
Abstract
Despite tremendous advances over the last 15 years in understanding fundamental mechanisms of apoptosis, this has failed to translate into improved cancer therapy for patients. However, there may now be light at the end of this long tunnel. Antiapoptotic Bcl-2 family members may be divided into two subclasses, one comprising Bcl-2, Bcl-XL and Bcl-w and the other Mcl-1 and Bcl2A1. Neutralization of both subclasses is required for apoptosis induction. Solution of the structure of antiapoptotic Bcl-2 family proteins has led to the design of novel small molecule inhibitors. Although many such molecules have been synthesized, rigorous verification of their specificity has often been lacking. Further studies have revealed that many putative Bcl-2 inhibitors are not specific and have other cellular targets, resulting in non-mechanism based toxicity. Two notable exceptions are ABT-737 and a related orally active derivative, ABT-263, which bind with high affinity to Bcl-2, Bcl-XL and Bcl-w and may prove to be useful tools for mechanistic studies. ABT-263 is in early clinical trials in lymphoid malignancies, small-cell lung cancer and chronic lymphocytic leukemia, and some patients have shown promising results. In in vitro studies, primary cells from patients with various B-cell malignancies are exquisitely sensitive to ABT-737, exhibiting novel morphological features of apoptosis including marked outer mitochondrial membrane rupture.
Similar content being viewed by others

Main
One of the cardinal features of cancer is a deregulation of apoptosis.1 Enormous progress has been made in the last 15 years in understanding the basic mechanisms of apoptosis, in particular, the role of Bcl-2 and the caspase family of proteins as critical regulators or executioners of apoptosis, respectively. However, so far there has been no progress in the translation of this knowledge into clinical application and improved cancer therapy.
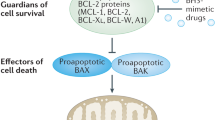
Apoptosis occurs following either triggering of cell surface death receptors (the extrinsic pathway) or perturbation of mitochondria (the intrinsic pathway).2, 3 Most drugs, including cancer chemotherapeutic agents, chemicals, irradiation as well as growth factor withdrawal, induce apoptosis by activation of the intrinsic pathway. Members of the Bcl-2 family control the integrity of the outer mitochondrial membrane (OMM) and thus are critical in determining the susceptibility of cells to apoptosis induced by the intrinsic pathway. The Bcl-2 family comprises antiapoptotic members, such as Bcl-2, Mcl-1, Bcl-XL and Bcl2A1 (Bfl-1/A1), multidomain proapoptotic members, such as Bax and Bak, and proapoptotic BH3-only proteins, including Bad, Bim, Puma, Bid, Bik, Noxa and Bmf.3, 4, 5, 6, 7 On receipt of a death signal, Bax and Bak can form oligomers in mitochondrial membranes leading to permeabilization of the OMM, release of cytochrome c and caspase activation, whereas antiapoptotic Bcl-2 members prevent this release by blocking activation of Bax and Bak. BH3-only proteins, which act upstream of Bax and Bak, are critical for cell death initiation, and their activity is tightly controlled by diverse transcriptional and post-translational mechanisms.3, 4, 6 BH3-only proteins selectively bind into the hydrophobic groove of antiapoptotic Bcl-2 family members leading to Bax/Bak activation, either by the direct or indirect activation model.4, 6, 8, 9, 10 Antiapoptotic Bcl-2 proteins can be divided into two groups, one comprising Bcl-2, Bcl-XL and Bcl-w and the other containing Mcl-1 and Bcl2A1; efficient apoptosis and effective therapy has been shown to require neutralization of both sets of antiapoptotic proteins.4, 6, 8, 9 Although the balance of pro- and antiapoptotic Bcl-2 family members is clearly important in determining cellular susceptibility to apoptosis, it is also necessary to consider the capacity of antiapoptotic Bcl-2 proteins to bind molecules, such as BH3-only proteins. For example, chronic lymphocytic leukemic (CLL) cells not only express high levels of Bcl-2, but also high levels of Bim, which is constitutively bound by Bcl-2.11
High levels of antiapoptotic Bcl-2 family members are associated with resistance of many tumors to chemotherapy.4, 12, 13 Furthermore, a comprehensive study of the NCI panel of 60 cell lines of human tumors of different origin showed a striking negative correlation between Bcl-XL levels and drug sensitivity to all categories of drugs tested except a subgroup including antimetabolites.14 Based on these and many other studies, antiapoptotic Bcl-2 family members offer a very attractive but challenging target for the development of anticancer agents. Antiapoptotic family members share up to four highly conserved domains, namely the BH1 (Bcl-2 homology), BH2, BH3 and BH4 domains. To date, structural studies have revealed a remarkable similarity between antiapoptotic (Bcl-XL, Bcl-2, Bcl-w and Mcl-1) and proapoptotic members (Bax, Bid and Bak) showing all these proteins to be helical bundles with a hydrophobic helix-turn-helix hairpin flanked by pairs of amphipathic helices. (Reviewed by Youle and Strasser6) The BH1, BH2 and BH3 domains form a hydrophobic pocket that binds BH3-peptides as well as the C-terminal anchor of Mcl-1, Bcl-w and Bax.6
Over the last few years, several small molecules have been synthesized as BH3 mimetics to induce apoptosis by inhibiting antiapoptotic Bcl-2 family members and thus being of potential value as anticancer agents (Figure 1). The design of small molecule inhibitors to interfere with protein–protein interactions has been a major challenge for the pharmaceutical industry. Some success has been achieved in cases where there are substantial protein surface indentations and, in relation to cancer chemotherapy, novel agents have been found that interfere with both p53-Mdm2 and antiapoptotic Bcl-2 interactions.15, 16 Thus, the hydrophobic pocket formed by antiapoptotic Bcl-2 family members has been the target of much innovative chemistry and several agents have either been synthesized or selected on the basis of in silico screens and proposed to act as selective inhibitors. The development of molecules required to interfere with Bcl-2 family members has required the synthesis of molecules of higher molecular weight (>800) than those generally synthesized as drugs. Such molecules may have different pharmacokinetics and routes of metabolism, for example, molecules of this size may be significantly excreted in the bile and undergo enterohepatic recirculation. Small molecules antagonizing Bcl-2 proteins include HA14-1, BH3I-1, antimycin A, chelerythrine, gossypol (NSC19048), apogossypol (NSC736630), TW-37, 4-(3-methoxy-phenylsulfonyl)-7-nitro-benzofurazan-3-oxide (MNB), TM12-06, obatoclax (GX15-070), ABT-737 and a related orally-active derivative, ABT-263 and pyrogallol-based molecules (Figure 1).16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 The synthesis of orally active Bcl-2 inhibitors, such as ABT-263, should provide greater flexibility in dosing regimens and ease of administration for patients.24, 28 Some of these molecules have been developed as far as early clinical trials with promising results (Wilson WH, 2007, abstract no. 1371).

Structures of some Bcl-2 inhibitors
The Specificity of Bcl-2 Inhibitors
Major differences are observed in both potency and selectivity of the various Bcl-2 inhibitors for different antiapoptotic Bcl-2 proteins (Table 1). Some reservations are clearly required in interpreting these data as they have been obtained from different studies sometimes using different methodologies to obtain the Ki values and also because of difficulties in obtaining precise values due to the high lipophilicity of some inhibitors, such as obatoclax or ABT-737. The data in Table 1 are potentially important for efficacy as well as toxicity of the inhibitors, as recent studies have highlighted the necessity to neutralize both sets of antiapoptotic Bcl-2 family members to induce apoptosis (see above).
By far the most potent small molecule inhibitors described so far are the Bad-like BH3 mimetics, ABT-737 and its orally active analog, ABT-263, which bind with very high affinity (Ki⩽1 nM) to Bcl-2, Bcl-XL and Bcl-w but with much lower affinity to Mcl-1 or Bcl2A1 (Table 1).24, 31 ABT-737 was discovered using nuclear magnetic resonance-based screening, chemical synthesis and structure-based design.16 Some inhibitors, such as obatoclax and BH3I-1 appear to be pan-Bcl-2 family inhibitors, although with much lower affinities (Table 1). Although compounds with micromolar affinities are unlikely to be useful as drugs because of the difficulty in obtaining therapeutically meaningful concentrations in vivo, they may serve as useful leads to generate second or third generation more potent inhibitors. Most of these compounds have been discovered by displacement of a BH3 peptide from Bcl-XL or Bcl-2, suggesting that they all target the hydrophobic groove. However, the different binding patterns indicate that different inhibitors target different sites of the hydrophobic groove and point to important functional differences between the individual Bcl-2 antagonists. Potentially, this may lead to combined use of different inhibitors targeting different regions of the groove. Recently the crystal structure of ABT-737 complexed with Bcl-XL has been described in which the chloro-biphenyl and thio-phenyl moieties of ABT-737 bind to the p2 and p4 pockets of Bcl-XL.35 Furthermore, some subtle differences were noted in the binding of ABT-737 to Bcl-XL when compared with binding of BH3 peptides.35 By mutational studies it had been shown that in addition to the hydrophobic groove, chelerythrine binds to the BH1-domain of Bcl-XL, which could also contribute to apoptosis induction.36 In an elegant study by Huang and co-workers,37 to test the selectivity of many of these putative selective Bcl-2 antagonists, their ability to kill either wild-type or Bax/Bak-deficient mouse embryonic fibroblasts was ascertained. Selective BH3 mimetics would be expected to kill wild-type fibroblasts but not those deficient in Bax and Bak. Of the agents tested HA14-1, BH3I-1, antimycin A, chelerythrine and gossypol killed wild-type and Bax/Bak-deficient fibroblasts with similar efficacy, whereas ABT-737 killed only the wild-type cells, demonstrating that of these chemicals only ABT-737 was a pure BH3 mimetic and was inducing cell death by specific inhibition of antiapoptotic Bcl-2 family members.37 As yet the specificity of other agents, such as obatoclax and TW-37, to kill Bax/Bak null fibroblasts has not been definitively ascertained, although two very recent reports indicate that obatoclax may be active in Bax/Bak null fibroblasts.24, 38 These studies suggest that a variety of Bcl-2 antagonists have additional targets besides inhibiting antiapoptotic Bcl-2 family members and these additional targets may lead to unpredicted, non-mechanism based toxicity.
The notion that it is necessary to neutralize both arms of the antiapoptotic Bcl-2 family raises important questions with regard to the specificity of the Bcl-2 inhibitors.
How valuable is it to have a specific inhibitor of Bcl-2, Bcl-XL and Bcl-w or would it be of more value to have a pan Bcl-2 inhibitor that, in addition, also inhibited Mcl-1 and Bcl2A1? Obatoclax is a pan Bcl-2 inhibitor whereas ABT-737 and ABT-263 are both Bad-like BH3 mimetics, which only inhibit Bcl-2, Bcl-XL and Bcl-w but do not inhibit Mcl-1 and Bcl2A1.16, 22, 24 Resistance to ABT-737 has been linked to high expression levels of Mcl-1 and in many instances this resistance can be overcome by treatment with an agent(s) that decreases Mcl-1, such as seliciclib, a cyclin-dependent kinase inhibitor.16, 37, 39, 40, 41, 42, 43 In contrast, obatoclax overcomes Mcl-1 mediated resistance to apoptosis partly by interfering with Mcl-1–Bak interactions.22 Thus, solely from an efficacy viewpoint, it would be preferable to use a pan rather than a more specific Bcl-2 inhibitor. However, issues of toxicity, both mechanism and non-mechanism based, have also to be considered. Owing to limited in vivo studies to date on most of the Bcl-2 inhibitors, relatively little is known about their potential toxicities, with the possible exceptions of ABT-737 and gossypol. ABT-737 is generally well tolerated in vivo but has been reported to cause a concentration-dependent and rapid decrease in circulating platelets and lymphocytes without affecting platelet aggregation.16, 44, 45 In dogs dosed with ABT-737, this rapid decrease in platelets returns to baseline within 3 days and is compatible with a mechanism involving platelet destruction rather than a mechanism involving decreased production from megakaryocytes as observed following conventional cytotoxic chemotherapy.24 As Bcl-XL is critical in limiting the proapoptotic activity of Bak in platelets, the observed thrombocytopenia appears to be because of inhibition of Bcl-XL resulting in activation of Bak.44 Older platelets contain less Bcl-XL than younger platelets and are more susceptible to ABT-737, which helps to explain the almost full recovery of platelets in mice dosed daily with ABT-737.44 Clearly, the more specific the inhibitor for individual Bcl-2 family members, the less non-mechanism based toxicity may be expected. Pan Bcl-2 inhibitors are more likely to exhibit mechanism-based toxicities than more specific inhibitors. For example, a pan Bcl-2 family inhibitor that inhibits Mcl-1 may induce toxicities in cells or tissues where there is an important constitutive function of Mcl-1.
Differential Sensitivity of Cell Lines and Some Primary Cells to ABT-737
There is an enormous variation in the sensitivity of different cell types to ABT-737, even considering the different times of exposure and the various methods used to assess apoptosis/cell death or clonogenicity (Table 2). These studies refer to sensitivity of the cells to ABT-737 alone. In many cell types, ABT-737 does not initiate apoptosis as a single agent but acts synergistically with other chemotherapeutic agents or radiation. Besides leukemia and lymphoma cells, ABT-737 has single agent activity in inducing apoptosis in multiple myeloma and small-cell lung cancer cell lines, whereas it is not very efficient in killing ovarian or pancreatic carcinoma cells (Table 2). As ABT-737 does not inhibit Mcl-1, factors influencing Mcl-1 levels may significantly affect the efficacy of ABT-737 and possibly other Bcl-2 inhibitors.37, 40, 41 To date only one study has described a possible role for Bcl2A1 in resistance to ABT-737, although one suspects that it may also play an important role in specific cell types or cellular environments.37
Studies from other laboratories as well as our own have recognized that primary cells from patients with CLL are very sensitive to ABT-737.11, 16, 52 In our studies, we have found that low nanomolar concentrations of ABT-737 induced extensive and rapid apoptosis in primary CLL cells with an EC50=7 nM as early as 4 h after exposure.52 Furthermore, we also found that primary cells from other B-cell malignancies, including mantle cell lymphoma, marginal zone lymphoma and follicular lymphoma, are all highly sensitive to ABT-737 with extensive and rapid apoptosis at nanomolar concentrations (Table 2).52 In this regard, the exquisite sensitivity of these primary malignant B cells to ABT-737 in these in vitro studies is compatible with the reported marked reduction in tumor burden observed in initial studies with ABT-263 of two patients with bulky SLL/CLL.28
These data also highlight major differences in the sensitivity of primary malignant B cells from patients compared with many of the more commonly used cell lines (see also data on mantle cell lymphoma cells described below). It is not clear how widespread such differences are and further study of primary tumor cells, particularly in other cell types, is required. Published data on AML and ALL cells do not show a remarkable difference in sensitivity between primary samples and cell lines; however, later time points were assessed in these studies. As yet the reason for the difference in primary malignant B-cells and cell lines is not clear. One possibility may be that the act of adapting cells to culture conditions presents some type of stress, and that this stress selects for more ‘robust’ variants that lose some of their susceptibility to apoptosis. Although we do not understand this difference between primary cells and cell lines, the data highlight potential problems of using cell lines to determine the susceptibility of cells to chemotherapeutic agents.
Bcl-2 Inhibitors as Tools for Mechanistic Studies
The majority of detailed mechanistic studies on Bcl-2 inhibitors have been carried out with ABT-737. Owing to its high affinity for Bcl-2 and Bcl-XL, ABT-737 is proving to be a very valuable tool for mechanistic studies. For example, in our recent study, we showed that ABT-737 induced apoptosis in CLL cells at low nanomolar concentrations as assessed by many different biochemical and morphological criteria including activation and oligomerization of Bax and Bak, loss of mitochondrial membrane potential, release of mitochondrial cytochrome c, activation of caspases, chromatin condensation and phosphatidylserine externalization.52 However in addition to all the classical changes associated with apoptosis, exposure of CLL cells to ABT-737 also resulted in mitochondrial matrix swelling and rupture of the OMM, changes much more commonly associated with necrosis than apoptosis.52 As there are no truly representative CLL cell lines, we were unable to compare primary CLL cells with an appropriate cell line. However, we were able to compare the ABT-737 sensitivity of primary mantle cell lymphoma cells from patients with mantle cell lymphoma cell lines. Low nanomolar concentrations of ABT-737 induced apoptosis in primary mantle cell lymphoma cells (EC50=11 nM at 4 h).52 Primary mantle cell lymphoma cells exposed to ABT-737 exhibited classical ultrastructural features of apoptosis, including chromatin condensation and margination of the chromatin against the nuclear envelope (Figure 2b). In the cytoplasm of these cells, mitochondrial matrix swelling and OMM breaks were also readily observed (Figure 2b inset) compared with control cells (Figure 2a). In contrast, exposure of Z-138, a mantle cell lymphoma cell line, required much higher concentrations of ABT-737 (EC50=∼3–10 μM at 4 h) to induce apoptosis. Furthermore examination of the ultrastructure revealed that ABT-737 induced extensive apoptosis in this cell line, as assessed by chromatin condensation and nucleolar disintegration (Figure 2d). Although some mitochondrial matrix swelling was observed, OMM breaks were not a significant feature of these cells (Figure 2d inset). In these cells, we also observed the presence of flocculent densities within the mitochondrial matrix, a feature more commonly associated with necrosis than apoptosis (Figure 2d inset).56 This was consistent with the presence of several lysed cells (Figure 2d). Similar morphologies were found in other mantle cell lymphoma cell lines studied (data not shown). Thus, in primary malignant B cells, particularly CLL and mantle cell lymphoma cells, mitochondrial matrix swelling and rupture of the OMM were prevalent ultrastructural features that were absent in several cell lines that we have studied, including Z-138. These studies therefore highlight an unexpected function of Bcl-2 in maintaining mitochondrial membrane integrity in ABT-737 sensitive primary lymphoma and CLL cells and suggest a role for Bcl-2 in fundamental morphological changes occurring during the transition from primary cells to derived cell lines.

Mantle cell lymphoma cell lines differ from primary cells in their sensitivity to ABT-737. (a) Untreated mantle cell lymphoma cells, examined immediately after isolation from patients, were characterized by irregular nuclei containing moderately condensed heterochromatin, which extended, as poorly defined projections, throughout the nucleoplasm. Their mitochondria (inset) were small, with well-developed cristae and occasional matrix granules. (b) Exposure of these cells to ABT-737 (30 nM) for 2 h resulted in marked chromatin condensation against the inner membrane of the nuclear envelope (arrowheads) and segregation of the nucleolus. In the cytoplasm of these cells, mitochondrial matrix swelling and OMM breaks (black arrows) were also readily observed (inset). (c) Z-138 cells, a mantle cell lymphoma line, exhibited much less marginal heterochromatin than primary cells but their mitochondria (inset) were similar. (d) Exposure of Z-138 cells for 4 h to ABT-737 (1 μM) induced chromatin condensation against the inner membrane of the nuclear envelope (arrowheads) together with nucleolar disintegration, ultrastructural changes associated with apoptosis. Swelling of the mitochondrial matrix was observed in many of these cells (inset), together with the presence of flocculent densities (white arrows) but OMM breaks were rare. Lysed cells and debris (*) were also common (bars=5 μm)
In another mechanistic study, ABT-737 was used to activate Bax and circumvent the loss of VDAC1.57 Activation of Bax is inhibited in cells lacking VDAC1, although Bak is still activated, but apoptosis could be restored using ABT-737. Thus, a novel pathway was delineated in which VDAC1 acts upstream of Bax but downstream of Bak activation in cisplatin-induced apoptosis. In another recent study, it was discovered that overexpression of Bcl-XL in hippocampal neurons resulted in an increased synapse formation. This was dependent on functional Drp1 expression and reverted by either knockdown of Bcl-XL or addition of ABT-737.58 Taken together, these examples show that ABT-737 is a powerful tool to target Bcl-2 or Bcl-XL and thereby give important insights into the functions of these proteins. This might be especially important in cell types that are difficult to transfect, as use of ABT-737 might complement or even circumvent knockdown experiments. However, unless non-mechanism based toxicities are excluded, mechanistic studies with Bcl-2 antagonists other than ABT-737 should be carefully considered and the interpretation of the data might be based on nonspecific effects rather than a direct interaction with Bcl-2 proteins.
Future Perspective
Previous attempts to target Bcl-2 therapeutically using antisense technology to inhibit protein translation so far have not significantly improved outcomes for cancer patients, although improved oligonucleotide design may enhance efficacy.59 The development of orally bioavailable Bcl-2 inhibitors, such as ABT-263, with the ability to inhibit specifically BH3-Bcl-2 protein–protein interactions at low nanomolar concentrations, potentially marks a significant development in cancer therapy. Phase I studies of ABT-263 are currently ongoing in both lymphoid malignancies and SCLC, both of which have been shown to be sensitive to the compound in vitro, although in the latter case only in cell lines and not in primary material.24, 28
Whether compounds such as ABT-263 find their way into routine clinical use will depend not only on efficacy but also on manageable, acceptable toxicities. Thrombocytopenia, for example, may limit the value of ABT-263 in patients with heavy bone marrow infiltration; the long-term toxicities of profound Bcl-2 suppression remain to be determined. Identification of proteins that mediate resistance to ABT-263 may allow the development of rational combinations. After many years of development, it now seems likely that the rational design of specific compounds that inhibit specific protein–protein interactions may lead to significant therapeutic advances.
Abbreviations
- OMM:
-
the outer mitochondrial membrane
- CLL:
-
chronic lymphocytic leukemia
References
Hanahan D, Weinberg RA . The hallmarks of cancer. Cell 2000; 100: 57–70.
Bratton SB, Cohen GM . Apoptotic death sensor: an organelle's alter ego? Trends Pharmacol Sci 2001; 22: 306–315.
Danial NN, Korsmeyer SJ . Cell death: critical control points. Cell 2004; 116: 205–219.
Adams JM, Cory S . The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 2007; 26: 1324–1337.
Cory S, Adams JM . The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2002; 2: 647–656.
Youle RJ, Strasser A . The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 2008; 9: 47–59.
Huang DC, Strasser A . BH3-Only proteins-essential initiators of apoptotic cell death. Cell 2000; 103: 839–842.
Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 2005; 17: 393–403.
Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev 2005; 19: 1294–1305.
Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 2007; 315: 856–859.
Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A . Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest 2007; 117: 112–121.
Miyashita T, Reed JC . Bcl-2 oncoprotein blocks chemotherapy-induced apoptosis in a human leukemia cell line. Blood 1993; 81: 151–157.
Sentman CL, Shutter JR, Hockenbery D, Kanagawa O, Korsmeyer SJ . bcl-2 inhibits multiple forms of apoptosis but not negative selection in thymocytes. Cell 1991; 67: 879–888.
Amundson SA, Myers TG, Scudiero D, Kitada S, Reed JC, Fornace Jr AJ . An informatics approach identifying markers of chemosensitivity in human cancer cell lines. Cancer Res 2000; 60: 6101–6110.
Fry DC, Vassilev LT . Targeting protein-protein interactions for cancer therapy. J Mol Med 2005; 83: 955–963.
Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005; 435: 677–681.
Becattini B, Kitada S, Leone M, Monosov E, Chandler S, Zhai D et al. Rational design and real time, in-cell detection of the proapoptotic activity of a novel compound targeting Bcl-X(L). Chem Biol 2004; 11: 389–395.
Chan SL, Lee MC, Tan KO, Yang LK, Lee AS, Flotow H et al. Identification of chelerythrine as an inhibitor of BclXL function. J Biol Chem 2003; 278: 20453–20456.
Degterev A, Lugovskoy A, Cardone M, Mulley B, Wagner G, Mitchison T et al. Identification of small-molecule inhibitors of interaction between the BH3 domain and Bcl-xL. Nat Cell Biol 2001; 3: 173–182.
Kitada S, Leone M, Sareth S, Zhai D, Reed JC, Pellecchia M . Discovery, characterization, and structure-activity relationships studies of proapoptotic polyphenols targeting B-cell lymphocyte/leukemia-2 proteins. J Med Chem 2003; 46: 4259–4264.
Mohammad R, Giri A, Goustin AS . Small-molecule inhibitors of Bcl-2 family proteins as therapeutic agents in cancer. Recent Patents Anticancer Drug Discov 2008; 3: 20–30.
Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy Madiraju SR et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci USA 2007; 104: 19512–19517.
Tang G, Yang CY, Nikolovska-Coleska Z, Guo J, Qiu S, Wang R et al. Pyrogallol-based molecules as potent inhibitors of the antiapoptotic Bcl-2 proteins. J Med Chem 2007; 50: 1723–1726.
Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 2008; 68: 3421–3428.
Tzung SP, Kim KM, Basanez G, Giedt CD, Simon J, Zimmerberg J et al. Antimycin A mimics a cell-death-inducing Bcl-2 homology domain 3. Nat Cell Biol 2001; 3: 183–191.
Wang G, Nikolovska-Coleska Z, Yang CY, Wang R, Tang G, Guo J et al. Structure-based design of potent small-molecule inhibitors of anti-apoptotic Bcl-2 proteins. J Med Chem 2006; 49: 6139–6142.
Wang JL, Liu D, Zhang ZJ, Shan S, Han X, Srinivasula SM et al. Structure-based discovery of an organic compound that binds Bcl-2 protein and induces apoptosis of tumor cells. Proc Natl Acad Sci USA 2000; 97: 7124–7129.
Wilson WH, Tulpule A, Levine AM, Dunleavy K, Krivoshik AP, Hagey AE et al. A phase 1/2a study evaluating the safety, pharmacokinetics and efficacy of ABT-263 in subjects with refractory or relapsed lymphoid malignancies. Blood 2007; 110: Abstract 1371.
Zhang M, Ling Y, Yang CY, Liu H, Wang R, Wu X et al. A novel Bcl-2 small molecule inhibitor 4-(3-methoxy-phenylsulfannyl)-7-nitro-benzofurazan-3-oxide (MNB)-induced apoptosis in leukemia cells. Ann Hematol 2007; 86: 471–481.
Bruncko M, Oost TK, Belli BA, Ding H, Joseph MK, Kunzer A et al. Studies leading to potent, dual inhibitors of Bcl-2 and Bcl-xL. J Med Chem 2007; 50: 641–662.
Zhai D, Jin C, Satterthwait AC, Reed JC . Comparison of chemical inhibitors of antiapoptotic Bcl-2-family proteins. Cell Death Differ 2006; 13: 1419–1421.
Verhaegen M, Bauer JA, Martin de la Vega C, Wang G, Wolter KG, Brenner JC et al. A novel BH3 mimetic reveals a mitogen-activated protein kinase-dependent mechanism of melanoma cell death controlled by p53 and reactive oxygen species. Cancer Res 2006; 66: 11348–11359.
Mohammad R, Young D, Chen B, Aboukameel A, Chen J, Nikolovska-Coleska Z et al. ApoG2, a potent, non-toxic small-molecule inhibitor of Bcl-2 family: a preclinical trial in lymphoma. Proc Amer Assoc Cancer Res 2006; 47: Abstract 1335.
Kutzki O, Park HS, Ernst JT, Orner BP, Yin H, Hamilton AD . Development of a potent Bcl-x(L) antagonist based on alpha-helix mimicry. J Am Chem Soc 2002; 124: 11838–11839.
Lee EF, Czabotar PE, Smith BJ, Deshayes K, Zobel K, Colman PM et al. Crystal structure of ABT-737 complexed with Bcl-xL: implications for selectivity of antagonists of the Bcl-2 family. Cell Death Differ 2007; 14: 1711–1713.
Zhang YH, Bhunia A, Wan KF, Lee MC, Chan SL, Yu VC et al. Chelerythrine and sanguinarine dock at distinct sites on BclXL that are not the classic BH3 binding cleft. J Mol Biol 2006; 364: 536–549.
van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006; 10: 389–399.
Konopleva M, Watt J, Contractor R, Tsao T, Harris D, Estrov Z et al. Mechanisms of antileukemic activity of the novel Bcl-2 homology domain-3 mimetic GX15-070 (obatoclax). Cancer Res 2008; 68: 3413–3420.
Chen S, Dai Y, Harada H, Dent P, Grant S . Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res 2007; 67: 782–791.
Dai Y, Grant S . Targeting multiple arms of the apoptotic regulatory machinery. Cancer Res 2007; 67: 2908–2911.
Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006; 10: 375–388.
Lin X, Morgan-Lappe S, Huang X, Li L, Zakula DM, Vernetti LA et al. ‘Seed’ analysis of off-target siRNAs reveals an essential role of Mcl-1 in resistance to the small-molecule Bcl-2/Bcl-X(L) inhibitor ABT-737. Oncogene 2007; 26: 3972–3979.
Tahir SK, Yang X, Anderson MG, Morgan-Lappe SE, Sarthy AV, Chen J et al. Influence of Bcl-2 family members on the cellular response of small-cell lung cancer cell lines to ABT-737. Cancer Res 2007; 67: 1176–1183.
Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S et al. Programmed anuclear cell death delimits platelet life span. Cell 2007; 128: 1173–1186.
Zhang H, Nimmer PM, Tahir SK, Chen J, Fryer RM, Hahn KR et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ 2007; 14: 943–951.
Chauhan D, Velankar M, Brahmandam M, Hideshima T, Podar K, Richardson P et al. A novel Bcl-2/Bcl-X(L)/Bcl-w inhibitor ABT-737 as therapy in multiple myeloma. Oncogene 2007; 26: 2374–2380.
Kline MP, Rajkumar SV, Timm MM, Kimlinger TK, Haug JL, Lust JA et al. ABT-737, an inhibitor of Bcl-2 family proteins, is a potent inducer of apoptosis in multiple myeloma cells. Leukemia 2007; 21: 1549–1560.
Trudel S, Stewart AK, Li Z, Shu Y, Liang SB, Trieu Y et al. The Bcl-2 family protein inhibitor, ABT-737, has substantial antimyeloma activity and shows synergistic effect with dexamethasone and melphalan. Clin Cancer Res 2007; 13: 621–629.
Kohl TM, Hellinger C, Ahmed F, Buske C, Hiddemann W, Bohlander SK et al. BH3 mimetic ABT-737 neutralizes resistance to FLT3 inhibitor treatment mediated by FLT3-independent expression of BCL2 in primary AML blasts. Leukemia 2007; 21: 1763–1772.
Del Gaizo Moore V, Schlis KD, Sallan SE, Armstrong SA, Letai A . BCL-2 dependence and ABT-737 sensitivity in acute lymphoblastic leukemia. Blood 2008; 111: 2300–2309.
Kuroda J, Kimura S, Andreeff M, Ashihara E, Kamitsuji Y, Yokota A et al. ABT-737 is a useful component of combinatory chemotherapies for chronic myeloid leukaemias with diverse drug-resistance mechanisms. Br J Haematol 2008; 140: 181–190.
Vogler M, Dinsdale D, Sun XM, Young KW, Butterworth M, Nicotera P et al. A novel paradigm for rapid ABT-737-induced apoptosis involving outer mitochondrial membrane rupture in primary leukemia and lymphoma cells. Cell Death Differ 2008; 15: 820–830.
Hann CL, Daniel VC, Sugar EA, Dobromilskaya I, Murphy SC, Cope L et al. Therapeutic efficacy of ABT-737, a selective inhibitor of BCL-2, in small cell lung cancer. Cancer Res 2008; 68: 2321–2328.
Witham J, Valenti MR, De-Haven-Brandon AK, Vidot S, Eccles SA, Kaye SB et al. The Bcl-2/Bcl-XL family inhibitor ABT-737 sensitizes ovarian cancer cells to carboplatin. Clin Cancer Res 2007; 13: 7191–7198.
Huang S, Sinicrope FA . BH3 mimetic ABT-737 potentiates TRAIL-mediated apoptotic signaling by unsequestering Bim and Bak in human pancreatic cancer cells. Cancer Res 2008; 68: 2944–2951.
Trump BF, Berezesky IK, Chang SH, Phelps PC . The pathways of cell death: oncosis, apoptosis, and necrosis. Toxicol Pathol 1997; 25: 82–88.
Tajeddine N, Galluzzi L, Kepp O, Hangen E, Morselli E, Senovilla L et al. Hierarchical involvement of Bak, VDAC1 and Bax in cisplatin-induced cell death. Oncogene 2008; 27: 4221–4232.
Li H, Chen Y, Jones AF, Sanger RH, Collis LP, Flannery R et al. Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci USA 2008; 105: 2169–2174.
Cheson BD . Oblimersen for the treatment of patients with chronic lymphocytic leukemia. Ther Clin Risk Manag 2007; 3: 855–870.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by A Verkhratsky
Rights and permissions
About this article
Cite this article
Vogler, M., Dinsdale, D., Dyer, M. et al. Bcl-2 inhibitors: small molecules with a big impact on cancer therapy. Cell Death Differ 16, 360–367 (2009). https://doi.org/10.1038/cdd.2008.137
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2008.137
Keywords
This article is cited by
-
Harnessing large language models (LLMs) for candidate gene prioritization and selection
Journal of Translational Medicine (2023)
-
Single-molecule fingerprinting of protein-drug interaction using a funneled biological nanopore
Nature Communications (2023)
-
secDrug: a pipeline to discover novel drug combinations to kill drug-resistant multiple myeloma cells using a greedy set cover algorithm and single-cell multi-omics
Blood Cancer Journal (2022)
-
Next generation proteomics with drug sensitivity screening identifies sub-clones informing therapeutic and drug development strategies for multiple myeloma patients
Scientific Reports (2021)
-
Depletion of Survivin suppresses docetaxel-induced apoptosis in HeLa cells by facilitating mitotic slippage
Scientific Reports (2021)
