996). Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). Journal of Biological Chemistry, 271, 20246–20249.
Maekawa, M., Ishizaki, T., Boku, S., Watanabe, N., Fujita, A., Iwamatsu, A., et al. (1999). Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science, 285, 895–898.
Narumiya, S., Ishizkai, T., & Uehata, M. (2000). Use and properties of ROCK-specific inhibitor. Methods Enzymol, 325, 273–284.
Watanabe, N., Kato, T., Fujita, A., Ishizaki, T., & Narumiya, S. (1999). Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nature Cell Biology, 1, 136–143.
Kosako, H., Yoshida, T., Matsumura, F., Ishizaki, T., Narumiya, S., & Inagaki, M. (2000). Rho-kinase/ROCK is involved in cytokinesis through the phosphorylation of myosin light chain and not ezrin/radixin/moesin proteins at the cleavage furrow. Oncogene, 19, 6059–6064.
Watanabe, S., Ando, Y., Yasuda, S., Hosoya, H., Watanabe, N., Ishizaki, T., et al. (2008). mDia2 Induces the Actin Scaffold for the Contractile Ring and Stabilizes Its Position during Cytokinesis in NIH 3T3 Cells. Molecular Biology of the Cell, 19, 2328–2338.
Ishizaki, T., Naito, M., Fujisawa, K., Maekawa, M., Watanabe, N., Saito, Y., et al. (1997). p160ROCK, a Rho-associated coiled-coil forming protein kinase, works downstream of Rho and induces focal adhesions. FEBS Letters, 404, 118–124.
Tsuji, T., Ishizaki, T., Okamoto, M., Higashida, C., Kimura, K., Furuyashiki, T., et al. (2002). ROCK and mDia1 antagonize in Rho-dependent Rac activation in Swiss 3T3 fibroblasts. Journal of Cell Biology, 157, 819–830.
Morii, N., & Narumiya, S. (1995). Preparation of native and recombinant Clostridium botulinum C3 ADP-ribosyltransferase and identification of Rho proteins by ADP-ribosylation. Methods Enzymol, 256, 196–206.
Sahai, E., & Marshall, C. J. (2002). ROCK and Dia have opposing effects on adherens junctions downstream of Rho. Nature Cell Biology, 4, 408–415.
Arakawa, Y., Bito, H., Furuyashiki, T., Tsuji, T., Takemoto-Kimura, S., Kimura, K., et al. (2003). Control of axon elongation via an SDF-1alpha/Rho/mDia pathway in cultured cerebellar granule neurons. Journal of Cell Biology, 161, 381–391.
Blumenstein, L., & Ahmadian, M. R. (2004). Models of the cooperative mechanism for Rho effector recognition: implications for RhoA-mediated effector activation. Journal of Biological Chemistry, 279, 53419–53426.
Rose, R., Weyand, M., Lammers, M., Ishizaki, T., Ahmadian, M. R., & Wittinghofer, A. (2005). Structural and mechanistic insights into the interaction between Rho and mammalian Dia. Nature, 435, 513–518.
Nobes, C. D., & Hall, A. (1999). Rho GTPases control polarity, protrusion, and adhesion during cell movement. Journal of Cell Biology, 144, 1235–1244.
Allen, W. E., Zicha, D., Ridley, A. J., & Jones, G. E. (1998). A role for Cdc42 in macrophage chemotaxis. Journal of Cell Biology, 141, 1147–1157.
Ridley, A. J., Schwartz, M. A., Burridge, K., Firtel, R. A., Ginsberg, M. H., Borisy, G., et al. (2003). Cell migration: integrating signals from front to back. Science, 302, 1704–1709.
Itoh, R. E., Kurokawa, K., Ohba, Y., Yoshizaki, H., Mochizuki, N., & Matsuda, M. (2002). Activation of rac and cdc42 video imaged by fluorescent resonance energy transfer-based single-molecule probes in the membrane of living cells. Molecular and Cellular Biology, 22, 6582–6591.
Etienne-Manneville, S., & Hall, A. (2001). Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKC. Cell, 106, 489–498.
Etienne-Manneville, S., & Hall, A. (2003). Cdc42 regulates GSK-3β and adenomatous polyposis coli to control cell polarity. Nature, 421, 753–756.
Gomes, E. R., Jani, S., & Gundersen, G. G. (2005). Nuclear movement regulated by Cdc42, MRCK, myosin, and actin flow establishes MTOC polarization in migrating cells. Cell, 121, 451–463.
Kraynov, V. S., Chamberlain, C., Bokoch, G. M., Schwartz, M. A., Slabaugh, S., & Hahn, K. M. (2000). Localized Rac activation dynamics visualized in living cells. Science, 290, 333–337.
Cory, G. O., & Ridley, A. J. (2002). Cell motility: braking WAVEs. Nature, 418, 732–733.
Worthylake, R. A., Lemoine, S., Watson, J. M., & Burridge, K. (2001). RhoA is required for monocyte tail retraction during transendothelial migration. Journal of Cell Biology, 154, 147–160.
Yoshinaga-Ohara, N., Takahashi, A., Uchiyama, T., & Sasada, M. (2002). Spatiotemporal regulation of moesin phosphorylation and rear release by Rho and serine/threonine phosphatase during neutrophil migration. Experimental Cell Research, 278, 112–122.
Worthylake, R. A., & Burridge, K. (2003). RhoA and ROCK promote migration by limiting membrane protrusions. Journal of Biological Chemistry, 278, 13578–13584.
Goulimari, P., Kitzing, T. M., Knieling, H., Brandt, D. T., Offermanns, S., & Grosse, R. (2005). Gα12/13 is essential for directed cell migration and localized Rho-Dia1 function. Journal of Biological Chemistry, 280, 42242–42251.
Fukuhara, S., Chikumi, H., & Gutkind, J. S. (2001). RGS-containing RhoGEFs: the missing link between transforming G proteins and Rho? Oncogene, 20, 1661–1668.
Yamana, N., Arakawa, Y., Nishino, T., Kurokawa, K., Tanji, M., Itoh, R. E., et al. (2006). The Rho-mDia1 pathway regulates cell polarity and focal adhesion turnover in migrating cells through mobilizing Apc and c-Src. Molecular and Cellular Biology, 26, 6844–6858.
Webb, D. J., Donais, K., Whitmore, L. A., Thomas, S. M., Turner, C. E., Parsons, J. T., et al. (2004). FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nature Cell Biology, 6, 154–161.
Arthur, W. T., Petch, L. A., & Burridge, K. (2000). Integrin engagement suppresses RhoA activity via a c-Src-dependent mechanism. Current Biology, 10, 719–722.
Arthur, W. T., & Burridge, K. (2001). RhoA inactivation by p190RhoGAP regulates cell spreading and migration by promoting membrane protrusion and polarity. Molecular Biology of the Cell, 12, 2711–2720.
Pertz, O., Hodgson, L., Klemke, R. L., & Hahn, K. M. (2006). Spatiotemporal dynamics of RhoA activity in migrating cells. Nature, 440, 1069–1072.
Sahai, E., & Marshall, C. J. (2002). RHO-GTPases and cancer. Nature Reviews Cancer, 2, 133–142.
Suwa, H., Ohshio, G., Imamura, T., Watanabe, G., Arii, S., Imamura, M., et al. (1998). Overexpression of the rhoC gene correlates with progression of ductal adenocarcinoma of the pancreas. British Journal of Cancer, 77, 147–152.
Clark, E. A., Golub, T. R., Lander, E. S., & Hynes, R. O. (2000). Genomic analysis of metastasis reveals an essential role for RhoC. Nature, 406, 532–535.
Ma, L., Teruya-Feldstein, J., & Weinberg, R. A. (2007). Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature, 449, 682–688.
Adamson, P., Paterson, H. F., & Hall, A. (1992). Intracellular localization of the P21rho proteins. Journal of Cell Biology, 119, 617–627.
van Golen, K. L., Wu, Z. F., Qiao, X. T., Bao, L., & Merajver, S. D. (2000). RhoC GTPase overexpression modulates induction of angiogenic factors in breast cells. Neoplasia, 2, 418–425.
Yoshioka, K., Imamura, F., Shinkai, K., Miyoshi, J., Ogawa, H., Mukai, M., et al. (1995). Participation of rhop21 in serum-dependent invasion by rat ascites hepatoma cells. FEBS Letter, 372, 25–28.
Itoh, K., Yoshioka, K., Akedo, H., Uehata, M., Ishizaki, T., & Narumiya, S. (1999). An essential part for Rho-associated kinase in the transcellular invasion of tumor cells. Nature Medicine, 5, 221–225.
Stetler-Stevenson, W. G., Aznavoorian, S., & Liotta, L. A. (1993). Tumor cell interactions with the extracellular matrix during invasion and metastasis. Annual Review of Cell Biology, 9, 541–573.
Wolf, K., Mazo, I., Leung, H., Engelke, K., von Andrian, U. H., Deryugina, E. I., et al. (2003). Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. Journal of Cell Biology, 160, 267–277.
Sahai, E., & Marshall, C. J. (2003). Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nature Cell Biology, 5, 711–719.
Wyckoff, J. B., Pinner, S. E., Gschmeissner, S., Condeelis, J. S., & Sahai, E. (2006). ROCK- and myosin-dependent matrix deformation enables protease-independent tumor-cell invasion in vivo. Current Biology, 16, 1515–1523.
Pinner, S., & Sahai, E. (2008). PDK1 regulates cancer cell motility by antagonising inhibition of ROCK1 by RhoE. Nature Cell Biology, 10, 127–137.
Linder, S., & Aepfelbacher, M. (2003). Podosomes: adhesion hot-spots of invasive cells. Trends in Cell Biology, 13, 376–385.
Burns, S., Thrasher, A. J., Blundell, M. P., Machesky, L., & Jones, G. E. (2001). Configuration of human dendritic cell cytoskeleton by Rho GTPases, the WAS protein, and differentiation. Blood, 98, 1142–1149.
West, M. A., Prescott, A. R., Eskelinen, E. L., Ridley, A. J., & Watts, C. (2000). Rac is required for constitutive macropinocytosis by dendritic cells but does not control its downregulation. Current Biology, 10, 839–848.
Zhang, D., Udagawa, N., Nakamura, I., Murakami, H., Saito, S., Yamasaki, K., et al. (1995). The small GTP-binding protein, rho p21, is involved in bone resorption by regulating cytoskeletal organization in osteoclasts. Journal of Cell Science, 108, 2285–2292.
Chellaiah, M. A., Soga, N., Swanson, S., McAllister, S., Alvarez, U., Wang, D., et al. (2000). Rho-A is critical for osteoclast podosome organization, motility, and bone resorption. Journal of Biological Chemistry, 275, 11993–12002.
Berdeaux, R. L., Diaz, B., Kim, L., & Martin, G. S. (2004). Active Rho is localized to podosomes induced by oncogenic Src and is required for their assembly and function. Journal of Cell Biology, 166, 317–323.
Rossman, K. L., Der, C. J., & Sondek, J. (2005). GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nature Reviews Molecular Cell Biology, 6, 167–180.
Khosravi-Far, R., Solski, P. A., Clark, G. J., Kinch, M. S., & Der, C. J. (1995). Activation of Rac1, RhoA, and mitogen-activated protein kinases is required for Ras transformation. Molecular and Cellular Biology, 15, 6443–6453.
Qiu, R. G., Chen, J., Kirn, D., McCormick, F., & Symons, M. (1995). An essential role for Rac in Ras transformation. Nature, 374, 457–459.
Qiu, R. G., Chen, J., McCormick, F., & Symons, M. (1995). A role for Rho in Ras transformation. Proceedings of the National Academy of Sciences of the United States of America, 92, 11781–11785.
Prendergast, G. C., Khosravi-Far, R., Solski, P. A., Kurzawa, H., Lebowitz, P. F., & Der, C. J. (1995). Critical role of Rho in cell transformation by oncogenic Ras. Oncogene, 10, 2289–2296.
Qiu, R. G., Abo, A., McCormick, F., & Symons, M. (1997). Cdc42 regulates anchorage-independent growth and is necessary for Ras transformation. Molecular and Cellular Biology, 17, 3449–3458.
Bodemann, B. O., & White, M. A. (2008). Ral GTPases and cancer: linchpin support of the tumorigenic platform. Nature Reviews Cancer, 8, 133−140.
Sahai, E., Alberts, A. S., & Treisman, R. (1998). RhoA effector mutants reveal distinct effector pathways for cytoskeletal reorganization, SRF activation and transformation. EMBO Journal, 17, 1350−1361.
Sahai, E., Ishizaki, T., Narumiya, S., & Treisman, R. (1999). Transformation mediated by RhoA requires activity of ROCK kinases. Current Biology, 9, 136−145.
Tran Quang, C., Gautreau, A., Arpin, M., & Treisman, R. (2000). Ezrin function is required for ROCK-mediated fibroblast transformation by the Net and Dbl oncogenes. EMBO Journal, 19, 4565–4576.
Sahai, E., Olson, M. F., & Marshall, C. J. (2001). Cross-talk between Ras and Rho signalling pathways in transformation favours proliferation and increased motility. EMBO Journal, 20, 755–766.
Yeatman, T. J. (2004). A renaissance for SRC. Nature Reviews Cancer, 4, 470–480.
Hamaguchi, M., & Hanafusa, H. (1987). Association of p60src with Triton X-100-resistant cellular structure correlates with morphological transformation. Proceedings of the National Academy of Sciences of the United States of America, 84, 2312–2316.
Liebl, E. C., & Martin, G. S. (1992). Intracellular targeting of pp60src expression: localization of v-src to adhesion plaques is sufficient to transform chicken embryo fibroblasts. Oncogene, 7, 2417–2428.
Fincham, V. J., Unlu, M., Brunton, V. G., Pitts, J. D., Wyke, J. A., & Frame, M. C. (1996). Translocation of Src kinase to the cell periphery is mediated by the actin cytoskeleton under the control of the Rho family of small G proteins. Journal of Cell Biology, 135, 1551–1564.
Fincham, V. J., & Frame, M. C. (1998). The catalytic activity of Src is dispensable for translocation to focal adhesions but controls the turnover of these structures during cell motility. EMBO Journal, 17, 81–92.
Fincham, V. J., Brunton, V. G., & Frame, M. C. (2000). The SH3 domain directs acto-myosin-dependent targeting of v-Src to focal adhesions via phosphatidylinositol 3-kinase. Molecular and Cellular Biology, 20, 6518–6536.
Timpson, P., Jones, G. E., Frame, M. C., & Brunton, V. G. (2001). Coordination of cell polarization and migration by the Rho family GTPases requires Src tyrosine kinase activity. Current Biology, 11, 1836–1846.
Sandilands, E., Cans, C., Fincham, V. J., Brunton, V. G., Mellor, H., Prendergast, G. C., et al. (2004). RhoB and actin polymerization coordinate Src activation with endosome-mediated delivery to the membrane. Developmental Cell, 7, 855–869.
Kaplan, K. B., Swedlow, J. R., Varmus, H. E., & Morgan, D. O. (1992). Association of p60c-src with endosomal membranes in mammalian fibroblasts. Journal of Cell Biology, 118, 321–333.
Sandilands, E., Brunton, V. G., & Frame, M. C. (2007). The membrane targeting and spatial activation of Src, Yes and Fyn is influenced by palmitoylation and distinct RhoB/RhoD endosome requirements. Journal of Cell Science, 120, 2555–2564.
Acknowledgement
This work was supported in part by Grants-in-Aid for Specially Promoted Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Narumiya, S., Tanji, M. & Ishizaki, T. Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev 28, 65–76 (2009). https://doi.org/10.1007/s10555-008-9170-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-008-9170-7