
Abstract
The role of nucleotides in intracellular energy provision and nucleic acid synthesis has been known for a long time. In the past decade, evidence has been presented that, in addition to these functions, nucleotides are also autocrine and paracrine messenger molecules that initiate and regulate a large number of biological processes. The actions of extracellular nucleotides are mediated by ionotropic P2X and metabotropic P2Y receptors, while hydrolysis by ecto-enzymes modulates the initial signal. An increasing number of studies have been performed to obtain information on the signal transduction pathways activated by nucleotide receptors. The development of specific and stable purinergic receptor agonists and antagonists with therapeutical potential largely contributed to the identification of receptors responsible for nucleotide-activated pathways. This article reviews the signal transduction pathways activated by P2Y receptors, the involved second messenger systems, GTPases and protein kinases, as well as recent findings concerning P2Y receptor signalling in C6 glioma cells. Besides vertical signal transduction, lateral cross-talks with pathways activated by other G protein-coupled receptors and growth factor receptors are discussed.
Similar content being viewed by others


Introduction
Pharmacological properties of P2Y receptors
Extracellular actions of adenine nucleotides were initially characterised in the cardiovascular system by Drury and Szent-Gyorgyi [1]. It took more than four decades before the concept of purinergic signalling was accepted, but now it is well established that nucleotides initiate and regulate a variety of biological processes, including neurotransmission, inflammation, regulation of blood pressure, platelet aggregation, cell growth and differentiation (Abbracchio et al. [2]; Burnstock and Williams [3]; Burnstock [4]; Ralevic and Burnstock [5]).
Nucleotides are released in the extracellular fluid by cell lysis, exocytosis, secretion of granules, efflux and upon cellular stress such as changes in osmolarity and mechanical perturbations. Once released, they mediate their effect by stimulation of nucleotide receptors.
Based on pharmacological properties, the first suggestion for the existence of ionotropic P2X receptors and metabotropic P2Y receptors was made by Burnstock and Kennedy [6]. After cloning, multiple subtypes of P2X and P2Y receptors were characterised unambiguously (Abbracchio and Burnstock [7] Burnstock and Williams [3]; Fredholm et al. [8]).
Up to now, the P2Y receptor family comprises at least eight subtypes, P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P2Y13 and the recently identified P2Y14 receptor (Ralevic and Burnstock, [5] Abbracchio et al. [9]; Communi et al. [10]; Hollopeter et al. [11]; Zhang et al. [12]). According to the agonist profile, P2Y receptors can be subdivided into receptors responding to adenine mono- and dinucleotides (P2Y1, P2Y11, P2Y12, P2Y13), and to uridine nucleotides (P2Y4, P2Y6), and receptors for adenine and uridine nucleotides (P2Y2). The pharmacological profile of the recently cloned P2Y14 receptor is distinct from the other P2Y receptors since UDP-glucose, UDP-galactose, UDP-glucuronic acid and UDP-N-acetylglucosamine are specific ligands of this receptor (Chambers et al. [13]). Natural P2Y receptor ligands do not exclusively bind to one receptor subtype. ADP is an agonist of P2Y1, P2Y12 and P2Y13 receptors, whereas ATP is a full agonist of P2Y2 and P2Y11, but a partial agonist or antagonist of P2Y1, P2Y12 and P2Y13 receptors. Although the pharmacological properties of P2Y receptors (Table 1) are well conserved between species, some remarkable differences have been observed. While UTP acts as an agonist of both human and rat P2Y4 receptors, ATP is a potent agonist of the rat P2Y4, but an antagonist of the human orthologue. Mutational analysis revealed that the second extracellular loop of the P2Y4 receptor is responsible for the opposing effect of ATP in both species (Herold et al. [14]). A similar phenomenon is observed when human and canine P2Y11 receptors were stably expressed in CHO-K1 and 1321N1 astrocytoma cells. Whereas the human P2Y11 is potently activated by adenosine triphosphate nucleotides, the canine orthologue displayed more selectivity towards the corresponding diphosphates. In this case, the nucleotide selectivity is due to differences in the amino acid sequence at the juxtaposition of TM6 and the third extracellular loop also reported to play an important role in agonist selectivity and signalling of other G protein-coupled receptors (GPCR) (Qi et al. [15]; Lawson and Wheatley [16]).
Despite their chemical stability, extracellular nucleotides are metabolised by several ecto-enzymes (Claes and Slegers [17]; Czajkowski and Baranska [18]; Goding et al. [19]; Zimmerman [20]). Extracellular hydrolysis complicates the evaluation of nucleotide-mediated effects on different cell types and can be overcome by the use of specific non-hydrolysable receptor agonists or ecto-enzyme inhibitors. Some P2Y receptor antagonists, such as pyridoxalphosphate-6-azophenyl-2′,4′-disulfonic acid (PPADS), suramin, reactive blue 2 and 4,4′-diisothiocyanatostilbene-2,2′-disulphonic acid (DIDS), are inhibitors of nucleotide hydrolyzing enzymes and are often used in studies of nucleotide-mediated signalling (Grobben et al. [21]). Nevertheless, care must be taken for the interpretation of experimental data since it is also shown that cells can internalise some of these molecules (Claes et al., [22]). Therefore specific P2Y receptor antagonists, developed for therapeutical purposes, have to be used to overcome the lack of specificity (Boeynaems et al. [23]; Lambrecht et al. [24]; Kam and Nethery [25]; Kim et al. [26]; Xu et al. [27]).
P2Y receptor expression on rat C6 glioma cells
Rat C6 glioma is a tumoral cell line of glial origin with oligodendrocytic, astrocytic and neuronal progenitor properties. Due to a point mutation in ‘phosphatase and tensin homologue deleted on chromosome ten’ (PTEN), the phosphatidylinositol 3-kinase (PI 3-K)/PKB signalling pathway is constitutively active and contributes to the proliferative and invasive properties of these cells (Kubiatowski et al. [28]; Roymans and Slegers [29]; Grobben et al. [30]). In addition, cell proliferation is sustained by secreted growth factors that stimulate growth factor receptors present on these cells. Such autocrine mechanisms are reported for IGFR, bFGFR and PDGFR (Okumura et al. [31]; Resnicoff et al. [32]; Strawn et al. [33]).
In C6 cells, an increase in cAMP by stimulation of the β-adrenergic receptor (β-AR) or by addition of membrane permeable cAMP analogues, e.g., dibutyryl cAMP (dBcAMP) or 8-chloro-cAMP, induces differentiation into an astrocyte type II (Roymans et al. [34]). During this process, cessation of cell growth is accompanied by a shift in intermediate filament synthesis from vimentin to glial fibrillary acidic protein (GFAP) (Backhovens et al. [35]). The latter protein is an astrocytic differentiation marker whose expression is regulated by cAMP at the transcriptional and translational level (Messens and Slegers [36]).
In our laboratory, the signalling pathways activated by extracellular nucleotides, and in particular those affecting cell proliferation and differentiation of C6 cells, were studied in detail. The presence of a P2Y receptor on these cells that negatively affects adenylate cyclase (AC) was postulated for more than a decade (Pianet et al. [37]; Boyer et al. [38]). This receptor is coupled to a Gi protein and has been denominated P2YAC-(Claes et al. [39]; Grobben et al. [40]) before its identification as the P2Y12 receptor initially cloned from blood platelets (Czajkowski et al. [41]; Hollopeter et al. [11]; Jin et al. [42]). C6 cells also express the phospholipase (PL)Cβ-coupled P2Y1, P2Y2, P2Y4 and P2Y6 receptors (Czajkowski et al. [41]; Nicholas et al. [43]; Tu et al. [44]; Claes and Slegers [17]). Recently, we also demonstrated the presence of P2Y13 mRNA (Van Kolen and Slegers [45]) implicating the expression of three ADP-activated receptors in these cells, i.e., P2Y1, P2Y12 and P2Y13. Although 2MeSADP is reported as a potent P2Y1, P2Y12 and P2Y13 agonist, stimulation with this compound inhibits AC, but induces no significant activation of PLC, indicating that the P2Y1 receptor is not activated by ADP in cells grown in chemically defined medium (Grobben et al. [40]). This is confirmed by Czajkowski et al. [46], who showed that, in cells cultivated in the presence of fetal calf serum, ADP signalling is predominantly determined by the P2Y1 receptor. However, upon serum deprivation, expression of the P2Y1 receptor is decreased and the P2Y12 receptor becomes the main activated receptor. Characterization of P2Y13 receptor function is complicated by the fact that P2Y12 and P2Y13 receptors have almost the same agonist profile (Table 1). The receptor antagonist N6-(2-methylthioethyl)-2-(3,3,3-trifluoropropylthio)-β,γ-dichloromethylene ATP (AR-C69931MX), often used as a specific P2Y12 antagonist, also blocks the P2Y13 receptor (Marteau et al. [47]). While the human and mouse P2Y13 receptor, like the P2Y12, is more potently activated by 2MeSADP than ADP, the rat P2Y13 receptor shows a higher selectivity for ADP (Fumagalli et al. [48]). In C6 cells, further distinction between the signalling of P2Y12 and P2Y13 receptors can be made by the use of PPADS, a P2Y13 antagonist without effect on P2Y12, and P1, P4-di(adenosine-5′) tetraphosphate (Ap4A), a P2Y13 antagonist that stimulates the P2Y12 receptor (Claes et al. [39]; Grobben et al. [40]; Marteau et al. [47]). In addition, recently synthesised PPADS derivatives pyridoxal-5′-phosphate-6-azo-(2-chloro-5-nitrophenyl)-2,4-disulphonate (MRS2211) and pyridoxal-5′-phosphate-6-azo-(4-chloro-3-nitrophenyl)-2,4-disulphonate (MRS2603) have no effect on the P2Y12 receptor but antagonise the P2Y13 receptor (Kim et al. [26]).
Although P2Y receptor expression in C6 cells depends on the cultivation conditions (Czajkowski et al. [46]), unpublished data of our laboratory revealed that induction of differentiation into astrocytes type II by dbcAMP (1 mM)- or (−)-isoproterenol (5 µM) does not significantly alter the expression of P2Y receptors. These observations are in accordance with previous studies on the expression of P2Y1, P2Y2, P2Y4, P2Y6, P2Y12, P2Y13 and P2Y14 receptors in glial cells and primary astrocytes (Bianco et al. [49]; Fumagalli et al. [48, 50]; Sasaki et al. [51]). The function of P2Y receptor expression in glial cells is still under investigation, but a number of studies point to an important role in the intercellular communication between astrocytes and neurons (Bezzi and Volterra, [52]). Another well documented effect of extracellular ATP is induction of reactive astrogliosis upon activation of ERK and cyclooxygenase (COX)-2 (Brambilla et al. [53]).
Similar to the observations made in C6 cells, functional responses of P2Y receptor subtypes in microglial cells depend on cultivation conditions. In N9 mouse brain microglia stimulation of expressed P2Y receptors induces Ca2+ mobilization but only P2Y6 and P2Y14 receptor-mediated responses are increased upon activation of microglia with lipopolysaccharide. The enhanced P2Y6 response is correlated with mRNA increase which was not the case for the P2Y14 receptor-mediated Ca2+ mobilization (Bianco et al. [49]). Furthermore, stimulation of microglial P2Y12/13 receptors induces membrane ruffling and chemotaxis towards injured neurons through Gi/o protein-mediated activation of Rac (Honda et al. [54]). The observations made in astrocytes and microglial cells emphasise the importance of P2Y receptors in brain signalling and identify these receptors as putative targets in defective neurotransmission, neuroimmune functioning, cell survival and cell proliferation in response to oxidative stress and brain injury.
P2Y receptor-activated signalling cascades
Second messengers
P2Y receptors are generally linked to PLC activation that catalyses the rapid hydrolysis of phosphatidylinositol 4,5-bisphosphate into the intracellular messenger inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG). Activation of PLC occurs by Giα- and/or Gqα-dependent mechanisms (Communi et al. [55]). Besides signalling through Gα subunits, intracellular Ca2+ concentration is also affected by Gβγ subunit-dependent interaction with voltage-gated Ca2+ channels. Several reports indicated modulation of K+ currents and PLCβ activation induced by distinct domains of Gβγ (Mirshahi et al. [56]). Co-expression studies performed in rat sympathetic neurons demonstrated that P2Y1, P2Y2 and P2Y6 receptors trigger the closing of N-type Ca2+ and M-type K+ channels, whereas P2Y4 receptor stimulation also displayed coupling to M-type K+ channels producing a less efficient inhibition of Ca2+ currents. In rat brain capillary endothelial cells, it was shown that the P2Y12 receptor inhibits ICa(N) and activates a G protein-coupled inward rectifier K+ (GIRK) channel. Interestingly, stimulation of the P2Y1 receptor also induces a K+ current that is rapidly followed by inactivation. Inhibition of ICa(N) by P2Y12 receptor stimulation is also reported in PC12 cells while in HEK 293 this inhibition is mediated by the P2Y13 receptor (Filippov et al. [57–60]; Simon et al. [61]; Wirkner et al. [62]). From co-expression studies of P2Y receptors with GIRK1 or GIRK2 in rat sympathetic neurons, it was concluded that P2Y receptors activate GIRK channels by the βγ subunits of Gi/o and inhibit these channels by the α subunits of Gq (Filippov et al. [63]).
P2Y-induced calcium release is followed by opening of voltage-independent Ca2+ channels. Although this response is observed in a variety of cell types, the physiological implications are miscellaneous. In this context, it has been reported that extracellular ATP induces a Ca2+ wave that propagates through neigh-bouring astrocytes by GAP junctions (Suadicani et al. [64]). In situations of increased neuronal activity or cell damage, ATP stimulates a Ca2+-dependent protonefflux from astrocytes. Acidification of the extracellular environment serves as a negative feedback mechanism for neurotransmitter release, but also increases blood flow by vasodilatation in cerebral arterioles (Dienel and Hertz [65]; Dixon et al. [66]). Although Ca2+ signalling is observed in a variety of cell types, the time dependence of the response is cell type specific. This is especially the case for the P2Y1 receptor which triggers persistent or transient Ca2+ responses when it is expressed in human 1321N1 astrocytoma or C6 glioma cells, respectively (Czajkowski et al. [41]; Sellers et al. [67]). A recent study also revealed that, in glial cells, prolongation of the P2Y1 receptor-induced Ca2+ response is regulated by interaction with the Na+/H+ exchanger regulatory factor type-2 which determines the signalling pathways that are ultimately activated in different cell types (Fam et al. [68]). Indeed, while transient P2Y1 receptor signalling increases proliferation in C6 cells, sustained signalling triggers apoptotic cascades in 1321N1 astrocytoma cells (Czajkowski et al. [46]; Sellers et al. [67]).
A well-known response to PLC-generated DAG and IP3/Ca2+ is the activation of classical PKCs that are involved in rapid internalisation and desensitisation of GPCRs through phosphorylation of residues localised in their cytoplasmic tail. In this regard, PKCβI is reported to attenuate phosphatidylinositol (PI)-hydrolysis induced by P2Y1 and P2Y2 receptors in endothelial cells (Chen and Lin [69]). In astrocytes, high frequency stimulation of the P2Y1 receptor by repeated addition of ATP causes rapid suppression of the P2Y1 receptor-induced Ca2+ response. This phenomenon, observed as Ca2+ oscillations, is mediated by protein kinase C-dependent phosphorylation of Thr339 in the carboxyterminus of the P2Y1 receptor (Fam et al. [70]). Besides modulation of receptor responsiveness, PKC signalling also affects long term effects. In the human osteoblastic HOBIT cell line, ATP increases expression of the early growth response protein-1 by a mechanism that requires a Ca2+-independent PKC isoform (Pines et al. [71]). In vascular smooth muscle cells, UDP stimulates cell cycle progression by a PLC- and PKCδ-dependent cascade (Hou et al. [72]). The same isoform is involved in ATP-mediated mitogenic signalling in astrocytes, but in these cells PKCδ activation does not involve PLC but requires PLD-dependent choline formation (Neary et al. [73]).
In addition to PLC-coupled receptors, a growing number of P2Y receptors have been shown to affect the activity of AC. Besides the existence of indirect mechanisms linked to an increase in cAMP (discussed in Communi et al. [55]), only the P2Y11 receptor is directly coupled to activation of AC and PLC while P2Y12, P2Y13 and P2Y14 receptors negatively affect cAMP synthesis (Chambers et al. [13]; Communi et al. [10]; Hollopeter et al. [11]; Zhang et al. [12]). Adenylate cyclase-dependent signalling is often mediated by the cAMP-regulated kinase PKA. Stimulation of the P2Y11 receptor with ATP is shown to activate human monocyte-derived dendritic cells by increased cAMP/PKA signalling (Wilkin et al. [74]). In bovine adrenocortical fasciculate cells, ADP and ATP increase cortisol production through PKA activation by an as yet unidentified Gs protein-coupled P2Y receptor (Nishi et al. [75]). Although in unstimulated cells the cytosolic cAMP concentration is already low, its further decrease by Gi protein-coupled receptors is sometimes sufficient to exert a significant inhibitory action towards PKA. Such a response is reported in microglial cells where ATP and ADP binding to P2Y12/13 receptors mediate chemotaxis by PKA-dependent translocation of β1 integrins to ruffling regions of the cell (Nasu-Tada et al. [76]).
Despite the fact that cells express a myriad of different GPCRs and downstream acting regulators, receptor stimulation promotes rapid and specific responses. In addition, multiple GPCRs, sharing the same second messenger cascade, can induce different cellular events in one cell type indicating that GPCR signal propagation requires physical interactions in a defined cellular compartment. An example of spatial organised signalling is the β-arrestin-dependent targeting of an activated receptor into clathrin-coated vesicles or enrichment in membrane microdomains (lipid rafts) formed by cholesterol and sphingolipids (Anderson [77]; DeFea et al. [78]). Modulation of receptor function by rafts is confirmed for an increasing number of GPCRs including P2Y receptors (Anderson [77]; Ostrom and Insel [79]). In endothelial cells, it is reported that P2Y receptor-induced vasodilatation is abolished by disruption of caveolae with methyl-β-cyclodextrin (Kaiser et al. [80]). In C6 glioma cells, signalling by P2Y2 and 5-hydroxytryptamine (HT)2A receptors is attenuated after knock-down of caveolin-1 by si-RNA. Moreover, interaction between the 5-HT2A receptor and caveolin-1 facilitates its interaction with Gαq. Since P2Y2 receptor mRNA is downregulated by caveolin-1 knock-down, further studies are required to demonstrate localisation of P2Y2 receptors in caveolae of C6 cells (Bhatnagar et al. [81]).
Small GTPases as molecular switches
The processing of extracellular stimuli by GPCRs often involves signalling by second messengers (cAMP, DAG, Ca2+) towards small GTPases and/or cross-talk with tyrosine kinases. Gq protein-coupled receptor signalling via PLCβ induces formation of DAG and IP3, Ca2+ mobilisation and activation of PKC ultimately leading to activation of proline-rich tyrosine kinase 2 (Pyk2). Pyk2 cooperates with Src to recruite Grb2 and SOS, a guanine nucleotide exchange factor (GEF) that activates Ras (Lev et al. [82]). Such a mechanism is reported for Ras-dependent ERK activation induced by the protease-activated receptor-1 in astrocytes (Wang and Reiser [83]). In PC12 cells, stimulation of the P2Y2 receptor also triggers tyrosine phosphorylation of Pyk2, but further signalling to Ras involves EGFR transactivation by Src (Soltoff et al. [84]). Tyrosine kinase-dependent Ras signalling is also reported for Gi protein-coupled receptors, but this proceeds through Gβγ subunit-mediated activation of PI 3-Kγ and Shc (Ellis et al. [85]; Lopez-Ilasaca et al. [86]).
GEFs can also be regulated in a tyrosine kinase-independent manner that proceeds through direct activation by cAMP, DAG and Ca2+ or by interaction with Gα subunits as observed for the Gq/11 protein-mediated activation of RhoA (Bhattacharya et al. [87]; Bos [88]; Lutz et al. [89]; Walker et al. [90]).
P2Y receptor signalling towards GTPases is involved in short term responses, such as stress fibre formation or modulation of cell adhesion, but also in long term responses like cell proliferation. Mitogenic Ras-dependent P2Y responses are reported for C6 and HEK293 cells where Ras is implicated in P2Y2 receptor-dependent signalling to the ERK pathway (Gao et al. [91]; Tu et al. [44]). On the other hand, increased proliferation of C6 cells by the P2Y12 receptor proceeds independently of Ras, but requires RhoA-dependent activation of ERK and Rho-associated coiled-coil-containing protein kinase (ROCK) (Grobben et al. [40]; Van Kolen and Slegers, unpublished data). Interestingly, when the P2Y12 receptor is expressed in CHO cells it activates ERK and RhoA/ROCK by independent mechanisms (Soulet et al. [92]).
Another example of cross-talk between P2Y receptors and GTPases is observed in blood platelets. As mentioned above, release of ADP and subsequent P2Y1 and P2Y12 receptor binding is essential for collagen-induced platelet aggregation. A crucial step for immediate and sustained aggregation of platelets is the activation of Rap1 that increases the affinity between integrin αIIbβ3 and fibrinogen. Knock-out studies revealed that ADP-induced GTP loading of Rap1 proceeds through both Gi and Gq signalling by P2Y12 and P2Y1 receptors, respectively. The mechanism initiated by the P2Y12 receptor is shown to be PI 3-K-dependent while P2Y1-mediated activation of Rap1 requires Ca2+ mobilisation (Woulfe et al. [93]; Greco et al. [94]; Larson et al. [95]; Lova et al. [96, 97]). Stimulation of the P2Y1 receptor also contributes to platelet shape changes by a Ca2+-independent pathway. RhoA and its effector ROCK are activated by ADP through G12/13 protein-dependent signalling of the P2Y1 receptor and contribute to rapid actin polymerization and shape changes (Paul et al. [98]). Signalling towards Rho GTPases is also important in other systems. In brain, ATP and ADP induce membrane ruffling and chemotaxis of microglial cells through Gi protein-dependent activation of Rac upon stimulation of P2Y12/13 receptors (Sasaki et al. [51]; Honda et al. [54]). Stress fibre formation in vascular smooth muscle cells is reported to be mediated by RhoA/ROCK signalling that becomes activated upon stimulation of P2Y1, P2Y2, P2Y4 and P2Y6 receptors (Sauzeau et al. [99]). In the latter study, information concerning the signalling towards RhoA is lacking. In a more recent study on endothelial cells, transactivation of VEGFR upon P2Y2 receptor stimulation and recruitment of the RhoGEF Vav is shown to be a possible mechanism to initiate RhoA-mediated cell adhesion (Seye et al. [100]).
Although several P2Y receptors activate RhoA, downstream signalling and physiological consequences are determined by celltype specific mechanisms leading to diverse responses.
ERK signalling
Several GPCRs are coupled to enhanced proliferation by multiple signal transduction pathways that phosphorylate ERK. Activation of this kinase requires Ras or GTPases of the RhoA family and is often modulated by second messenger-activated pathways, although cross-talk with growth factor receptors also triggers ERK signalling.
In neurons Gs protein-mediated activation of AC increases ERK phosphorylation by a PKA/Rap1/B-Raf cascade. In contrast, induction of cAMP synthesis decreases ERK phosphorylation in C6 cells and astrocytes by a negative action of PKA on the Ras/c-Raf1 interaction, or by Rap1-mediated inhibition of c-Raf1. These observations led to the hypothesis that an increase in cAMP stimulates MEK/ERK signalling in B-Raf expressing cells but inhibits this cascade in B-Raf negative cells (Dugan et al. [101]). In both cases, PKA activation has a central role and mediates its effects through Src and Rap1 activation (Stork and Schmitt [102]). Although the majority of cAMP-dependent effects can be explained by this hypothesis, a few exceptions are reported. In some B-Raf positive cells, an increase in cAMP is shown to inhibit B-Raf, suggesting that regulation of this kinase by cAMP also depends on other cell type specific factors. One model suggests the involvement of 14-3-3 proteins acting as scaffolding proteins to shield B-Raf and Raf1 from PKA phosphorylation (Qiu et al. [103]). Other studies indicated that regulation of ERK by cAMP involves multiple cell type specific mechanisms. In COS cells overexpressing β-AR1 or β-AR2, stimulation of these receptors activate AC through a Gs protein-dependent mechanism as expected. However, PKA also phosphorylates these receptors and induces a switch from Gs to Gi/o protein binding to β-AR resulting in activation of ERK upon receptor stimulation (Martin et al. [104]). Modulation of the ERK cascade by cAMP can also occur independently of PKA. In this context, cAMP binds Epac1 or Epac2, “exchange protein directly activated by cAMP,” GEFs that activate Rap1 and Rap2 (de Rooij et al. [105, 106]; Kawasaki et al. [107]). Several examples of Gs protein-mediated activation of ERK through Epac are reported (Laroche-Joubert et al. [108]; Lin et al. [109]). Another PKA-independent mechanism of ERK phosphorylation is the Gsβγ/Src-mediated activation of Ras (Schmitt and Stork [110]).
Gq and some Gi/o protein-coupled receptors activate PLCβ and trigger formation of IP3 and DAG, resulting in Ca2+ release and PKC activation, respectively. Ca2+ increase can activate ERK through Pyk2 that activates Ras as mentioned above. Otherwise, Ca2+-dependent modulation of Ras activity is also mediated by Ras guanine nucleotide-releasing factor (RasGRF), a GEF that contains Ca2+- and DAG-binding domains, (Ebinu et al. [111]) or by Ca2+/calmodulin-dependent kinases CaMK-II and CaMK-IV (reviewed in Agell et al. [112] and Walker et al. [90]). Increase of intracellular calcium and DAG formation also results in activation of cPKCs while DAG formation alone is sufficient to activate nPKCs. Increase in PKC activity modulates the ERK cascade through Ras by inhibition of RasGAPs and/or stimulation of RasGEFs. In addition, PKC can activate Raf independently of Ras. Indeed, it is shown that PKCα phosphorylates Raf at Ser499 (Kolch et al. [113]). However, mutation of this serine residue into alanine does not affect Raf activity in response to phorbol esters (Yip-Schneider et al. [114]). More convincing data were obtained when constitutively active PKC was expressed in rat 6 fibroblasts. These cells display Ras-independent signalling towards ERK by direct phosphorylation of Raf by PKCɛ. Since activation of Ras is required in several systems this interaction is cell type-dependent (Cacace et al. [115]; Ueffing et al. [116]). Direct phosphorylation of Raf by PKC is also involved in ERK activation by the Gi protein-coupled leukotriene (LT)D4 receptor in intestinal epithelial cells. Although stimulation of this receptor also triggers a parallel PKC-independent activation of Ras, transfection experiments confirmed that Ras is dispensable for LTD4 receptor-mediated ERK activation (Paruchuri et al. [117]). When constitutive active point mutants of PKCα, PKCδ and PKCɛ were introduced in COS cells, only PKCδ activated the ERK cascade (Ueda et al. [118]), indicating that involvement of PKC isoforms in ERK signalling vary among different cell types. This is also confirmed by the observation that, in platelets, cPKCs are involved in thrombin-induced MEK and ERK activation independently of Ras or Raf (Nadal-Wollbold et al. [119]).
Gi/o protein-coupled receptors that are not linked to PLC activation can also modulate mitogenic signalling through Gβγ-dependent activation of PI 3-Kγ. Signalling from PI 3-Kγ to ERK proceeds through Shc/Grb2/SOS/Ras (Lopez-Ilasaca et al. [86]). An increasing number of reports point to the involvement of PKCKζ in Gi protein-dependent phosphorylation of ERK. The first observation was made in CHO cells where stimulation of the LPA receptor triggers MEK/ERK signalling via a PI 3-Kγ-dependent activation of PKCζ not abrogated by transfection with dominant negative Ras (Takeda et al. [120]). In addition, a recent report indicated that angiotensin II-induced ERK activation in rat vascular smooth muscle cells requires interaction between Ras and PKCζ (Zhao et al. [121]). PKCζ-dependent activation of ERK is mediated by interaction with MEK, a property shared by other PKC isoforms (Schönwasser et al. [122]), or by regulation of Raf1. Studies performed in rat embryonic hippocampal cells indicated that PKCζ can phosphorylate the Raf kinase inhibitory protein (RKIP) resulting in dissociation of the Raf1/RKIP complex (Corbit et al. [123]). In addition, co-immunoprecipitation experiments in COS cells showed that modulation of c-Raf1 by PKCζ is also regulated by 14-3-3 scaffolding proteins (Van Der Hoeven et al. [124]).
Initial studies concerning P2Y receptor-mediated activation of ERK were made in astrocytes where this cascade was shown to be involved in cell proliferation and process elongation (Neary and Zhu [125]; King et al. [126]).
Although ATP triggers pertussis toxin insensitive IP3 and Ca2+ responses in astrocytes, these are not required for the signalling towards ERK which depends on rapid membrane translocation of PKCδ upon phosphatidylcholine hydrolysis by PLD (Neary et al. [73]). In PC12 cells, stimulation of the P2Y2 receptor also induces PKCδ-dependent ERK phosphorylation, although this mechanism requires Ca2+ and Pyk2 for the association of Shc and Grb2 to the receptor and for subsequent activation of SOS/Ras/Raf/MEK/ERK (Soltoff et al. [84]). Many reports showed that P2Y receptor-mediated ERK signalling requires PKC activation (Graham et al. [127]; Huwiler and Pfeilschifter [128]; Erlinge [129]), but a PKC-independent mechanism is reported in thyroid FRTL-5 cells (Tornquist et al. [130]). In 1321N1 astrocytoma cells, stimulation of the P2Y6 receptor with UDP activates PKCα, ɛ and ζ which are correlated with ERK phosphorylation (Kim et al. [131]). Although the use of general PKC inhibitors 3-[1-(dimethylaminopropyl) indol-3-yl]-4-(indol-3-yl)maleimide hydrochloride (GF109203X) and 12-(2-cyanoethyl)-6,7,12,13-tetrahydro-13-methyl-5-oxo-5H-indolo(2,3-a)pyrrolo(3,4-c)-carbazole (Gö 6976) diminished ERK signalling, the lack of specificity of these compounds makes it difficult to determine the contribution of each of these PKC isoforms in the mechanism of ERK activation (Way et al. [132]).
Enhanced proliferation by a P2Y receptor-mediated stimulation of the ERK pathway has been reported in a large number of cell types such as human mesangial cells, vascular smooth muscle cells and primary astrocytes (Huwiler and Pfeilschifter [128]; Harper et al. [133]). Transient ERK activation by P2Y1, P2Y2 or P2Y12 receptor stimulation also increases cell proliferation in C6 cells indicating that activation of several P2Y receptor subtypes can converge into the same physiological response (Table 2) (Tu et al. [44]; Claes et al. [39]; Czajkowski et al. [46]).
In addition to mitogenesis, P2Y receptor signalling towards ERK elicits other physiological processes including cell survival, inflammation and reactive gliosis. In human lung microvascular endothelial cells, hyperoxia-induced release of ATP results in cell survival through ERK and PI 3-K signalling cascades activated by P2Y2 and/or P2Y6 receptors, while stimulation of the ERK cascade by the P2Y6 receptor protects 1321N1 astrocytoma cells from TNFα-induced apoptosis (Ahmad et al. [134]; Kim et al. [131]). Rapid ERK1/2 and p38 MAPK activation plays an important role in P2Y2 receptor-dependent primary granule release from human neutrophils (Meshki et al. [135]). A similar phenomenon is observed in articular chondrocytes where ATP acts as a pro-inflammatory mediator by increasing arachidonic acid production and release of prostaglandin E2 through a P2Y2 receptor-dependent activation of p38 and ERK1/2 (Berenbaum et al. [136]). In primary astrocytes, P2Y receptor-mediated ERK activation by ATP is shown to induce reactive astrogliosis, a phenomenon that occurs upon brain injury and is characterised by astroglial proliferation, cellular hypertrophy and up regulation of GFAP. This effect is mediated by an ERK-dependent increase in the expression of COX-2 (Brambilla et al. [53]). In C6 cells, P2Y12 and P2Y2 receptor-induced activation of ERK is coupled to an enhanced cell proliferation, while a negative modulation of GFAP synthesis by the P2Y12 receptor is reported (Claes et al. [39]; Tu et al. [44]; Van Kolen and Slegers [45]). These differences are probably due to the fact that in C6 cells induction of GFAP expression is not correlated with an enhanced proliferation but requires growth arrest.
In summary, most P2Y receptors are coupled to ERK phosphorylation, but the signalling mechanism and the physiological effect of this pathway are cell type specific and are determined by the cellular context.
PI 3-K/PKB signalling
PKB/Akt is involved in a large variety of cellular processes including glucose metabolism, mitogenesis, differentiation, survival and motility (Brazil et al. [137]). This member of the AGC protein kinase superfamily is recruited to the plasmamembrane upon PI 3-K-mediated PIP3 formation, but is also controlled in a PI 3-K-independent, but calmodulin-dependent, fashion upon intracellular Ca2+ mobilisation by stimulation of neuronal NMDA receptors (Cantley [138]; Leevers et al. [139]; Woodgett [140]; Yano et al. [141]).
Modulation of PKB activity is reported for a variety of GPCR ligands including adrenergics, cannabinoids, carbachol, glutamate, histamine, nucleotides and thrombin (Dickenson [142]; Franke et al. [143], Iacovelli et al. [144] Murga et al. [145]; Sanchez et al. [146]). Due to the existence of multiple phosphoinositide-dependent cascades, regulation of PKB signalling by GPCRs varies among the studied systems.
In HEK293 cells, stimulation of β-AR with (−)-isoproterenol activates PKB via Gsβγ, Src, Ras and PI 3-K (Schmitt and Stork [110]; Bommakanti et al. [147]) while activation of AC by Gαs exerts differential effects on PKB activity. In cells expressing Epac, cAMP activates PI 3-K/PKB via Rap1 while, in other cells, cAMP activates PKA that exerts a negative action on PI 3-K and PKB (Mei et al. [148]; Wang et al. [149]).
Gi protein-mediated activation of PKB can occur through the coupling of the Gβγ subunit to the catalytic subunit of PI 3-K or via growth factor receptor trans-activation. Although only p110β was initially reported to be activated by Gβγ subunits, this feature is also observed for the p110β isoform (Kurosu et al. [150]; Stoyanov et al. [151]). This mechanism is reported in Vero cells where stimulation with LPA activates Ras upon increase in p110β lipid kinase activity (Yart et al. [152]). Gi protein-mediated transactivation of growth factor receptors is reported in HaCaT, A-431, and HEK293 cells where stimulation of the angiotensin type I receptor by mechanical stress induces transactivation of EGFR leading to activation of the PI 3-K/PKB cascade and protection of these cells from apoptosis (Kippenberger et al. [153]).
In 1321N1 astrocytoma cells, PLCβ activation by the Gq protein-coupled muscarinic M3 receptor also triggers PI 3-K activation through ErbB3 transactivation, but this mechanism requires Ca2+ mobilisation (Tang et al. [154]). In contrast, some reports showed an inhibitory pathway from Gq protein-coupled receptors towards PI 3-K by direct interaction between Gα-subunits released from heterotrimeric G proteins and p110α, as reported for the α1A-AR in rat-1 fibroblasts (Ballou et al. [155, 156]), or by inhibition of insulin receptor substrate-1-associated PI 3-K activity in 1321N1 astrocytoma cells by carbachol, histamine or thrombin. These observations reveal opposing effects of muscarinic receptor stimulation on PI 3-K activity mediated by insulin and ErbB3 receptors in these cells (Batty et al. [157]).
Modulation of PI 3-K/PKB signalling is also reported for a few P2Y receptors. In bovine adventitial fibroblasts, ATP is shown to induce proliferation through parallel but independent ERK and PI 3-K signalling cascades that contribute to mTOR and p70S6K phosphorylation (Gerasimovskaya et al. [158]). In rat mesangial cells, stimulation of the P2Y2 receptor with ATP or UTP activates PKB by a PDK-1-dependent mechanism while, in C6 cells, ADP activates PI 3-K/PKB by the Gi protein-coupled P2Y12 receptor but inhibits PI 3-K by stimulation of the Gq/G11/12 protein-coupled P2Y1 receptor (Table 2) (Van Kolen and Slegers [45]; Czajkowski et al. [46]; Huwiler et al. [159]). Although most effects of P2Y-mediated activation of PI 3-K signalling are known to be related to cell proliferation, differentiation and survival, this signalling cascade is also involved in other processes. In this regard, it can be mentioned that P2Y12 receptor-mediated PI 3-K/PKB activation modulates proliferation and differentiation of C6 cells, but also plays an important role in ADP-induced platelet aggregation (Van Kolen and Slegers [45]; Czajkowski et al. [46]; Chen et al. [160]; Kim et al. [161]).
P2Y receptor-integrated G protein-coupled receptor and receptor tyrosine kinase signalling cascades
G protein-coupled receptor cross-talk
Complementary to vertical downstream signalling upon GPCR stimulation, these receptors also mediate lateral signalling by cross-talk with other receptors (reviewed in Cordeaux and Hill [162]). In human platelets, it was reported that P2Y12 receptor activation potentiates P2Y1 receptor-mediated Ca2+ signalling, while the P2Y1 receptor negatively regulates this action (Hardy et al. [163]). In renal mesangial cells, P2Y receptors activated by ATP and UTP induce a rapid desensitisation of the sphingosine-1-phosphate (S1P) receptor by PKC-dependent phosphorylation (Xin et al. [164]). A more complex interplay is observed between P2Y receptors and 5-HT receptor subtypes. Studies performed in CHO cells stably expressing 5-HT1A receptors revealed that the responsiveness of this receptor is reduced by a PLD/PKC-dependent phosphorylation upon short (<5 min) pre-treatment with ATP, while the agonist efficacy of the overexpressed 5-HT1B receptor is not altered. Alternatively, longer treatment with ATP alone attenuates 5-HT1B signalling by a mechanism that requires activation of phospholipase A2 (PLA2) (Berg et al. [165]). Furthermore, stimulation of P2Y receptors can also modulate the release of transmitter molecules, including dopamine, glutamate and serotonin (Bezzi and Voltera [52]; Krugel et al. [166]; Nedergaard et al. [167]). A recently discovered mechanism of GPCR cross-talk is the assembly of a heteromeric receptor complex displaying the pharmacological profile of one receptor and the signalling properties of the other. Such an interaction is reported in HEK293 cells overexpressing A1 and P2Y1 receptors. The heteromeric A1-P2Y1 receptor complex inhibits AC through Gi/o protein, but displays P2Y1 receptor-like pharmacological properties (Yoshioka et al. [168]).
P2Y receptor-mediated transactivation
Many studies reveal that GPCRs and growth factor receptors share a number of signalling modules (e.g., Raf/MEK/ERK, PI 3-K/PDK/PKB) to transduce their effects. In the past decade, it has become clear that the signalling pathways of both receptor systems are interconnected. Stimulation of a GPCR can induce a rapid tyrosine phosphorylation of RTKs. This transactivation mechanism is reported for many GPCRs and proceeds through the Gβγ subunit-dependent activation of Src. Src in turn activates RTKs by phosphorylation of specific tyrosines located in their intracellular domains or induction of matrix metalloproteases-dependent release of growth factor receptor ligands, e.g., release of heparin-bound EGF (Luttrell and Luttrell [169]).
Another target for signal integration of GPCRs and RTKs are docking proteins. Although these proteins contain phospho-tyrosine binding domains that interact with phosphorylated tyrosine residues of RTKs, stimulation of GPCRs can induce growth factor receptor-independent phosphorylation of docking proteins by Src (Bisotto and Fixman [170]).
In addition to GPCR-dependent phosphorylation of RTKs, the opposite activation mechanism is also reported. Binding of PDGF to its cognate receptor induces association of PDGFR with the Gi protein-coupled S1P receptor. Subsequently, Src is recruited to this complex by Gβγ subunits and phosphorylates Grb-2 associated binder-1 resulting in dynamin II-induced “pinching off” of vesicles involved in endocytosis of PDGF-S1P signalling complexes and subsequent activation of ERK1/2 (Waters et al. [171]).
Cross-talk between RTKs and P2Y receptors is reported in Müller glial cells where ATP exerts its mitogenic effect through transactivation of EGF and PDGF receptors resulting in ERK-dependent enhanced proliferation. In these cells, ATP-induced activation of ERK was abolished by treatment with the RTK autophosphorylation inhibitor tyrphostin (AG1478) (Milenkovic et al. [172]). In rat striatal astrocytes, ATP and bFGF activate ERK and induce astrogliosis by a mechanism that is insensitive to RTK inhibition (Abbracchio et al. [173]; Bolego et al. [174]; Neary et al. [175]). More recently, mechanistic studies performed in 1321N1 astrocytoma cells reveal that the human P2Y2 receptor interacts with Src and Pyk2, probably by its proline-rich putative SH3 binding sites (PXXP). This interaction is implicated in P2Y2 receptor-induced transactivation of EGF, PDGF and VEGF receptors (Liu et al. [176]; Seye et al. [100]). Src inhibition abolishes growth factor receptor transactivation and ERK phosphorylation. Although the rat P2Y2 receptor lacks PXXP motives, tyrosine kinase-dependent activation of ERK upon P2Y2 receptor stimulation is reported in a few rat cell lines, including C6 and PC12 cells (Soltoff et al. [84]; Tu et al. [44]). In the latter cases, P2Y2 receptordependent activation of Pyk2 is mediated by PKC and Ca2+ suggesting that the PXXP sequence is dispensable for P2Y2 receptor-induced tyrosine phosphorylation of Pyk2 and downstream signalling towards ERK. Moreover, P2Y2 mutants lacking PXXP-motives are still able to activate ERK demonstrating the existence of other pathways towards phosphorylation of ERK (Liu et al. [176]). Observations made in human endothelial cells, where UTP-induced signalling to ERK was shown to depend on Ca2+, PKC and integrin-mediated cell anchorage, already pointed to a pathway distinct from the classical Ras/Raf/MEK/ERK cascade (Short et al. [177]). Human and mouse P2Y2 receptors contain a RGD sequence which allows activation of ERK by interaction with αVβ3/β5 integrins followed by Go protein coupling. Since these proteins also mediate cell adhesion and chemotaxis, the observed P2Y2/αVβ3/β5-interaction also points to a possible function of P2Y2 receptors in inflammatory responses (Erb et al. [178]).
It is clear that, in analogy with other GPCRs, crosstalk between P2Y and growth factor receptors may occur at different levels of the signal transduction pathway depending on receptor subtypes and on the studied system. For the P2Y2 receptor, additional transactivation mechanisms are facilitated by the presence of signalling motives (e.g., PXXP or RGD) that allow direct interaction with other signalling components (Src, integrins).
P2Y receptor-activated signal transduction pathways in C6 glioma cells
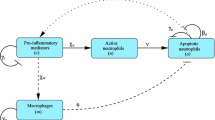
As mentioned above, the final outcome of nucleotide-mediated signalling is influenced by ecto-enzymes (Claes and Slegers [17]; Czajkowski and Baranska [18]; Grobben et al. [21, 179]). ATP and ADP hydrolysis to adenosine results in growth inhibition by a mechanism that is not yet fully understood. When nucleotide hydrolysis is prevented, ATP, ADP and ApnA (in particular Ap3A and Ap4A) increase cell proliferation more than two-fold. Stimulation with 2MeSADP, a P2Y agonist not hydrolysed by the ecto-enzymes present on the plasma membrane of C6 cells, also results in growth enhancement and inhibition of β-AR-induced differentiation into astrocyte type II (Claes et al. [39]; Van Kolen and Slegers [45]). The pathways involved in the P2Y receptor-dependent effects on growth and differentiation of these cells are presented in Figure 1.

Overview of P2Y receptor-mediated signalling cascades in C6 cells. Green and red lines represent stimulatory (green arrows) and inhibitory (red squares) actions respectively. Dashed lines are incomplete characterised pathways. P2Y2 receptor stimulation enhances ERK-dependent proliferation through a PLC-dependent pathway while P2Y12 receptor stimulation enhances cell proliferation by RhoA- and PKCζ-dependent activation of ERK (Claes et al. [39]; Grobben et al. [40]; Tu et al. [44]; Van Kolen and Slegers, unpublished data). P2Y12 receptor stimulation also inhibits cAMP-dependent induction of differentiation by reactivation of PKB which requires Src/Pyk2 complex formation and Rap1 activation. Formation of the Src/Pyk2 complex requires Ca2+ and PLD2 which is constitutively active (Claes et al. [22]; Van Kolen and Slegers [45]; Van Kolen et al. [185]). Cyclic AMP-dependent inhibition of PKB and ERK is suggested to depend on inhibition of Rap1 (Wang et al. [149]). The negative modulation of PI 3-K by the P2Y1 receptor is only displayed in the presence of serum (Czajkowski et al. [46])
Nucleotides stimulate several purinergic receptors that activate the ERK cascade by at least two distinct mechanisms. The P2Y2 receptor, stimulated by UTP and ATP, enhances ERK phosphorylation through a PLCβ/PKC/Ras/Raf/MEK cascade that is attenuated by inhibition of tyrosine kinases and Ca2+ chelation by BAPTA-AM (Tu et al. [44]). The Ca2+-dependence of the P2Y2 receptor-mediated activation of ERK suggests the involvement of a cPKC (α, βI, βII or γ). It is also shown that ADP stimulates the P2Y1 receptor and activates ERK through a Ca2+-dependent mechanism (Czajkowski et al. [46]), likely by a similar mechanism as reported for the P2Y2 receptor (Tu et al. [44]). In addition, it has been shown that ADP can activate ERK by stimulation of the P2Y12 receptor through a RhoA- and PKC-dependent pathway that does not require Ca2+, Ras or tyrosine kinase activation (Grobben et al. [40]). The fact that Ca2+ removal does not affect P2Y12 receptor-mediated ERK activation excludes the involvement of cPKCs. Stimulation of the P2Y12 receptor does not induce PI-turnover, but nPKCs might be involved since alternative activation mechanisms, based on Ser/Thr and Tyr phosphorylation, have been reported (Steinberg [180]; Parekh et al. [181]). Data from our laboratory suggest an important role for PKCζ in P2Y12 receptor-dependent activation of ERK. The fact that no cross-talk between ERK and PI 3-K is observed in C6 cells indicates that PKCζ exerts its actions independently of PI 3-K via a RhoA-dependent mechanism (Grobben et al. [40]; Van Kolen and Slegers, [199]). Although P2Y receptors use different mechanisms to activate ERK, they all converge to increased cell proliferation by enhanced synthesis of c-Myc, c-Jun and c-Fos (Zhang et al. [182]). Progression through the G1/S phase of the cell cycle is due to a decreased expression of p27Kip and increased expression of cyclinD.
While stimulation of ERK signalling by P2Y receptors has been known for several years, the coupling with PI 3-K activation was discovered more recently. When C6 cells are grown in the presence of serum, P2Y1 receptor signalling predominates and is shown to inhibit PI 3-K (Czajkowski et al. [46]). Upon serum deprivation, P2Y1 receptor expression decreases while P2Y12 becomes the main ADP-stimulated receptor that enhances the activity of PI 3-K by a Gi proteindependent mechanism. These observations demonstrate that, in addition to autocrine growth factor receptor signalling, the constitutive PI 3-K activity in C6 cells is modulated by P2Y1 and P2Y12 receptor expression. Another cross-talk at the level of PI 3-K/PKB is observed for P2Y12 and β-AR. Increase in cAMP upon stimulation of the latter receptor transiently inhibits PKB phosphorylation. Stimulation of the P2Y12 receptor, which negatively affects AC, does not only counteract this inhibition but even enhances PKB activity in comparison to unstimulated cells, suggesting that P2Y12 receptor-mediated PI 3-K/PKB activation is not only due to its inhibitory effect on AC (Van Kolen and Slegers [45]; Czajkowski et al. [46]; Baranska et al. [183]). In addition to their opposing effects on PI 3-K/PKB signalling, unpublished data of our laboratory revealed similar modulation of ERK signalling by P2Y12 and β-AR. Whether the P2Y12 receptor-mediated reversal of ERK inhibition is involved in the inhibition of β-AR-induced GFAP synthesis remains to be determined. The observation that stimulation of the cells with UTP activates ERK, but fails to inhibit the β-AR-induced growth arrest and GFAP synthesis, suggests that ERK activation alone is not sufficient to counteract differentiation (Claes et al. [39]; Tu et al. [44]). Conversely, transfection of C6 cells with constutively active PKB prevented (−)-isoproterenol-induced differentiation indicating that inhibition of PKB signalling is required for cAMP-dependent induction of differentiation. Apparently this observation is in contrast with data showing that cAMP-dependent induction of differentiation requires PI 3-K activity which is not inhibited upon a 48-h treatment with dbcAMP (Roymans et al. [34]). This might be explained by the fact that induction of differentiation by stimulation of β-AR proceeds through transient inhibition of PKB while recovery of this activity is required to prohibit cell death. This hypothesis is confirmed by a recent study where sustained inhibition of PI 3-K/PKB by cannabinoids is shown to induce apoptosis in C6 cells (Table 2) (Ellert-Miklaszewska et al. [184]). Taken together, P2Y12 receptor stimulation inhibits cAMP-dependent induction of differentiation by a transient increase in PI 3-K/PKB activity. Ca2+ chelation inhibits the basal PKB activity and P2Y12 receptor-mediated increase in PKB phosphorylation. Although C6 cells also express the P2Y2 receptor, stimulation with UTP does not enhance the activity of PI 3-K/PKB, which may be explained by a differential coupling to G protein subtypes. P2Y2 receptor-mediated signalling proceeds through Gq proteins while the activation of PDK is Gi protein-dependent (Table 2) (Tu et al. [44]; Huwiler et al. [159]). The lack of Gi protein coupling of the P2Y2 receptor in C6 cells might be a consequence of compartimentalisation into caveolae as reported for some Gq protein-coupled receptors (Bhatnagar et al. [81]).
Although experiments in CHO cells reveal that P2Y12 receptor-induced ERK activation requires PI 3-Kγ (Soulet et al. [92]), experiments performed with LY294002 or Wortmannin excluded cross-talk between both cascades in C6 cells (Grobben et al. [40]). These differences in signalling mechanisms can be explained by the fact that the latter PI 3-K-isoform is only moderately expressed in C6 cells (Van Kolen and Slegers [45]). The exact mechanism of P2Y12 receptor-induced PI 3-K/PKB activation is not fully understood, but recent data revealed that Src and Pyk2 are involved in P2Y12 receptor signalling to PI 3-K (Van Kolen et al., [185]). A similar pathway is observed in PC12 cells where Src, in complex with Pyk2 and PLD2, activates PI 3-K in response to H2O2 (Banno et al., [186]). Since PLD2 is constitutively active in C6 cells (Bobeszko et al. [187]), a significant role for this enzyme in PI 3-K/Akt signalling is suggested. Although Soulet et al. [92] reported that transactivation of PDGFR is involved in PI 3-K activation by the P2Y12 receptor in CHO cells, the use of receptor kinase inhibitors indicated that PDGFR and EGFR are not transactivated by the P2Y12 receptor in C6 cells. Alternatively, a Rap1-mediated activation of PI 3-K by the P2Y12 receptor cannot be excluded. Indeed, PI 3-K is postulated as a downstream effector of Rap1 that is inhibited by an increase in cAMP concentration (Wang et al. [149]). Data from our laboratory indicated a rapid P2Y12 receptor-induced activation of Rap1 that was abolished by Ca2+ chelation and inhibition of Src/Pyk2 complex formation but not by PI 3-K inhibition (Van Kolen et al. [185]). These results positioned Rap1 downstream of Src/Pyk2 but upstream of PI 3-K. In addition, this mechanism involves Gβγ protein subunits and Ca2+-dependent activation of Pyk2 that requires association to IGF-IR and PLD2 to interact with Src. Although Src and Pyk2 are shown to activate Ras/Raf/MEK/ERK in primary astrocytes (Wang and Reiser [83]), this mechanism did not contribute to P2Y12 receptor-mediated ERK activation in C6 cells pointing to a physical separation of both cascades (Grobben et al. [40]; Van Kolen and Slegers, [199]). Indeed, the formation of a Pyk2/Src/PLD2/IGFI-R complex may contribute to compartmentalisation of this signalling pathway that requires intact lipid rafts to be active (Van Kolen et al. [185]). In contrast, in blood platelets Rap1, but also Pyk2 activation by the P2Y12 receptor, depends on PI 3-K activity but is insensitive to Ca2+ chelation (Lova et al. [96, 97]; Koziak et al. [188]). These findings indicate that different cell specific pathways are involved in P2Y12 receptor-mediated activation of PI 3-K/PKB and additional research is required to allow full characterisation of these signalling cascades.
Conclusions
At present, nucleotides are known to regulate a variety of biological processes related to vascular-, immunological- and intestinal functioning. In vitro studies on glial and neuronal cells implicated the P2Y receptor-activated signalling pathways in regulation of cell motility, proliferation, chemotaxis and protection against oxidative stress. Furthermore, investigations on tumoral cells demonstrated that stimulation of P2Y receptors contribute to tumorigenesis by increasing cell proliferation through ERK and PKB signalling pathways activated by independent mechanisms. From these observations, a role of these receptors as potential targets in clinical applications emerges.
P2Y receptors modulate these physiological functions by activation of GTPases and direct or indirect activation of protein kinases. Characterisation of the involved receptor(s) and elucidation of P2Y receptorinduced activation of defined pathways needs to be improved by synthesis of specific P2Y agonists and antagonists.
Studies on P2Y receptor-mediated signalling, discussed in this review, demonstrate that besides vertical signal transduction, lateral cross-talk between growth factor receptors and GPCRs extends the signalling properties of a defined receptor subset. It also becomes clear that signal transduction pathways activated by P2Y receptors largely depend on the cell type and their environment. On the one hand, cellular specificity is determined by differential expression of signalling proteins, but on the other hand also depends on the assembly of signalling modules. Besides specific protein-protein interactions, intracellular compartmentalisation (e.g., lipid rafts, clathrin-coated vesicles) also contributes to the specificity of receptor signalling. Identification of the signalling modules and cellular compartmentalisation will provide more insight into the P2Y receptor-activated signalling cascades.
Abbreviations
- AC:
-
adenylate cyclase
- Ap3A:
-
P1,P3-di(adenosine-5′)triphosphate
- Ap4A:
-
P1,P4-di(adenosine-5′)tetraphosphate
- AR:
-
adrenergic receptor
- COX:
-
cyclooxygenase
- DAG:
-
diacylglycerol
- ERK:
-
extracellular signal-regulated kinase
- GFAP:
-
glial fibrillary acidic protein
- GPCR:
-
G protein-coupled receptor
- HT:
-
hydroxytryptamine
- IP3 :
-
inositol (1,4,5)-triphosphate
- PAP:
-
adenosine-3′,5′-biphosphate
- PI:
-
phosphatidylinositol
- PL:
-
phospholipase
- PI 3-K:
-
phosphatidylinositol 3-kinase
- PPADS:
-
pyridoxalphosphate-6-azophenyl-2′-4′-disulphonate
- Pyk2:
-
proline-rich tyrosine kinase 2
- RKIP:
-
Raf kinase inhibitory protein
- RTK:
-
receptor tyrosine kinase
References
Drury AN, Szent-Gyorgyi A (1929) The physiological activity of adenine compounds with especial reference to their action upon the mammalian heart. J Physiol 68:213–37
Abbracchio MP, Saffrey MJ, Höpker V, Burnstock G (1994) Modulation of astroglial cell proliferation by analogues of adenosine and ATP in primary cultures of rat striatum. Neuroscience 59:67–6
Burnstock G, Williams M (2000) P2 purinergic receptors: modulation of cell function and therapeutic potential. J Pharmacol Exp Ther 295:862–69
Burnstock G (1993) Physiological and pathological roles of purines: an update. Drug Dev Res 28:195–06
Ralevic V, Burnstock G (1998) Receptors for purines and pyrimidines. Pharmacol Rev 50:413–92
Burnstock G, Kennedy C (1985) Is there a basis for distinguishing two types of P2-purinoceptors? Gen Pharmacol 16:433–40
Abbracchio MP, Burnstock G (1994) Purinoceptors: are there families of P2X and P2Y purinoceptors? Pharmacol Ther 64:445–75
Fredholm BB, Abbracchio MP, Burnstock G, Daly JW, Harden TK, Jacobson KA, Leff P, Williams M (1994) Nomenclature and classification of purinoceptors. Pharmacol Rev 46:143–56
Abbracchio MP, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Miras-Portugal MT, King BF, Gachet C, Jacobson KA, Weisman GA, Burnstock G (2003) Characterization of the UDP-glucose receptor (re-named here the P2Y14 receptor) adds diversity to the P2Y receptor family. Trends Pharmacol Sci 24:52–5
Communi D, Gonzalez NS, Detheux M, Brézillon S, Lannoy V, Parmentier M, Boeynaems JM (2001) Identification of a novel human ADP receptor coupled to Gi. J Biol Chem 276:41479–1485
Hollopeter G, Jantzen HM, Vincent D, Li G, England L, Ramakrishnan V, Yang RB, Nurden P, Nurden A, Julius D, Conley PB (2001) Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature 409:202–07
Zhang FL, Luo L, Gustafson E, Palmer K, Qiao X, Fan X, Yang S, Laz TM, Bayne M, Monsma F Jr (2002) P2Y13: identification and characterization of a novel Gαi-coupled ADP receptor from human and mouse. J Pharmacol Exp Ther 301:705–13
Chambers JK, Macdonald LE, Sarau HM, Ames RS, Freeman K, Foley JJ, Zhu Y, McLaughlin MM, Murdock P, McMillan L, Trill J, Swift A, Aiyar N, Taylor P, Vawter L, Naheed S, Szekeres P, Hervieu G, Scott C, Watson JM, Murphy AJ, Duzic E, Klein C, Bergsma DJ, Wilson S, Livi GP (2000) A G protein-coupled receptor for UDP-glucose. J Biol Chem 275:10767–0771
Herold CL, Qi AD, Harden TK, Nicholas RA (2004) Agonist versus antagonist action of ATP at the P2Y4 receptor is determined by the second extracellular loop. J Biol Chem 279:11456–1464
Qi A-D, Zambon AC, Insel PA, Nicholas RA (2001) An arginine/glutamine difference at the juxtaposition of transmembrane domain 6 and the third extracellular loop contributes to the markedly different nucleotide selectivities of human and canine P2Y11 receptors. Mol Pharmacol 60:1375–382
Lawson Z, Wheatley M (2004) The third extracellular loop of G-protein-coupled receptors: more than just a linker between two important transmembrane helices. Biochem Soc Trans 32:1048–050
Claes P, Slegers H (2004) P2Y receptor affects the proliferation and differentiation of glial and neuronal cells: a focus on rat C6 glioma cells. Curr Neuropharmacol 2:207–20
Czajkowski R, Baranska J (2002) Cross-talk between the ATP and ADP nucleotide receptor signalling pathways in glioma C6 cells. Acta Biochim Pol 49:877–89
Goding JW, Grobben B, Slegers H (2003) Physiological and pathophysiological functions of the ecto-nucleotide pyrophosphatase/phosphodiesterase family. Biochim Biophys Acta 1638:1–9
Zimmermann H (2000) Extracellular metabolism of ATP and other nucleotides. Naunyn-Schmiedeberg’s Arch Pharmacol 362:299–09
Grobben B, Claes P, Roymans D, Esmans EL, Van Onckelen H, Slegers H (2000) Ecto-nucleotide pyrophosphatase modulates the purinoceptor-mediated signal transduction and is inhibited by purinoceptor antagonists. Br J Pharmacol 130:139–45
Claes P, Van Kolen K, Roymans D, Blero D, Vissenberg K, Erneux C, Verbelen JP, Esmans EL, Slegers H (2004) Reactive blue 2 inhibition of cyclic AMP-dependent differentiation of rat C6 glioma cells by purinergic receptor-independent inactivation of phosphatidylinositol 3-kinase. Biochem Pharmacol 67:1489–498
Boeynaems JM, van Giezen H, Savi P, Herbert JM (2005) P2Y receptor antagonists in thrombosis. Curr Opin Investig Drugs 6:275–82
Lambrecht G, Braun K, Damer M, Ganso M, Hildebrandt C, Ullmann H, Kassack MU, Nickel P (2002) Structure-activity relationships of suramin and pyridoxal-5′-phosphate derivatives as P2 receptor antagonists. Curr Pharm Des 8:2371–399
Kam PC, Nethery CM (2003) The thienopyridine derivatives (platelet adenosine diphosphate receptor antagonists), pharmacology and clinical developments. Anesthesia 58:28–5
Kim YC, Lee JS, Sak K, Marteau F, Mamedova L, Boeynaems JM, Jacobson KA (2005) Synthesis of pyridoxal phosphate derivatives with antagonist activity at the P2Y13 receptor. Biochem Pharmacol 70:266–74
Xu B, Stephens A, Kirschenheuter G, Greslin AF, Cheng X, Sennelo J, Cattaneo M, Zighetti ML, Chen A, Kim SA, Kim HS, Bischofberger N, Cook G, Jacobson KA (2002) Acyclic analogues of adenosine bisphosphates as P2Y receptor antagonists: phosphate substitution leads to multiple pathways of inhibition of platelet aggregation. J Med Chem 45:5694–709
Kubiatowski T, Jang T, Lachyankar MB, Salmonsen R, Nabi RR, Quesenberry PJ, Litofsky NS, Ross AH, Recht LD (2001) Association of increased phosphatidylinositol 3-kinase signaling with increased invasiveness and gelatinase activity in malignant gliomas. J Neurosurg 95:480–88
Roymans D, Slegers H (2001) Phosphatidylinositol 3-kinases in tumor progression. Eur J Biochem 268:487–98
Grobben B, De Deyn PP, Slegers H (2002) Rat C6 glioma as experimental model system for the study of glioblastoma growth and invasion. Cell Tissue Res 310:257–70
Okumura N, Takimoto K, Okada M, Nakagawa H (1989) C6 glioma cells produce basic fibroblast growth factor that can stimulate their own proliferation. J Biochem 106:904–09
Resnicoff M, Sell C, Rubini M, Coppola D, Ambrose D, Baserga R, Rubin R (1994) Rat glioblastoma cells expressing an anti-sense RNA to the insulin-like growth factor-1 (IGF-1) receptor are nontumorigenic and induce regression of wild-type tumors. Cancer Res 54:2218–222
Strawn LM, Mann E, Elliger SS, Chu LM, Germain LL, Niederfellner G, Ullrich A, Shawver LK (1994) Inhibition of glioma cell growth by a truncated platelet-derived growth factor-β receptor. J Biol Chem 269:21215–1222
Roymans D, Vissenberg K, De Jonghe C, Grobben B, Claes P, Verbelen JP, Van Broeckhoven C, Slegers H (2001) Phosphatidylinositol 3-kinase activity is required for the expression of glial fibrillary acidic protein upon cAMP-dependent induction of differentiation in rat C6 glioma. J Neurochem 76:610–18
Backhovens H, Gheuens J, Slegers H (1987) Expression of glial fibrillary acidic protein in rat C6 glioma relates to vimentin and is independent of cell-cell contact. J Neurochem 49:348–54
Messens J, Slegers H (1992) Synthesis of glial fibrillary acidic protein in rat C6 glioma in chemically defined medium: cyclic AMP-dependent transcriptional and translational regulation. J Neurochem 58:2071–080
Pianet I, Merle M, Labouesse J (1989) ADP and, indirectly, ATP are potent inhibitors of cAMP production in intact isoproterenol-stimulated C6 glioma cells. Biochem Biophys Res Commun 163:1150–157
Boyer JL, Lazarowski ER, Chen XH, Harden TK (1993) Identification of a P2Y-purinergic receptor that inhibits adenylyl cyclase. J Pharmacol Exp Ther 267:1140–146
Claes P, Grobben B, Van Kolen K, Roymans D, Slegers H (2001) P2YAC-receptor agonists enhance the proliferation of rat C6 glioma cells through activation of the p42/44 mitogen-activated protein kinase. Br J Pharmacol 134:402–08
Grobben B, Claes P, Van Kolen K, Roymans D, Fransen P, Sys SU, Slegers H (2001) Agonists of the P2YAC-receptor activate MAP kinase by a ras-independent pathway in rat C6 glioma. J Neurochem 78:1325–338
Czajkowski R, Lei L, Sabala P, Baranska J (2002) ADP-evoked phospholipase C stimulation and adenylyl cyclase inhibition in glioma C6 cells occur through two distinct nucleotide receptors, P2Y1 and P2Y12. FEBS Lett 513:179–83
Jin J, Tomlinson W, Kirk IP, Kim YB, Humphries RG, Kunapuli SP (2001) The C6-2B glioma cell P2YAC is pharmacologically and molecularly identical to the platelet P2Y12 receptor. Br J Pharmacol 133:521–28
Nicholas RA, Lazarowski ER, Watt WC, Li Q, Boyer J, Harden TK (1996) Pharmacological and second messenger signalling selectivities of cloned P2Y receptors. J Auton Pharmacol 16:319–23
Tu MT, Luo SF, Wang CC, Chien CS, Chiu CT, Lin CC, Yang CM (2000) P2Y2 receptor-mediated proliferation of C6 glioma cells via activation of Ras/Raf/MEK/MAPK pathway. Br J Pharmacol 129:1481–489
Van Kolen K, Slegers H (2004) P2Y12 receptor stimulation inhibits β-adrenergic receptor-induced differentiation by reversing the cyclic AMP-dependent inhibition of protein kinase B. J Neurochem 89:442–53
Czajkowski R, Banachewicz W, Ilnytska O, Drobot LB, Baranska J (2004) Differential effects of P2Y1 and P2Y12 nucleotide receptors on ERK1/ERK2 and phosphatidylinositol 3-kinase signalling and cell proliferation in serum-deprived and nonstarved glioma C6 cells. Br J Pharmacol 141:497–07
Marteau F, Le Poul E, Communi D, Communi D, Labouret C, Savi P, Boeynaems JM, Gonzalez NS (2003) Pharmacological characterization of the human P2Y13 receptor. Mol Pharmacol 64:104–12
Fumagalli M, Trincavelli L, Lecca D, Martini C, Ciana P, Abbracchio MP (2004) Cloning, pharmacological characterisation and distribution of the rat G-protein-coupled P2Y13 receptor. Biochem Pharmacol 68:113–24
Bianco F, Fumagalli M, Pravettoni E, D’Ambrosi N, Volonte C, Matteoli M, Abbracchio MP, Verderio C (2005) Pathophysiological roles of extracellular nucleotides in glial cells: differential expression of purinergic receptors in resting and activated microglia. Brain Res Rev 48:144–56
Fumagalli M, Brambilla R, D’Ambrosi N, Volonte C, Matteoli M, Verderio C, Abbracchio MP (2003) Nucleotide-mediated calcium signaling in rat cortical astrocytes: role of P2X and P2Y receptors. Glia 43:203–18
Sasaki Y, Hoshi M, Akazawa C, Nakamura Y, Tsuzuki H, Inoue K, Kohsaka S (2003) Selective expression of Gi/o-coupled ATP receptor P2Y12 in microglia in rat brain. Glia 44:242–50
Bezzi P, Voltera A (2001) A neuron-glia signalling network in the active brain. Curr Opin Neurobiol 11:387–94
Brambilla R, Neary JT, Cattabeni F, Cottini L, D’Ippolito G, Schiller PC, Abbracchio MP (2002) Induction of COX-2 and reactive gliosis by P2Y receptors in rat cortical astrocytes is dependent on ERK1/2 but independent of calcium signaling. J Neurochem 83:1285–296
Honda S, Sasaki Y, Ohsawa K, Imai Y, Nakamura Y, Inoue K, Kohsaka S (2001) Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J Neurosci 21:1975–982
Communi D, Janssens R, Suarez-Huerta N, Robaye B, Boeynaems JM (2000) Advances in signalling by extracellular nucleotides: the role and transduction mechanisms of P2Y receptors. Cell Signal 12:351–60
Mirshahi T, Mittal V, Zhang H, Linder ME, Logothetis DE (2002) Distinct sites on G protein βγ subunits regulate different effector functions. J Biol Chem 277:36345–6350
Filippov AK, Webb TE, Barnard EA, Brown DA (1998) P2Y2 nucleotide receptors expressed heterologously in sympathetic neurons inhibit both N-type Ca2+ and M-type K+ currents. J Neurosci 18:5170–179
Filippov AK, Webb TE, Barnard EA, Brown DA (1999) Dual coupling of heterologously-expressed rat P2Y6 nucleotide receptors to N-type Ca2+ and M-type K+ currents in rat sympathetic neurons. Br J Pharmacol 126:1009–017
Filippov AK, Brown DA, Barnard EA (2000) The P2Y1 receptor closes the N-type Ca2+ channel in neurons, with both adenosine triphosphates and diphosphates as potent agonists. Br J Pharmacol 129:1063–066
Filippov AK, Simon J, Barnard EA, Brown DA (2003) Coupling of the nucleotide P2Y4 receptor to neuronal ion channels. Br J Pharmacol 138:400–06
Simon J, Filippov AK, Göransson S, Wong YH, Frelin C, Michel AD, Brown DA, Barnard EA (2002) Characterization and channel coupling of the P2Y12 nucleotide receptor of brain capillary endothelial cells. J Biol Chem 277:31390–1400
Wirkner K, Schweigel J, Gerevich Z, Franke H, Allgaier C, Barsoumian EL, Draheim H, Illes P (2004) Adenine nucleotides inhibit recombinant N-type calcium channels via G protein-coupled mechanisms in HEK 293 cells; involvement of the P2Y13 receptor-type. Br J Pharmacol 141:141–51
Filippov AK, Fernandez-Fernandez JM, Marsh SJ, Simon J, Barnard EA, Brown DA (2004) Activation and inhibition of neuronal G protein-gated inwardly rectifying K+ channels by P2Y nucleotide receptors. Mol Pharmacol 66:468–77
Suadicani SO, Flores CE, Urban-Maldonado M, Beelitz M, Scemes E (2004) Gap junction channels coordinate the propagation of intercellular Ca2+ signals generated by P2Y receptor activation. Glia 48:217–29
Dienel GA, Hertz L (2005) Astrocytic contributions to bioenergetics of cerebral ischemia. Glia 50:362–88
Dixon SJ, Yu R, Panupinthu N, Wilson JX (2004) Activation of P2 nucleotide receptors stimulates acid efflux from astrocytes. Glia 47:367–76
Sellers LA, Simon J, Lundahl TS, Cousens DJ, Humphrey PP, Barnard EA (2001) Adenosine nucleotides acting at the human P2Y1 receptor stimulate mitogen-activated protein kinases and induce apoptosis. J Biol Chem 276:16379–6390
Fam SR, Paquet M, Castleberry AM, Oller H, Lee CJ, Traynelis SF, Smith Y, Yun CC, Hall RA (2005) P2Y1 receptor signaling is controlled by interaction with the PDZ scaffold NHERF-2. Proc Natl Acad Sci USA 102:8042–047
Chen BC, Lin WW (1999) PKCbetaI mediates the inhibition of P2Y receptor-induced inositol phosphate formation in endothelial cells. Br J Pharmacol 127:1908–914
Fam SR, Gallagher CJ, Kalia LV, Salter MW (2003) Differential frequency dependence of P2Y1- and P2Y2- mediated Ca2+ signaling in astrocytes. J Neurosci 23:4437–444
Pines A, Romanello M, Cesaratto L, Damante G, Moro L, D’andrea P, Tell G (2003) Extracellular ATP stimulates the early growth response protein 1 (Egr-1) via a protein kinase C-dependent pathway in the human osteoblastic HOBIT cell line. Biochem J 373:815–24
Hou M, Harden TK, Kuhn CM, Baldetorp B, Lazarowski E, Pendergast W, Moller S, Edvinsson L, Erlinge D (2002) UDP acts as a growth factor for vascular smooth muscle cells by activation of P2Y6 receptors. Am J Physiol Heart Circ Physiol 282:H784–H792
Neary JT, Kang Y, Bu Y, Yu E, Akong K, Peters CM (1999) Mitogenic signaling by ATP/P2Y purinergic receptors in astrocytes: involvement of a calcium-independent protein kinase C, extracellular signal-regulated protein kinase pathway distinct from the phosphatidylinositol-specific phospholipase C/calcium pathway. J Neurosci 19:4211–220
Wilkin F, Duhant X, Bruyns C, Suarez-Huerta N, Boeynaems JM, Robaye B (2001) The P2Y11 receptor mediates the ATP-induced maturation of human monocyte-derived dendritic cells. J Immunol 166:7172–177
Nishi H, Kato F, Masaki E, Kawamura M (2002) ADP-sensitive purinoceptors induce steroidogenesis via adenylyl cyclase activation in bovine adrenocortical fasciculata cells. Br J Pharmacol 137:177–84
Nasu-Tada K, Koizumi S, Inoue K (2005) Involvement of beta1 integrin in microglial chemotaxis and proliferation on fibronectin: different regulations by ADP through PKA. Glia 52:98–07
Anderson RG (1998) The caveolae membrane system. Annu Rev Biochem 67:199–25
DeFea KA, Zalevsky J, Thoma MS, Déry O, Mullins RD, Bunnet NW (2000) β-Arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J Cell Biol 148:1267–281
Ostrom RS, Insel P (2004) The evolving role of lipid rafts and caveolae in G protein-coupled receptor signaling: implications for molecular pharmacology. Br J Pharmacol 143:235–45
Kaiser RA, Oxhorn BC, Andrews G, Buxton IL (2002) Functional compartmentation of endothelial P2Y receptor signaling. Circ Res 23:292–99
Bhatnagar A, Sheffler DJ, Kroeze WK, Compton-Toth BA, Roth BL (2004) Caveolin-1 interacts with 5-HT2A serotonin receptors and profoundly modulates the signaling of selected Gαq-coupled protein receptors. J Biol Chem 279:34614–4623
Lev S, Moreno H, Martinez R, Canoll P, Peles E, Musacchio JM, Plowman GD, Rudy B, Schlessinger J (1995) Protein tyrosine kinase Pyk2 involved in Ca2+-induced regulation of ion channel and MAP kinase functions. Nature 376:737–45
Wang H, Reiser G (2003) The role of the Ca2+-sensitive tyrosine kinase Pyk2 and Src in thrombin signalling in rat astrocytes. J Neurochem 84:1349–357
Soltoff SP, Avraham H, Avraham S, Cantley LC (1998) Activation of P2Y2 receptors by UTP and ATP stimulates mitogen-activated kinase activity through a pathway that involves related adhesion focal tyrosine kinase and protein kinase C. J Biol Chem 273:2653–660
Ellis CA, Malik AB, Gilchrist A, Hamm H, Sandoval R, Voyno-Yasenetskaya T, Tiruppathi C (1999) Thrombin induces proteinase-activated receptor-1 gene expression in endothelial cells via activation of Gi-linked Ras/mitogen-activated protein kinase pathway. J Biol Chem 274:13718–3727
Lopez-Ilasaca M, Crespo P, Pellici PG, Gutkind JS, Wetzker R (1997) Linkage of G protein-coupled receptors to the MAPK signaling pathway through PI 3-kinase γ. Science 275:394–97
Bhattacharya M, Babwah AV, Ferguson SS (2004) Small GTP-binding protein-coupled receptors. Biochem Soc Trans 32:1040–044
Bos JL (2005) Linking Rap to cell adhesion. Curr Opin Cell Biol 17:123–28
Lutz S, Freichel-Blomquist A, Yang Y, Rumenapp U, Jakobs KH, Schmidt M, Wieland T (2005) The guanine nucleotide exchange factor p63RhoGEF, a specific link between Gq/11-coupled receptor signaling and RhoA. J Biol Chem 280:11134–1139
Walker SA, Cullen PJ, Taylor JA, Lockyer PJ (2003) Control of Ras cycling by Ca2+. FEBS Lett 546:6–0
Gao Z, Chen T, Weber MJ, Linden J (1999) A2B adenosine and P2Y2 receptors stimulate mitogen-activated protein kinase in human embryonic kidney-293 cells. Cross-talk between cyclic AMP and protein kinase C pathways. J Biol Chem 274:5972–980
Soulet C, Sauzeau V, Plantavid M, Herbert JM, Pacaud P, Payrastre B, Savi P (2004) Gi-dependent and -independent mechanisms downstream of the P2Y12 ADP-receptor. J Thromb Haemost 2:135–46
Woulfe D, Jiang H, Mortensen R, Yang J, Brass LF (2002) Activation of Rap1B by Gi family members in platelets. J Biol Chem 277:23382–3390
Greco F, Sinigaglia F, Balduini C, Torti M (2004) Activation of the small GTPase Rap2B in agonist-stimulated human platelets. J Thromb Haemost 2:2223–230
Larson MK, Chen H, Kahn ML, Taylor AM, Fabre JE, Mortensen RM, Conley PB, Parise LV (2003) Identification of P2Y12-dependent and -independent mechanisms of glycoprotein activation in platelets. Blood 101:1409–415
Lova P, Paganini S, Sinigaglia F, Balduini C, Torti M (2002) A Gi-dependent pathway is required for activation of the small GTPase Rap1B in human platelets. J Biol Chem 277:12009–2015
Lova P, Paganini S, Hirsch E, Barberis L, Wymann M, Sinigaglia F, Balduini C, Torti M (2003) A selective role for phosphatidylinositol 3,4,5-trisphosphate in the Gi-dependent activation of platelet Rap1B. J Biol Chem 278:131–38
Paul BZ, Daniel JL, Kunapuli SP (1999) Platelet shape change is mediated by both calcium-dependent and -independent signaling pathways: role of p160 Rho-associated coiled-coil-containing protein kinase in platelet shape change. J Biol Chem 274:28293–8300
Sauzeau V, Le Jeune H, Cario-Toumaniantz C, Vaillant N, Gadeau AP, Desgranges C, Scalbert E, Chardin P, Pacaud P, Loirand G (2000) P2Y1, P2Y2, P2Y4, and P2Y6 receptors are coupled to Rho and Rho kinase activation in vascular myocytes. Am J Physiol Heart Circ Physiol 278:H1751–H1761
Seye CI, Yu N, Gonzalez FA, Erb L, Weisman GA (2004) The P2Y2 nucleotide receptor mediates vascular cell adhesion molecule-1 expression through interaction with VEGF receptor-2 (KDR/Flk-1). J Biol Chem 279:35679–5986
Dugan LL, Kim JS, Zhang Y, Bart RD, Sun Y, Holtzman DM, Gutmann DH (1999) Differential effects of cAMP in neurons and astrocytes. Role of B-raf. J Biol Chem 274:25842–5848
Stork PJ, Schmitt JM (2002) Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol 12:258–66
Qiu W, Zhuang S, von Lintig FC, Boss GR, Pilz RB (2000) Cell type-specific regulation of B-Raf kinase by cAMP and 14-3-3 proteins. J Biol Chem 275:31921–1929
Martin NP, Whalen EJ, Zamah MA, Pierce KL, Lefkowitz RJ (2004) PKA-mediated phosphorylation of the β1-adrenergic receptor promotes Gs/Gi switching. Cell Signal 16:1397–403
de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL (1998) Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396:474–77
de Rooij J, Rehmann H, van Triest M, Cool RH, Wittinghofer A, Bos JL (2000) Mechanism of regulation of the Epac Family of cAMP-dependent RapGEFs. J Biol Chem 275:20829–0836
Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya N, Matsuda M, Housman DE, Graybiel AM (1998) A family of cAMP-binding proteins that directly activates Rap1. Science 282:2275–279
Laroche-Joubert N, Marsy S, Michelet S, Imbert-Teboul M, Doucet A (2002) Protein kinase A-independent activation of ERK and H,K-ATPase by cAMP in native kidney cells: role of Epac I. J Biol Chem 277:18598–8604
Lin SL, Johnson-Farley NN, Lubinsky DR, Cowen DS (2003) Coupling of neuronal 5-HT7 receptors to activation of extracellular-regulated kinase through a protein kinase A-independent pathway that can utilize Epac. J Neurochem 87:1076–085
Schmitt JM, Stork PJ (2002) Gα and Gβγ require distinct Src-dependent pathways to activate Rap1 and Ras. J Biol Chem 277:43024–3032
Ebinu JO, Bottorff DA, Chan EY, Stang SL, Dunn RJ, Stone JC (1998) RasGRP, a Ras guanyl nucleotide-releasing protein with calcium- and diacylglycerol-binding motifs. Science 280:1082–086
Agell N, Bachs O, Rocamora N, Villalonga P (2002) Modulation of the Ras/Raf/MEK/ERK pathway by Ca2+ and calmodulin. Cell Signal 14:649–54
Kolch W, Heidecker G, Kochs G, Hummel R, Vahidi H, Mischak H, Finkenzeller G, Marmé D, Rapp UR (1996) Protein kinase Cα activates Raf-1 by direct phosphorylation. Nature 364:249–53
Yip-Schneider MT, Miao W, Lin A, Barnard DS, Tzivion G, Marshall MS (2000) Regulation of the Raf-1 kinase domain by phosphorylation and 14-3-3 association. Biochem J 351:151–59
Cacace AM, Ueffing M, Philipp A, Han EK, Kolch W, Weinstein IB (1996) PKC epsilon functions as an oncogene by enhancing activation of the Raf kinase. Oncogene 13:2517–526
Ueffing M, Lovric J, Philipp A, Mischak H, Kolch W (1997) Protein kinase C-ɛ associates with the Raf-1 kinase and induces the production of growth factors that stimulate Raf-1 activity. Oncogene 15:2921–927
Paruchuri S, Hallberg B, Juhas M, Larsson C, Sjolander A (2002) Leukotriene D4 activates MAPK through a Ras-independent but PKC-dependent pathway in intestinal epithelial cells. J Cell Sci 115:1883–893
Ueda Y, Hirai S, Osada S, Suzuki A, Mizuno K, Ohno S (1996) Protein kinase Cδ activates the MEK-ERK pathway in a manner independent of Ras and dependent on Raf. J Biol Chem 271:23512–3519
Nadal-Wollbold F, Pawlowski M, Levy-Toledano S, Berrou E, Rosa JP, Bryckaert M (2002) Platelet ERK2 activation by thrombin is dependent on calcium and conventional protein kinases C but not Raf-1 or B-Raf. FEBS Lett 531:475–82
Takeda H, Matozaki T, Takada T, Noguchi T, Yamao T, Tsuda M, Ochi F, Fukunaga K, Inagaki K, Kasuga M (1999) PI 3-kinase γ and protein kinase C-ζ mediate RAS-independent activation of MAP kinase by a Gi protein-coupled receptor. EMBO J 18:386–95
Zhao Y, Liu J, Li L, Liu L, Wu L (2005) Role of Ras/PKCζ/MEK/ERK1/2 signaling pathway in angiotensin II-induced vascular smooth muscle cell proliferation. Regul Pept 128:43–0
Schönwasser DC, Marais RM, Marshall CJ, Parker PJ (1998) Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel and atypical protein kinase C isotypes. Mol Cell Biol 18:790–98
Corbit KC, Trakul N, Eves EM, Diaz B, Marshall M, Rosner MR (2003) Activation of Raf-1 signaling by protein kinase C through a mechanism involving Raf kinase inhibitory protein. J Biol Chem 278:13061–3068
Van Der Hoeven PC, Van Der Wal JC, Ruurs P, Van Blitterswijk WJ (2000) Protein kinase C activation by acidic proteins including 14-3-3. Biochem J 347:781–85
Neary JT, Zhu Q (1994) Signalling by ATP receptors in astrocytes. Neuroreport 5:1617–620
King BF, Neary JT, Zhu Q, Wang S, Norenberg MD, Burnstock G (1996) P2 purinoceptors in rat cortical astrocytes: expression, calcium-imaging and signalling studies. Neuroscience 74:1187–196
Graham A, McLees A, Kennedy C, Gould GW, Plevin R (1996) Stimulation by the nucleotides, ATP and UTP of mitogen-activated protein kinase in EAhy 926 endothelial cells. Br J Pharmacol 117:1341–347
Huwiler A, Pfeilschifter J (1994) Stimulation by extracellular ATP and UTP of the mitogen-activated protein kinase cascade and proliferation of rat mesangial cells. Br J Pharmacol 113:1455–463
Erlinge D (1998) Extracellular ATP: a growth factor for vascular smooth muscle cells. Gen Pharmacol 31:1–
Tornquist K, Ekokoski E, Dugue B (1996) Purinergic agonist ATP is a comitogen in thyroid FRTL-5 cells. J Cell Physiol 166:241–48
Kim SG, Gao ZG, Soltysiak KA, Chang TS, Brodie C, Jacobson KA (2003) P2Y6 nucleotide receptor activates PKC to protect 1321N1 astrocytoma cells against tumor necrosis factor-induced apoptosis. Cell Mol Neurobiol 23:401–18
Way KJ, Chou E, King GL (2000) Identification of PKC-isoform-specific biological actions using pharmacological approaches. Trends Pharmacol Sci 21:181–87
Harper S, Webb TE, Charlton SJ, Ng LL, Boarder MR (1998) Evidence that P2Y4 receptors are involved in the regulation of rat aortic smooth muscle cells by UTP and ATP. Br J Pharmacol 124:703–10
Ahmad S, Ahmad A, Ghosh M, Leslie CC, White CW (2004) Extracellular ATP-mediated signaling for survival in hyperoxia-induced oxidative stress. J Biol Chem 279:16317–6325
Meshki J, Tuluc F, Bredetean O, Ding Z, Kunapuli SP (2004) Molecular mechanism of nucleotide-induced primary granule release in human neutrophils: role for the P2Y2 receptor. Am J Physiol Cell Physiol 286:264–71
Berenbaum F, Humbert L, Bereziat G, Thirion S (2003) Concomitant recruitment of ERK1/2 and p38 MAPK signalling pathway is required for activation of cytoplasmic phospholipase A2 via ATP in articular chondrocytes. J Biol Chem 278:13680–3687
Brazil DP, Park J, Hemmings BA (2002) PKB binding proteins: getting in on the Akt. Cell 111:293–03
Cantley LC (2002) The phosphoinositide 3-kinase pathway. Science 296:1655–657
Leevers SJ, Vanhaesebroeck B, Waterfield MD (1999) Signaling through phosphoinositide 3-kinases: the lipids take centre stage. Curr Opin Cell Biol 11:219–25
Woodgett JR (2005) Recent advances in the protein kinase B signaling pathway. Curr Opin Cell Biol 17:150–57
Yano S, Tokumitsu H, Soderling TR (1998) Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature 396:584–87
Dickenson JM (2002) Stimulation of protein kinase B and p70 S6 kinase by the histamine H1 receptor in DDT1MF-2 smooth muscle cells. Br J Pharmacol 135:1967–976
Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN (1995) The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell 81:727–36
Iacovelli L, Bruno V, Salvatore L, Melchiorri D, Gradini R, Caricasole A, Barletta E, De Blasi A, Nicoletti F (2002) Native group-III metabotropic glutamate receptors are coupled to the mitogen-activated protein kinase/phosphatidylinositol-3-kinase pathways. J Neurochem 82:216–23
Murga C, Laguinge L, Wetzker R, Cuadrado A, Gutkind JS (1998) Activation of Akt/protein kinase B by G protein-coupled receptors. A role for α and βγ subunits of heterotrimeric G proteins acting through phosphatidylinositol-3-OH kinase γ. J Biol Chem 273:19080–9085
Sanchez MG, Ruiz-Llorente L, Sanchez AM, Diaz-Laviada I (2003) Activation of phosphoinositide 3-kinase/PKB pathway by CB1 and CB2 cannabinoid receptors in prostate PC-3 cells. Involvement in Raf-1 stimulation and NGF induction. Cell Signal 15:851–59
Bommakanti RK, Vinayak S, Simonds WF (2000) Dual regulation of Akt/Protein Kinase B by heterotrimeric G protein subunits. J Biol Chem 275:38870–8876
Mei FC, Qiao J, Tsygankova OM, Meinkoth JL, Quilliam LA, Cheng X (2002) Differential signaling of cyclic AMP: opposing effects of exchange protein directly activated by cyclic AMP and cAMP-dependent protein kinase on protein kinase B activation. J Biol Chem 277:11497–1504
Wang L, Liu F, Adamo ML (2001) Cyclic AMP inhibits extracellular signal-regulated kinase and phosphatidylinositol 3-kinase/Akt pathways by inhibiting Rap1. J Biol Chem 276:37242–7249
Kurosu H, Maehama T, Okada T, Yamamoto T, Hoshino S, Fukui Y, Ui M, Hazeki O, Katada T (1997) Heterodimeric phosphoinositide 3-kinase consisting of p85 and p110β is synergistically activated by the βγ subunits of G proteins and phosphotyrosyl peptide. J Biol Chem 272:24252–4256
Stoyanov B, Volinia S, Hanck T, Rubio I, Loubtchenkov M, Malek D, Stoyanova S, Vanhaesebroeck B, Dhand R, Nürnberg B, Gierschik P, Seedorf K, Hsuan JJ, Waterfield MD, Wetzker R (1995) Cloning and characterization of a G protein-activated human phosphoinositide-3 kinase. Science 269:690–93
Yart A, Roche S, Wetzker R, Laffargue M, Tonks N, Mayeux P, Chap H, Raynal P (2002) A function for phosphoinositide 3-kinase β lipid products in coupling βγ to Ras activation in response to lysophosphatidic acid. J Biol Chem 277:21167–1178
Kippenberger S, Loitsch S, Guschel M, Muller J, Knies Y, Kaufmann R, Bernd A (2005) Mechanical stretch stimulates protein kinase B/Akt phosphorylation in epidermal cells via angiotensin II type 1 receptor and epidermal growth factor receptor. J Biol Chem 280:3060–067
Tang X, Batty IH, Downes CP (2002) Muscarinic receptors mediate phospholipase C-dependent activation of protein kinase B via Ca2+, ErbB3, and phosphoinositide 3-kinase in 1321N1 astrocytoma cells. J Biol Chem 277:338–44
Ballou LM, Cross ME, Huang S, McReynolds EM, Zhang BX, Lin RZ (2000) Differential regulation of the phosphatidylinositol 3-kinase/Akt and p70 S6 kinase pathways by the α1A-adrenergic receptor in rat-1 fibroblasts. J Biol Chem 275:4803–809
Ballou LM, Lin H-Y, Fan G, Jiang Y-P, Lin RZ (2003) Activated Gαq inhibits p110α phosphatidylinositol 3-kinase and Akt. J Biol Chem 278:23472–3479
Batty IH, Fleming IN, Downes CP (2004) Muscarinic-receptor-mediated inhibition of insulin-like growth factor-1 receptor-stimulated phosphoinositide 3-kinase signalling in 1321N1 astrocytoma cells. Biochem J 379:641–51
Gerasimovskaya EV, Tucker DA, Weiser-Evans M, Wenzlau JM, Klemm DJ, Banks M, Stenmark KR (2005) Extracellular ATP-induced proliferation of adventitial fibroblasts requires phosphoinositide 3-kinase, Akt, mammalian target of rapamycin, and p70 S6 kinase signaling pathways. J Biol Chem 280:1838–848
Huwiler A, Rolz W, Dorsch S, Ren S, Pfeilschifter J (2002) Extracellular ATP and UTP activate the protein kinase B/Akt cascade via the P2Y2 purinoceptor in renal mesangial cells. Br J Pharmacol 136:520–29
Chen J, De S, Damron DS, Chen WS, Hay N, Byzova TV (2004) Impaired platelet responses to thrombin and collagen in AKT-1-deficient mice. Blood 104:1703–710
Kim S, Jin J, Kunapuli SP (2004) Akt activation in platelets depends on Gi signaling pathways. J Biol Chem 279:4186–195
Cordeaux Y, Hill SJ (2002) Mechanisms of cross-talk between G-protein-coupled receptors. Neurosignals 11:45–7
Hardy AR, Jones ML, Mundell SJ, Poole AW (2004) Reciprocal cross-talk between P2Y1 and P2Y12 receptors at the level of calcium signaling in human platelets. Blood 104:1745–752
Xin C, Ren S, Pfeilschifter J, Huwiler A (2004) Heterologous desensitization of the sphingosine-1-phosphate receptors by purinoceptor activation in renal mesangial cells. Br J Pharmacol 143:581–89
Berg KA, Evans KL, Cropper JD, Clarke WP (2003) Temporal regulation of agonist efficacy at 5-hydroxytryptamine 5-HT1A and 5-HT1B receptors. J Pharmacol Exp Ther 304:200–05
Krugel U, Kittner H, Illes P (2001) Mechanisms of adenosine 5′-triphosphate-induced dopamine release in the rat nucleus accumbens in vivo. Synapse 39:222–32
Nedergaard M, Takano T, Hansen AJ (2002) Beyond the role of glutamate as a neurotransmitter. Nat Rev Neurosci 3:748–55
Yoshioka K, Saitoh O, Nakata H (2001) Heteromeric association creates a P2Y-like adenosine receptor. Proc Natl Acad Sci USA 98:7617–622
Luttrell DK, Luttrell LM (2004) Not so strange bedfellows: G-protein-coupled receptors and Src family kinases. Oncogene 23:7969–978
Bisotto S, Fixman ED (2001) Src-family tyrosine kinases, phosphoinositide 3-kinase and Gab1 regulate extracellular signal-regulated kinase 1 activation induced by the type A endothelin-1 G-protein-coupled receptor. Biochem J 360: 77–5
Waters CM, Connell MC, Pyne S, Pyne NJ (2005) c-Src is involved in regulating signal transmission from PDGFβ receptor-GPCR(s) complexes in mammalian cells. Cell Signal 17:263–77
Milenkovic I, Weick M, Wiedemann P, Reichenbach A, Bringmann A (2003) P2Y receptor-mediated stimulation of Muller glial cell DNA synthesis: dependence on EGF and PDGF receptor transactivation. Invest Ophthalmol Vis Sci 44:1211–220
Abbracchio MP, Brambilla R, Ceruti S, Cattabeni F (1999) Signalling mechanisms involved in P2Y receptor-mediated reactive astrogliosis. Prog Brain Res 120:333–42
Bolego C, Ceruti S, Brambilla R, Puglisi L, Cattabeni F, Burnstock G, Abbracchio MP (1997) Characterization of the signalling pathways involved in ATP and basic fibroblast growth factor-induced astrogliosis. Br J Pharmacol 121:1692–699
Neary JT, Kang Y, Shi YF (2004) Signalling from nucleotide receptors to protein kinase cascades in astrocytes. Neurochem Res 29:2037–042
Liu J, Liao Z, Camden J, Griffin KD, Garrad RC, Santiago-Perez LI, Gonzalez FA, Seye CI, Weisman GA, Erb L (2004) Src homology 3 binding sites in the P2Y2 nucleotide receptor interact with Src and regulate activities of Src, proline-rich tyrosine kinase 2, and growth factor receptors. J Biol Chem 279:8212–218
Short SM, Boyer JL, Juliano RL (2000) Integrins regulate the linkage between upstream and downstream events in G protein-coupled receptor signaling to mitogen-activated protein kinase. J Biol Chem 275:12970–2977
Erb L, Liu J, Ockerhausen J, Kong Q, Garrad RC, Griffin K, Neal C, Krugh B, Santiago-Perez LI, Gonzalez FA, Gresham HD, Turner JT, Weisman GA (2001) An RGD sequence in the P2Y2 receptor interacts with αVβ3 integrins and is required for Go-mediated signal transduction. J Cell Biol 153:491–01
Grobben B, Anciaux K, Roymans D, Stefan C, Bollen M, Esmans EL, Slegers H (1999) An ecto-nucleotide pyrophosphatase is one of the main enzymes involved in the extracellular metabolism of ATP in rat C6 glioma. J Neurochem 72:826–34
Steinberg SF (2004) Distinctive activation mechanisms and functions for protein kinase Cδ. Biochem J 384:449–59
Parekh DB, Ziegler W, Parker PJ (2000) Multiple pathways control protein kinase C phosphorylation. EMBO J 19:496–03
Zhang W, Turner DJ, Segura BJ, Cowles R, Mulholland MW (2000) ATP induces c-fos expression in C6 glioma cells by activation of P2Y receptors. J Surg Res 94:49–5
Baranska J, Czajkowski R, Sabala P (2004) Cross-talks between nucleotide receptor-induced signaling pathways in serum-deprived and non-starved glioma C6 cells. Adv Enzyme Regul 44:219–32
Ellert-Miklaszewska A, Kaminska B, Konarska L (2005) Cannabinoids down-regulate PI3K/Akt and Erk signalling pathways and activate proapoptotic function of Bad protein. Cell Signal 17:25–7
Van Kolen K, Gilany K, Moens L, Esmans EL, Slegers H (2006) P2Y12 receptor signalling towards PKB proceeds through IGF-I receptor cross-talk and requires activation of Src, Pyk2 and Rap1. Cell Signal 18:1169–181
Banno Y, Ohguchi K, Matsumoto N, Koda M, Ueda M, Hara A, Dikic I, Nozawa Y (2005) Implication of phospholipase D2 in oxidant-induced phosphoinositide 3-kinase signaling via Pyk2 activation in PC12 cells. J Biol Chem 280:16319–6324
Bobeszko M, Krzeminski P, Pomorski P, Dygas A, Baranska J (2004) Expression and regulation of phospholipase D isoforms in sphingosine and phorbol ester-stimulated glioma C6 cells. Biochem Biophys Res Commun 317:689–96
Koziak K, Kaczmarek E, Park SY, Fu Y, Avraham S, Avraham H (2001) RAFTK/Pyk2 involvement in platelet activation is mediated by phosphoinositide 3-kinase. Br J Haematol 114:134–40
Boyer JL, Zohn IE, Jacobson KA, Harden TK (1994) Differential effects of P2-purinoceptor antagonists on phospholipase C- and adenylyl cyclase-coupled P2Y-purinoceptors. Br J Pharmacol 113:614–20
Boyer JL, Romero-Avila T, Schachter JB, Harden TK (1996) Identification of competitive antagonists of the P2Y1 receptor. Mol Pharmacol 50:1323–329
Communi D, Govaerts C, Parmentier M, Boeynaems JM (1997) Cloning of a human purinergic P2Y Receptor coupled to phospholipase C and adenylyl cyclase. J Biol Chem 272:31969–1973
Korcok J, Raimundo LN, Du X, Sims SM, Dixon SJ (2005) P2Y6 nucleotide receptors activate NF-KB and increase survival of osteoclasts. J Biol Chem 280:16909–6915
Muller CE (2002) P2-pyrimidinergic receptors and their ligands. Curr Pharm Des 8:2353–569
Skelton L, Cooper M, Murphy M, Platt A (2003) Human immature monocyte-derived dendritic cells express the G protein-coupled receptor GPR105 (KIAA0001, P2Y14) and increase intracellular calcium in response to its agonist, uridine diphosphoglucose. J Immunol 171:1941–949
Yerxa BR, Sabater JR, Davis CW, Stutts MJ, Lang-Furr M, Picher M, Jones AC, Cowlen M, Dougherty R, Boyer J, Abraham WM, Boucher RC (2002) Pharmacology of INS37217 [P(1)-uridine 5′)-P(4)-(2′-deoxycytidine 5′)tetraphosphate, tetrasodium salt], a next-generation P2Y2 receptor agonist for the treatment of cystic fibrosis. J Pharmacol Exp Ther 302:871–80
Jacobson KA, Costanzi S, Ohno M, Joshi BV, Besada P, Xu B, Tchilibon S (2004) Molecular recognition at purine and pyrimidine nucleotide (P2) receptors. Curr Top Med Chem 4:805–19
von Kügelgen I (2006) Pharmacological profiles of cloned mammalian P2Y-receptor subtypes. Pharmacol. Ther. (in press doi: 10.1016/j.pharmathera.2005.08.014)
Belcheva MM, Haas PD, Tan Y, Heaton VM, Coscia J (2002) The fibroblast growth factor receptor is at the site of convergence between μ-opioid receptor and growth factor signaling pathways in rat C6 glioma cells. J Pharmacol Exp Ther 303:909–18
Van Kolen K, Slegers H (2006) Atypical PKC ζ is involved in RhoA-dependent mitogenic signaling by the P2Y12 receptor in C6 cells. FEBS J 273:1843–854
Acknowledgment
This work was supported by grants from the Fund for Scientific Research Flanders (HS) and BOF-NOI (HS). K.V.K. is a fellow of the Institute of Scientific Technology (IWT).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Van Kolen, K., Slegers, H. Integration of P2Y receptor-activated signal transduction pathways in G protein-dependent signalling networks. Purinergic Signalling 2, 451–469 (2006). https://doi.org/10.1007/s11302-006-9008-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-006-9008-0