
Abstract
Modulation of tumor suppressor activities may provide new opportunities for cancer therapy. Here we show that disruption of the gene Ppm1d encoding Wip1 phosphatase activated the p53 and p16 (also called Ink4a)–p19 (also called ARF) pathways through p38 MAPK signaling and suppressed in vitro transformation of mouse embryo fibroblasts (MEFs) by oncogenes. Disruption of the gene Cdkn2a (encoding p16 and p19), but not of Trp53 (encoding p53), reconstituted cell transformation in Ppm1d-null MEFs. In vivo, deletion of Ppm1d in mice bearing mouse mammary tumor virus (MMTV) promoter–driven oncogenes Erbb2 (also called c-neu) or Hras1 impaired mammary carcinogenesis, whereas reduced expression of p16 and p19 by methylation-induced silencing or inactivation of p38 MAPK correlated with tumor appearance. We conclude that inactivation or depletion of the Wip1 phosphatase with resultant p38 MAPK activation suppresses tumor appearance by modulating the Cdkn2a tumor-suppressor locus.
Similar content being viewed by others


Main
Wip1 is a member of the PP2C family of evolutionarily conserved protein phosphatases. Originally described as a p53-regulated gene1, Ppm1d was later implicated as a negative regulator of p53 through its ability to attenuate p38 MAPK activity, thereby mediating inactivation of p53 after cellular stresses2. The importance of this negative feedback loop was further illustrated by the findings that Wip1 complements several oncogenes for cell transformation in vitro and that human PPM1D is amplified and overexpressed in certain types of cancers, including human primary breast cancer3,4, neuroblastoma5 and ovarian clear cell adenocarcinoma6. Wip1 directly inactivates p38 MAPK by dephosphorylating a threonine residue that is required for full enzyme activation2. In turn, the p38 MAPK pathway has been implicated as a negative regulator of cell cycle progression by several mechanisms, which implies that Wip1 is also an important regulator of cell cycle progression. p38 MAP kinase inhibits expression of D-type cyclins at transcriptional and post-translational levels7,8, phosphorylates and induces degradation of the Cdc25A phosphatase9, inhibits the Cdc25B phosphatase through phosphorylation of 14-3-3 sites10 and phosphorylates the p53 tumor suppressor on two activating sites (Ser33 and Ser46) in the N-terminal region, which contribute to p53-mediated apoptosis2,11. Together, these p38 MAPK targets cooperate to activate cell cycle checkpoints, which implies that defects in p38 MAPK regulation or function may perturb cell cycle progression leading to increased tumorigenesis. Indeed, inactivation of p38α MAPK or MKK3 and MKK6 by gene targeting markedly increased tumorigenesis3,12.
Whether or not p38 MAPK is the only target for Wip1, several recent studies suggest that, similar to p38 deficiency3,12, PPM1D positively regulates cell proliferation and thus behaves as an oncogene3,4, whereas depletion of Wip1 substantially reduced cell proliferation rates and activated apoptosis5. These findings argue that Wip1 may be a promising new target for treating certain types of tumors. Using knockout mice with a disruption in Ppm1d (ref. 13), here we explore this possibility by characterizing the ability of Ppm1d-null MEFs to undergo transformation in vitro and by analyzing mammary gland cancer–prone mouse strains crossed with Ppm1d-null mice.
Results
Ppm1d-null MEFs are resistant to transformation
To investigate whether Wip1 deficiency interfered with oncogene-induced transformation of primary MEFs, we prepared wild-type and Ppm1d-null MEFs from embryos obtained from interbreeding Ppm1d+/− mice. Preliminary examination of MEFs established from Ppm1d-null mice showed a reduced proliferation rate with features of premature senescence. Passage 2–3 Ppm1d-null MEFs had a substantial proliferative defect, and proliferation stopped after passage 6–7 (Fig. 1a). Because cultures of such short lifespan would be impossible to analyze thoroughly, we transformed wild-type and Ppm1d-null MEFs with the oncogenes Ad5 E1A 12S and Hras1. The proliferation rates of these transformed, Ppm1d-null MEFs were not different from those of wild-type cells (Fig. 1b), and the levels of expression for E1A and Hras1 were similar for both cell types (data not shown). We then asked whether Ppm1d-null cells expressing oncogenes were tumorigenic in vivo. We injected multiple nude mice with each set of MEFs transformed with E1A and Hras1 and analyzed tumor formation after 28 d. All mice injected with transformed wild-type MEFs developed tumors, but only 2 of 15 sites injected with transformed Ppm1d-null MEFs did so (Fig. 1c), and these tumors were at least 10 times smaller than those produced by wild-type cells (data not shown). To investigate whether this effect was specific for cells transformed with E1A and Hras1, we also analyzed tumor formation after injecting cells infected with retroviruses expressing either Hras1 V12 and Myc or Hras1 V12 and Erbb2. None of the sites injected with transformed Ppm1d-null MEFs developed tumors, whereas all sites injected with similarly transformed wild-type cells did (Fig. 1c).

(a) Second-passage MEFs were established from either wild-type (WT) or Ppm1d-null mice, and the number of cells per dish was determined every third day before passage. (b) Early-passage (passage 2 or 3) wild-type (WT) or Ppm1d-null MEFs were infected with retroviruses expressing E1A and Hras1, and cell proliferation was analyzed using the MTS reagent. (c) BALB/CanNCr-nu male mice at 6–8 weeks of age were injected subcutaneously with 106 wild-type (right side) or Ppm1d-null (left side) MEFs expressing different pairs of complementary oncogenes as indicated and tumor appearance after 4 weeks was monitored. The number of mice that developed tumors from oncogene-expressing Ppm1d-null MEFs and from wild-type MEFs (WT) is given (tumor incidence). Five mice were injected with each cell type, and all sites injected with oncogene-expressing wild-type MEFs developed tumors.
Levels of p53, p16 and p19 in Ppm1d-null MEFs
Because Wip1 regulates p38 MAPK activation, we reasoned that disruption of Ppm1d might result in constitutive activation of p38 MAPK and consequent activation of p53. Indeed, we observed more phosphorylation at the activating sites of p38 MAPK in Ppm1d-null MEFs transformed with E1A and Hras1 than in similarly transformed wild-type cells (Fig. 2b). To investigate the mechanism by which Ppm1d deficiency protected nude mice from tumor formation by MEFs infected with different oncogenes, we next investigated whether Ppm1d-null cells had greater p53 activity. We transiently transfected wild-type or Ppm1d-null MEFs transformed with E1A and Hras1 with chloramphenicol acetyltransferase (CAT) reporter plasmids containing either the PG13 p53 response element or a mutated responsive element (MG15). After 2 d, we collected the cells and analyzed CAT activity. Ppm1d-null cells had greater p53-mediated transactivation based on the reporter assays (Fig. 2a). We also observed greater expression of the p53-regulated gene products p21 (also called Waf1) and Mdm2, as well as of p53 itself, in Ppm1d-null cells compared with wild-type cells. We then extended our analysis to determine whether other cell cycle inhibitors were differentially expressed in Ppm1d-null and wild-type MEFs transformed with E1A and Hras1. Levels of p27 (also called Kip1) were only slightly higher in Ppm1d-null cells, but the levels of p16 and p19 were substantially higher than in wild-type cells (Fig. 2b). Because the primary targets of the analyzed cell cycle inhibitors are cyclin-dependent kinases, we checked whether the activities of the kinases Cdk2, Cdk4 and Cdk6 were attenuated in Ppm1d-null cells. Although we observed more p21 in Ppm1d-null cells, there were no substantial differences in Cdk2 activity between wild-type and Ppm1d-null cells (Fig. 2c). We observed no difference in Cdk6 activity, but Cdk4-associated activity was substantially lower in Ppm1d-null MEFs transformed with E1A and Hras1 than in similarly transformed wild-type cells (Fig. 2c).

(a) Activation of p53-mediated transcription was analyzed in wild-type (WT) and Ppm1d-null MEFs expressing E1A and Hras1 using CAT reporters linked to a basal promoter containing the wild-type (PG13-CAT) or mutated (MG15-CAT) p53-response elements. CAT assays were carried out as described11; an arrow marks the position of acetylated chloramphenicol. The levels of p53 in nuclear extracts35 were determined using western immunoblotting. (b) Levels of the indicated proteins involved in cell cycle control were analyzed in wild-type (WT) and Ppm1d-null MEFs expressing E1A and Hras1. The levels of p19 protein were analyzed in nuclear extracts. (c) Kinase activities for Cdk2, Cdk4 and Cdk6 were determined after immunoprecipitation with appropriate antibodies; histone H1 (HH1) served as the substrate for Cdk2 kinase and glutathione S-transferase–pRb (GST-Rb) for Cdk4 and Cdk6. Cdk levels, determined by western immunoblotting, also are shown. WT, wild-type. (d) Wild-type (WT) and Ppm1d-null MEFs expressing E1A and Hras1 were transfected with either p16-LUC36 or p19-CAT37, and activities of the reporters were analyzed 24 h later. The relative increase in activity in extracts from Ppm1d-null MEFs relative to wild-type MEFs is plotted.
We next determined whether accumulation of p16 and p19 occurs at the transcriptional or post-translational level. We observed no differences in the half-lives of either protein (data not shown), but we noted substantially greater transcriptional activity from both the p16 and p19 promoters in Ppm1d-null MEFs relative to wild-type MEFs using promoter-linked assays with either luciferase (for p16) or CAT (for p14, the human homolog of mouse p19) reporters (Fig. 2d). These data suggest that greater expression of p16 and p19 resulted from transcriptional induction in Ppm1d-null cells.
The p16-p19 pathway controls resistance to transformation
Our biochemical analyses indicated that either the p53 or the p16-p19 pathways could be responsible for the inability of Ppm1d-null MEFs to undergo transformation in vitro. Although activity of Cdk2 was not affected in Ppm1d-null MEFs transformed with E1A and Hras1 (Fig. 2c), p53 may have other activities that contribute to its tumor-suppressing properties. To address this issue systematically, we crossed Ppm1d-null and Trp53-null mice and analyzed the susceptibility of MEFs from the resulting mice to transformation. We reasoned that if activation of the p53 tumor-suppressor pathway was responsible for the inability of Ppm1d-null MEFs to be transformed, then inactivation of p53 would allow subsequent tumor formation in nude mice by Ppm1d−/− Trp53−/− cells expressing different oncogenes. Analysis of MEFs from Ppm1d−/− Trp53−/− mice showed a loss of the premature senescence (Fig. 3a) seen in Ppm1d-null cells with wild-type Trp53 (Fig. 1a). Ppm1d−/− Trp53−/− MEFs also seemed to be immortalized, as they continued to grow in culture after multiple passages, similar to Trp53−/− MEFs (Fig. 3a). Whereas transformation could be induced with a single oncogene in Trp53-null cells14, injecting nude mice with Ppm1d−/− Trp53−/− MEFs expressing Hras1, Myc or Erbb2 did not result in tumor formation (Fig. 3b). None of the 15 mice injected with Ppm1d−/− Trp53−/− MEFs expressing these oncogenes (five mice for each oncogene) developed tumors within 4 weeks, whereas all the mice injected with Trp53-null cells expressing the same oncogenes developed tumors within this period.

(a) Second-passage MEFs were established from wild-type (WT), Trp53-null or Ppm1d−/− Trp53−/− mice, and cell growth was determined using the MTS protocol as described above. (b) BALB/CanNCr-nu male mice at 6–8 weeks of age were injected subcutaneously with 106 Trp53−/− (right side) or Ppm1d−/− Trp53−/− (left side) MEFs expressing different oncogenes (indicated above), and tumor formation was analyzed 3 weeks later. (c) BALB/CanNCr-nu male mice at 6–8 weeks of age were injected subcutaneously with 106 Cdkn2a−/− (right side) or Ppm1d−/− Cdkn2a−/− (left side) MEFs expressing different oncogenes (indicated above), and tumor formation was analyzed 3 weeks later. (d) BALB/CanNCr-nu male mice at 6–8 weeks of age were injected subcutaneously with 106 p16Ink4a−/− (right side) or Ppm1d−/− p16Ink4a−/− (left side) MEFs that expressed one of two pairs of complementary oncogenes, E1A and Hras1 or Hras1 and Myc. Tumors were weighed 4 weeks later.
We next asked whether induction of the Cdkn2a locus and accumulation of p16 and p19 is the primary mechanism of tumor resistance of Ppm1d-null MEFs. We crossed knockout mice bearing a deletion of exons 2 and 3 of Cdkn2a (ref. 15; removing both p16 and p19) with Ppm1d-null mice and established MEFs from Ppm1d−/− Cdkn2a−/− mice. As for Ppm1d−/− Trp53−/− mice, disruption of p16 and p19 was also sufficient to overcome the premature induction of senescence observed in Ppm1d-null cells (data not shown). In contrast to Ppm1d−/− Trp53−/− cells, however, MEFs from Ppm1d−/− Cdkn2a−/− mice regained the ability to undergo oncogene-induced transformation (Fig. 3c). All nude mice injected with Ppm1d−/− Cdkn2a−/− MEFs expressing Hras1 (n = 10), Myc (n = 5) or Erbb2 (n = 5) developed tumors within 3 weeks. We obtained similar data with Ppm1d+/+ Cdkn2a−/− MEFs transformed with the same oncogenes (Fig. 3c). These data suggest that activation of the p16-p19 pathway is responsible for the tumor-resistant phenotype of Ppm1d-null MEFs in vitro.
To dissect further the pathway that renders Ppm1d-null MEFs resistant to oncogene-induced transformation, we crossed Ppm1d-null mice with Cdkn2a-ablated mice proficient for expression of only the portion of Cdkn2a encoding p19Arf (hereafter referred to as p19Arf) but not the portion of the gene encoding p16Ink4a (hereafter referred to as p16Ink4a), and established double-null Ppm1d−/− p16Ink4a−/− MEFs. If p16 is essential in preventing transformation of Ppm1d-null MEFs, then the Ppm1d−/− p16Ink4a−/− MEFs should form tumors in nude mice, as was observed for Ppm1d−/− Cdkn2a−/− cells (Fig. 3c). We infected p16Ink4a−/− MEFs and Ppm1d−/− p16Ink4a−/− MEFs with viruses expressing E1A and Hras1 or Myc and Hras1 and injected each set of oncogene-expressing cells into nude mice (Fig. 3d). All the sites injected with transformed p16Ink4a−/− MEFs developed tumors (five for each pair of oncogenes). Tumors also developed in mice injected with Ppm1d−/− p16Ink4a−/− MEFs expressing E1A and Hras1 (five injected mice) but not in mice injected with Ppm1d−/− p16Ink4a−/− MEFs expressing Hras1 and Myc (none of five mice). Furthermore, the tumors produced by Ppm1d−/− p16Ink4a−/− MEFs expressing E1A and Hras1 were at least three times smaller than tumors produced by p16Ink4a−/− MEFs. Therefore, both the p16-dependent and p19-dependent pathways contribute to the transformation-resistant phenotype of Ppm1d-null MEFs, and p19 has a primary role in protection from transformation in the presence of Hras1 and Myc.
Ppm1d-disrupted mice are resistant to mammary cancer
We next investigated whole tissues to determine whether similar changes are found in vivo. The level of activating p38 MAPK phosphorylation was markedly higher in several tissues of Ppm1d−/− mice compared with those of wild-type mice, including mammary gland, spleen and kidney (Fig. 4a). We observed little difference in the levels of p21 or p27 in any of these tissues, but the level of p16 was substantially higher in mammary gland tissue from Ppm1d−/− mice compared with wild-type mice (Fig. 4a). To address the mechanism by which p16 accumulation in mammary gland epithelial cells could block tumor formation, we next examined phosphorylation of pRb on Ser780. The activity of Cdk4 is inhibited by its association with p16 (ref. 16). Ser780 of pRb is specifically phosphorylated by the Cdk4 kinase and thus represents a direct physiological marker for Cdk4 activity in vivo17. Consistent with greater expression of p16, we observed a marked decrease in the level of pRb phosphorylation at Ser780 in mammary gland tissue from Ppm1d−/− females (Fig. 4b). This implies that the in vivo activity of Cdk4 in Ppm1d-null cells is reduced. In contrast to MEFs, only a slight increase in p19Arf mRNA was observed in mammary gland tissues from Ppm1d-null mice compared with wild-type mice (Fig. 4c).

(a) The levels of the indicated proteins were determined in the indicated tissues from wild-type (WT) and Ppm1d-null (KO) mice by western immunoblotting. (b) Phosphorylation of pRb protein at the Cdk4-dependent site Ser780 was determined in mammary tissue from wild-type (WT) or Ppm1d-null (KO) mice using an antibody specific to phosphorylated pRb. (c) Analysis of p19ARF mRNA in mammary tissue from wild-type (WT) and Ppm1d-null (KO) mice was carried out using a quantitative dot-blotting procedure38.
Next we investigated whether ablation of Ppm1d protects mice from certain types of mammary cancer in vivo, as has been reported for cyclin D1 deficiency18,19. We crossed Ppm1d−/− mice with three different strains of mammary gland cancer–prone mice; these breast cancer models are based on MMTV promoter–driven expression of Erbb2, Hras1 or Wnt1. We intercrossed Ppm1d+/− mice carrying each of these oncogenes to yield females with three different genotypes: Ppm1d+/+, Ppm1d+/− or Ppm1d−/−. All females were kept virgin during the observation period. We found that absence of Ppm1d did not prevent mammary gland cancers induced by Wnt1, nor was the frequency of Wnt1-induced tumors or the survival time of mice that developed these tumors substantially affected by the presence or absence of functional Ppm1d (Fig. 5a). In contrast, Ppm1d-null mice were considerably more resistant to tumor formation induced by Erbb2 or Hras1 (Fig. 5a). During the 16-month observation period, all 28 Ppm1d+/+ MMTV-Erbb2 mice died of mammary gland cancer. Among Ppm1d+/− MMTV-Erbb2 females, 30 of 33 mice developed tumors, which was not significantly different from the proportion of wild-type mice that developed tumors (P = 0.306), whereas almost 50% (14 of 29, P < 0.0001) of Ppm1d−/− MMTV-Erbb2 females remained tumor-free. Likewise, all wild-type (n = 12) and Ppm1d+/− (n = 14) females carrying the MMTV-Hras1 oncogene died of mammary carcinomas, whereas nearly 70% of the Ppm1d−/− MMTV-Hras1 females remained free of tumors during the 13-month observation period (P = 0.0005 relative to wild-type). Examination of the median lifespan for Ppm1d−/− MMTV-Erbb2 and Ppm1d−/− MMTV-Hras1 females showed a statistically significant increase in lifespan in both groups compared with wild-type or heterozygous littermates (Fig. 5a). These data suggest that Ppm1d deficiency overcomes the tumor-promoting activity of both Erbb2 and Hras1 in vivo.

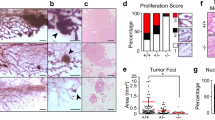
(a) The occurrence of mammary cancers in wild-type (WT), Ppm1d+/− and Ppm1d−/− transgenic mice bearing different oncogenes was analyzed over 16 months. We used the mammary gland cancer–prone mouse models MMTV-Erbb2, MMTV-Hras1 and MMTV-Wnt1. (b) Mitotic index analyses (left two panels) were based on cells positive for phosphorylated histone H3 in formalin-fixed and paraffin-embedded sections from tumors that arose in Ppm1d−/− MMTV-Erbb2 mice. In vivo apoptosis analyses (middle panels) in formalin-fixed and paraffin-embedded tumor sections were accomplished using a BrdUTP-TdT FragEL kit (Oncogene Research Products). Nuclei were counterstained with Methyl Green; arrows point to phosphorylated histone H3–positive or apoptotic nuclei that had incorporated BrdUTP. The morphology of tumors that arose in wild-type (WT) MMTV-Erbb2 and Ppm1d−/− MMTV-Erbb2 mice was examined after staining with hematoxylin and eosin (H&E, right two panels). Representative areas are shown.
Although many Ppm1d−/− MMTV-Erbb2 and Ppm1d−/− MMTV-Hras1 females remained free of mammary carcinomas during the observation period, tumors did develop in some mice. To assess the presence of microscopic lesions, we examined normal mammary tissues from three MMTV-Erbb2 and three MMTV-Hras1 mice with breast cancer. Five of six mammary tissue samples from wild-type mice and four of six mammary tissue samples from Ppm1d-null had low-grade lesions that were either diffuse or multifocal in appearance. We observed no differences between samples from wild-type and Ppm1d-null mice. We next analyzed the proliferative ability of cells in the tumors that arose in Ppm1d−/− MMTV-Erbb2 mice. Staining for phosphorylated histone H3 showed that the mitotic index was lower in tumors from Ppm1d−/− MMTV-Erbb2 mice (0.8 ± 0.2% compared with 2.6 ± 0.2% for wild-type mice; Fig. 5b), whereas the number of apoptotic cells was higher in tumors from Ppm1d−/− MMTV-Erbb2 (1.9 ± 0.3%) than in those from Ppm1d+/+ MMTV-Erbb2 mice (1.1+/−0.2%; Fig. 5b). Thus histological evidence suggests that, even when tumors form in Ppm1d−/− mice, the cells in these tumors have a lower proliferation potential.
Tumor development and loss of p19 and p16
To investigate the level at which cells emerged from the Ppm1d deficiency-mediated block to tumor formation, we analyzed the levels of different, cell cycle–related proteins in mammary tumors from Ppm1d−/− MMTV-Erbb2 mice by western immunoblotting. Delayed tumor appearance in Ppm1d-null mice could result from a loss of oncogene expression; however, our analysis did not reveal any difference in Erbb2 expression between transgene-expressing Ppm1d−/− and Ppm1d+/+ mice (Fig. 6a). All tumors from Ppm1d−/− MMTV-Erbb2 mice retained high levels of phosphorylation at the p38 MAPK activating site (Fig. 6b), implying that deregulation occurred downstream of p38 MAPK. Among the analyzed proteins (Fig. 6b), we found no significant differences in the levels of p53, p21 or p27. In marked contrast, the levels of p16 were substantially reduced in two (Fig. 6b) of six tumors from Ppm1d−/− MMTV-Erbb2 females and p16 was undetectable in the other four samples (Fig. 6b). In addition, the levels of cyclin D1 were substantially higher in tumor samples from Ppm1d−/− MMTV-Erbb2 mice. Analysis of Cdk4 activity in these tumors, based on phosphorylation of Ser780 in pRb, showed that all tumor samples from Ppm1d−/− MMTV-Erbb2 mice had regained high levels of Cdk4 kinase activity that were comparable to the levels observed in tumors from Ppm1d+/+ MMTV-Erbb2 mice (Fig. 6b). As we could not detect p19 protein using western blotting, we analyzed p19Arf mRNA using northern blotting (Fig. 6c).The p19Arf mRNA level was reduced in one sample and undetectable in another tumor (Fig. 6c). Sequencing of cDNA obtained by reverse transcription of p19Arf mRNA showed the wild-type sequence in all other tumor samples (data not shown). Confirming these results, p19Arf mRNA levels, determined by real-time PCR, were substantially lower in only two of six tumors that arose in Ppm1d−/− MMTV-Erbb2 mice, whereas p16Ink4a levels were very low in all these samples (Supplementary Fig. 1 online). Because p16 expression was more frequently lost in these tumors, we conclude that p16 has a more important role in preventing tumor development in the absence of Wip1 in this context. A role for p19 in preventing tumor formation is not excluded, however, especially in other cellular contexts.

(a) Expression of the onco-transgene in normal (N) mammary glands and tumor (T) samples from Ppm1d+/+ (WT) MMTV-Erbb2 and Ppm1d−/− MMTV-Erbb2 mice was analyzed by RT-PCR as described19. (b) Individual tumors from either wild-type (WT; lanes 1–3) or Ppm1d-null (lanes 4–9) mice expressing MMTV-Erbb2 were analyzed for the indicated proteins by western blotting. Phosphorylation status of the pRb protein at Ser780 in these tumors was determined using an antibody specific to phosphorylated pRb. (c) p19ARF mRNA levels in individual tumors from either wild-type (WT; lanes 1–3) or Ppm1d-null (lanes 4–9) mice expressing MMTV-Erbb2 were analyzed by northern blotting. (d) A 120-bp segment overlapping the p16Ink4a translational start site was amplified from bisulfite-treated tumor DNA with primers specific for either the unmethylated (U) or methylated (M) allele. The asterisk indicates a nonspecific band seen in PCR reactions using primers for the unmethylated allele. (e) Expression of p16 in normal and tumor cell cultures established from Ppm1d−/− MMTV-Erbb2 females after treatment of the cultures with 0 μM (control), 1 μM or 3 μM DAC for 24 h. Actin served as a loading control.
Both Erbb2- and Hras1-driven transformation of mammary epithelial cells requires cyclin D1 and Cdk4 activity18,19, which is inhibited by p16. The appearance of tumors in Ppm1d−/− MMTV-Erbb2 mice strongly correlated with low levels of p16 (Fig. 6b), and a common mechanism of suppressing p16 expression in tumors is methylation-induced silencing of its promoter20. To determine if this was the case for tumors that arose in Ppm1d−/− MMTV-Erbb2 mice, we adapted a PCR-based method to detect methylation of CpG sites flanking the translation start site of mouse p16. We detected methylation in each of the three tumors that were examined but not in the normal tissues from these Ppm1d−/− MMTV-Erbb2 mice (Fig. 6d). Next, we established primary cultures from several tumors, treated them with increasing concentrations of the DNA methylation inhibitor 5-aza-2′-deoxycytidine (DAC) and measured levels of p16 24 h later. Exposure of these cells to DAC restored p16 protein in the primary tumor cultures to levels comparable to those observed in cells obtained from normal mammary gland tissues of Ppm1d−/− MMTV-Erbb2 mice (Fig. 6e). These data suggest that inhibition of p16 expression and development of mammary carcinomas in Ppm1d−/− MMTV-Erbb2 mice may result from methylation-induced silencing of the p16 promoter.
Inactivation of p38 MAPK leads to tumors in Ppm1d−/− mice
Although Wip1 may have several possible targets, p38 MAPK is thought to be an important one with respect to cell proliferation. p38 MAPK is an important mediator of tumor suppression, as inactivation of p38α by gene targeting or ablation of activation through disruption of the upstream kinases, MKK3 and MKK6, facilitated tumorigenesis3,12. If p38 MAPK functions as the relevant tumor suppressor in Ppm1d-null mice, then inactivation of p38 MAPK should reverse the tumor-resistant phenotype. To address this possibility, we first inactivated p38 MAPK in MEFs expressing E1A and Hras1 with the specific p38 MAPK inhibitor21 SB203580 at 10 μM. This treatment substantially reduced the levels of both p16 and p19 in Ppm1d-null MEFs (Fig. 7a). Similarly, injection of SB203580 (15 mg per kg body weight) substantially reduced p16 levels in the mammary gland tissues of Ppm1d-null mice within 24 h (Fig. 7b).

(a) MEFs expressing E1A and Hras1 from wild-type (WT) or Ppm1d-null mice were treated overnight with 10 μM SB203580 (lane 2 and 4), a chemical inhibitor of p38 MAPK, and the levels of p16 and p19 were analyzed by western immunoblotting. Lanes 1 and 3 were mock-treated with water; p38 was used as a loading control. (b) Wild-type (WT) and Ppm1d-null females were injected intraperitoneally with 15 mg SB203580 hydrochloride per kg body weight (lanes 2 and 4), and the levels of p16 in mammary tissues were analyzed the next day. Lanes 1 and 3 were controls (mammary tissues from mice injected with water). (c) Ppm1d−/− MMTV-Erbb2 females were injected with either 15 mg SB203580 hydrochloride per kg body weight or an equivalent volume of water every second day for 2 months, and tumor appearance was monitored. Injection of 2-month-old wild-type MMTV-Erbb2 mice (n = 4) with SB203580 did not result in the appearance of tumors within 60 d (data not shown).
Next we examined the effect of p38 MAPK inhibition on tumorigenesis in Ppm1d-null mice carrying MMTV-Erbb2. These mice were resistant to tumor formation (Fig. 4), possibly as a result of constitutive activation of p38 MAPK. We injected Ppm1d−/− MMTV-Erbb2 females with either water (n = 4) or 15 mg SB203580 hydrochloride per kg body weight (n = 4) at 12 months of age every second day for 2 months. As early as 30 days after beginning the injections, mice treated with the inhibitor developed mammary gland tumors, and after 2 months, three of four mice had mammary gland carcinomas (Fig. 7c), compared with none of the mice injected with water (P = 0.04). Thus, constitutive activation of p38 MAPK in the absence of Wip1 may account for the tumor-resistant phenotype of Ppm1d−/− MMTV-Erbb2 mice.
Discussion
Inhibiting positive regulators of cell proliferation, activating tumor-suppressor pathways and inducing apoptosis are primary approaches for intervention in modern cancer therapy. Among positive regulators of proliferation, Wip1 was found to complement different oncogenes in the transformation of wild-type MEFs3. Depletion of Wip1 in PPM1D-amplified human neuroblastoma cells arrested cell proliferation and activated apoptosis5. Here we show that Ppm1d deficiency renders non–Ppm1d-amplified cells resistant to transformation both in vitro and in vivo. Disruption of Ppm1d not only removed this newly recognized mediator of cell transformation, but also induced several tumor-suppressor pathways, including those mediated through p16-p19 and p53. Consistent with the marked accumulation of p16 in Ppm1d-null cells, Cdk4 activity was substantially inhibited, resulting in decreased phosphorylation of pRb at Ser780 (Fig. 4b). But enhanced p16 expression did not seem to be the only mechanism of resistance to transformation induced by Ppm1d inactivation in MEFs, as the growth of tumors arising in nude mice from Ppm1d−/− p16Ink4a−/− MEFs expressing Hras1 and Myc was still suppressed (Fig. 3d). Thus, enhanced expression of both p16 and p19 contributes to the transformation-resistant phenotype of Ppm1d-null MEFs (Fig. 3d). These mechanisms are independent of p53, as double-null Ppm1d−/− Trp53−/− MEFs were not tumorigenic in nude mice (Fig. 3b).
Because transformation of wild-type MEFs by several oncogenes does not require cyclin D1 or Cdk4 activity19, it is not surprising that increased levels of p16 alone would not render Ppm1d-null MEFs fully resistant to oncogene-induced transformation; thus, our data are compatible with the additional involvement of p19 (Fig. 3d). In contrast, transformation of mammary gland epithelial cells by Erbb2 or Hras1 requires activation of the cyclin D1–dependent pathway; thus, enhanced expression of p16 in the absence of Wip1 may protect mammary epithelial cells from transformation. The Wip1 phosphatase is expressed in mouse mammary gland tissues22, but disruption of Ppm1d does not affect normal mammary gland development (Supplementary Fig. 2 online). Here we show that Ppm1d-null mice are impaired in mammary tumorigenesis induced by Erbb2 or Hras1 but not Wnt1, as reported for cyclin D1–deficient mice18,19. A primary implication of our findings is that transformation caused by overexpression or amplification of cyclin D1, Erbb2 or Hras1 will be attenuated by depletion or inactivation of Wip1. CCND1 (encoding cyclin D1) is amplified in ∼20% of human breast cancers23, and overexpression of cyclin D1 has been reported for >50% of human mammary carcinomas24,25. Likewise, amplification or overexpression of Erbb2 has been documented in ∼50% of human breast cancers26, and PPM1D is amplified in up to 18% of primary human breast tumors3,12. Although a certain percentage of primary human mammary carcinomas may not respond to depletion of Wip1 activity owing to mutation of p16Ink4a or methylation-induced silencing of its promoter20,27, an anti-Wip1-directed therapy still may be effective for treatment of most mammary tumors. Furthermore, the tumors that did develop in Ppm1d−/− MMTV-Erbb2 mice had lower growth rates and higher cellular apoptosis than comparable tumors in wild-type mice (Fig. 5b,c). This result indicates that even after elimination of p16 expression as the primary block to tumor development, secondary mechanisms that contribute to tumor resistance in Ppm1d−/− mice still operated. Thus, Ppm1d deficiency through activation of p38 MAPK could target other molecules that may negatively contribute to proliferation, such as inhibition of Cdc25 phosphatases9,10,28 or induction of p19. Thus, we propose that p38 MAPK–dependent modulation of cell cycle regulators including expression of the Cdkn2a locus, through inactivation or depletion of Wip1, will be a powerful approach to treating human primary breast cancers as well as other human malignancies.
Methods
Mice.
All animal protocols used in this study were approved by the National Cancer Institute Animal Safety and Use Committee. We crossed transgenic mice bearing MMTV promoter–driven oncogenes with Ppm1d-null mice to yield Ppm1d heterozygotes carrying Erbb2 (ref. 29), Hras1 (ref. 30) or Wnt1 (ref. 31). We then intercrossed these mice to yield experimental groups that were Ppm1d+/+, Ppm1d+/− and Ppm1d−/− and carried each oncogene. We used only female mice for analyses and kept all females as virgins throughout the entire observation period. We monitored mice by palpation twice weekly for tumors. We obtained Cdkn2a−/− mice15 and p16Ink4a−/− mice32 from the Mouse Models of Human Cancers Consortium repository. Trp53-null mice were described previously33.
Western blotting and in vitro kinase reactions.
We prepared cell lysates as described11, and separated 50–100 μg of protein per lane by SDS-PAGE and transferred it to Immobilon-P membranes (Millipore). We probed the immunoblots with antibodies to cyclin D1 (72-13G, Santa Cruz Biotechnology), p21 (Ab-4, Oncogene Research Products), Mdm2 (Mouse/Human MDM2 Antibody Sampler Kit, Oncogene Research Products), p53 (Ab-7, Oncogene Research Products), p16 (M-156, Santa Cruz Biotechnology), p27 (F8, Santa Cruz Biotechnology), phosphorylated p38 (Cell Signaling Technology), p38 (C20, Santa Cruz Biotechnology), Cdk2 (M2, Santa Cruz Biotechnology), Cdk4 (H-303, Santa Cruz Biotechnology), Cdk6 (C21, Santa Cruz Biotechnology), actin (Ab-1, Oncogene Research Products), pRb (C15, Santa Cruz Biotechnology) or pRb phosphorylated at Ser780 (Cell Signaling Technology). As secondary antibodies, we used peroxidase-conjugated IgGs (Jackson ImmunoResearch Laboratories) followed by enhanced chemiluminescence detection (Amersham Biosciences). For in vitro kinase reactions, we immunoprecipitated the kinase using the appropriate antibody and carried out kinase reactions as described11 using either glutathione S-transferase–pRb (for Cdk4 and Cdk6) or histone H1 for Cdk2 as substrates.
Cell growth and retrovirus infection.
We prepared wild-type and Ppm1d-null MEFs from 13.5-d-old embryos and cultured them using a 3T3 protocol. We plated one million cells in a 10-cm tissue culture dish and determined the number of cells per dish after 3 d using the MTS reagent (Promega). We then replated one million cells in another dish and repeated this procedure until the cells stopped growing.
The pBabe-puro-Hras1V12 retrovirus, which expresses the activated Hras1 cDNA, was provided by S. Lowe (Cold Spring Harbor Laboratory); the pBabe-puro-NeuT retrovirus, expressing the activated rat Erbb2 cDNA with a mutation causing the amino acid substitution V664E, was provided by P. Sicinski (Dana Farber Cancer Institute). Retroviruses containing adenovirus E1A and Myc were described previously19. We transfected Phoenix-Eco cells with different retroviruses and infected MEFs as described3. We prepared mammary gland epithelial cells from aseptically removed mammary gland tumors, which were minced in Dulbecco's modified minimal Eagle's medium containing 1.0 mg ml−1 collagenase (type III, Sigma Chemical), 10% fetal bovine serum, 5 μg ml−1 bovine insulin, 10 ng ml−1 mouse epidermal growth factor, 100 units ml−1 penicillin and 100 μg ml−1 streptomycin and then incubated at 37 °C overnight. The next day, we passed cells through a 19-gauge needle twice, centrifuged them at 100g for 10 min and resuspended the cell pellets in Dulbecco's modified minimal Eagle's medium without collagenase.
Tumorigenicity assay.
To evaluate tumorigenicity, we resuspended one million cells in 0.5 ml of phosphate-buffered saline and injected them subcutaneously into the flanks of 6-week-old to 8-week-old Balb/CanNCr-nu male mice. In all experiments, we injected wild-type MEFs on the right side and Ppm1d-nulls on the left. We monitored tumor formation in mice and killed them 28 d after injection.
DNA methylation analysis.
We isolated genomic DNA from mammary tumors and the normal mammary tissue of Ppm1d−/− MMTV-Erbb2 mice using the Wizard Genomic DNA Purification Kit (Promega) according to the manufacturer's instructions. We carried out sodium bisulfite modification, which converts nonmethylated cytosine in DNA to uracil, as described34. We denatured 1 μg of genomic DNA in a volume of 50 μl with NaOH (final concentration, 0.2 M) for 10 min at 37 °C. We added 30 μl of 10 mM hydroquinone (Sigma Chemical) and 520 μl of 3 M sodium bisulfite (Sigma Chemical), pH 5.0 (prepared fresh), and mixed. The solution was overlayed with mineral oil and incubated at 50 °C for 16 h. We recovered modified DNA using the Wizard DNA cleanup kit (Promega) according to the manufacturer's instructions and then denatured it for 5 min with NaOH (final concentration 0.3 M) at room temperature. We precipitated the DNA with one volume isopropanol and 2 μl of 5 mg ml−1 glycogen (Ambion), incubated it at −20 °C for 30 min, centrifuged it at 14,000g for 30 min at 4 °C and then washed it in 70% ethanol. We then air-dried the DNA for 10 min and resuspended it in 50 μl of water.
We carried out methylation-specific PCR as described34. We added 2 μl (∼50 ng) of modified DNA to a PCR mix containing 0.2 mM dNTPs, 1.5 mM MgCl2, 5% DMSO, 1× Taq buffer (GeneChoice), 0.5 units Taq polymerase (GeneChoice) and 0.5 μM primers. Primer sequences are available on request. We heated reactions at 94 °C for 3 min followed by 35 cycles of 94 °C for 1 min, 58 °C for 30 s and 72 °C for 30 s. We carried out a final extension at 72 °C for 3 min and then cooled the reaction to 4 °C. We separated 15 μl of the PCR reaction by electrophoresis in a 2.5% agarose gel that subsequently was stained with 0.5 μg ml−1 ethidium bromide. We visualized bands by ultraviolet illumination and documented them using an Eagle Eye system (Stratagene).
Real-time PCR analysis of p16Ink4a and p19ARF mRNA expression.
We extracted RNA using the Trizol reagent (Invitrogen) from tumors that arose in wild-type MMTV-Erbb2 and Ppm1d−/− MMTV-Erbb2 mice. We prepared cDNA using the Thermoscript RT-PCR system (Invitrogen) with random hexamers using 1 μg of total RNA. We diluted samples to a final volume of 100 μl with distilled water and then carried out real-time PCR using 12.5 μl of 2× SYBR green mixture (Bio-Rad), 5.5 μl of distilled water, 5 μl of diluted cDNA and 1 μl of each 5 μM forward and reverse primers. The primer sequences for p16Ink4a, p19Arf and Gapd are available on request; the PCR product lengths were 150 bp, 111 bp, and 83 bp, respectively. The annealing temperatures for real-time PCR for p16Ink4a and p19Arf were 55 °C and 57 °C, respectively, for 10 s with an extension at 72 °C for 10 s. We carried out reactions in triplicate. We analyzed data by taking the average of the threshold values for Gapd and subtracting it from the average of the values for the target gene. We then subtracted these ΔCt values for Ppm1d−/− MMTV-Erbb2 tumor samples from the ΔCt values from wild-type MMTV-Erbb2 tumor samples (giving a ΔΔCt value). We calculated the relative induction as 2ΔΔCt.
Note: Supplementary information is available on the Nature Genetics website.
References
Fiscella, M. et al. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc. Natl. Acad. Sci. USA 94, 6048–6053 (1997).
Takekawa, M. et al. p53-inducible Wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J. 19, 6517–6526 (2000).
Bulavin, D.V. et al. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat. Genet. 31, 210–215 (2002).
Li, J. et al. Oncogenic properties of PPM1D located within a breast cancer amplification epicenter at 17q23. Nat. Genet. 31, 133–134 (2002).
Saito-Ohara, F. et al. PPM1D is a potential target for 17q gain in neuroblastoma. Cancer Res. 63, 1876–1883 (2003).
Hirasawa, A. et al. Association of 17q21-q24 gain in ovarian clear cell adenocarcinomas with poor prognosis and identification of PPM1D and APPBP2 as likely amplification targets. Clin. Cancer Res. 9, 1995–2004 (2003).
Casanovas, O. et al. Osmotic stress regulates the stability of cyclin D1 in a p38SAPK2-dependent manner. J. Biol. Chem. 275, 35091–35097 (2000).
Lavoie, J.N., L'Allemain, G., Brunet, A., Müller, R. & Pouysségur, J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J. Biol. Chem. 271, 20608–20616 (1996).
Goloudina, A. et al. Regulation of human Cdc25A stability by serine 75 phosphorylation is not sufficient to activate a S-phase checkpoint. Cell Cycle 2, 473–478 (2003).
Bulavin, D.V. et al. Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 411, 102–107 (2001).
Bulavin, D.V. et al. Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J. 18, 6845–6854 (1999).
Brancho, D. et al. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 17, 1969–1978 (2003).
Choi, J. et al. Mice deficient for the wild-type p53-induced phosphatase gene (Wip1) exhibit defects in reproductive organs, immune function, and cell cycle control. Mol. Cell. Biol. 22, 1094–1105 (2002).
Lowe, S.W., Jacks, T., Housman, D.E. & Ruley, H.E. Abrogation of oncogene-associated apoptosis allows transformation of p53-deficient cells. Proc. Natl. Acad. Sci. USA 91, 2026–2030 (1994).
Serrano, M. et al. Role of the INK4a locus in tumor suppression and cell mortality. Cell 85, 27–37 (1996).
Sherr, C.J. & Roberts, J.M. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 9, 1149–1163 (1995).
Kitagawa, M. et al. The consensus motif for phosphorylation by cyclin D1-Cdk4 is different from that for phosphorylation by cyclin A/E-Cdk2. EMBO J. 15, 7060–7069 (1996).
Bowe, D.B., Kenney, N.J., Adereth, Y. & Maroulakou, I.G. Suppression of Neu-induced mammary tumor growth in cyclin D1 deficient mice is compensated for by cyclin E. Oncogene 21, 291–298 (2002).
Yu, Q., Geng, Y. & Sicinski, P. Specific protection against breast cancers by cyclin D1 ablation. Nature 411, 1017–1021 (2001).
Rocco, J.W. & Sidransky, D. p16(MTS-1/CDKN2/INK4a) in cancer progression. Exp. Cell Res. 264, 42–55 (2001).
Lee, J.C., Kassis, S., Kumar, S., Badger, A. & Adams, J.L. p38 mitogen-activated protein kinase inhibitors—mechanisms and therapeutic potentials. Pharmacol. Ther. 82, 389–397 (1999).
Choi, J., Appella, E. & Donehower, L.A. The structure and expression of the murine wildtype p53-induced phosphatase 1 (Wip1) gene. Genomics 64, 298–306 (2000).
Dickson, C. et al. Amplification of chromosome band 11q13 and a role for cyclin D1 in human breast cancer. Cancer Lett. 90, 43–50 (1995).
Bartkova, J. et al. Cyclin D1 protein expression and function in human breast cancer. Int. J. Cancer 57, 353–361 (1994).
Gillett, C. et al. Amplification and overexpression of cyclin D1 in breast cancer detected by immunohistochemical staining. Cancer Res. 54, 1812–1817 (1994).
Bièche, I. & Lidereau, R. Genetic alterations in breast cancer. Genes Chromosomes Cancer 14, 227–251 (1995).
Nielsen, N.H., Roos, G., Emdin, S.O. & Landberg, G. Methylation of the p16Ink4a tumor suppressor gene 5′-CpG island in breast cancer. Cancer Lett. 163, 59–69 (2001).
Bulavin, D.V., Amundson, S.A. & Fornace, J.A.J. p38 and Chk1 kinases: different conductors for the G2/M checkpoint symphony. Curr. Opin. Genet. Dev. 12, 92–97 (2002).
Muller, W.J., Sinn, E., Pattengale, P.K., Wallace, R. & Leder, P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell 54, 105–115 (1988).
Sinn, E. et al. Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell 49, 465–475 (1987).
Tsukamoto, A.S., Grosschedl, R., Guzman, R.C., Parslow, T. & Varmus, H.E. Expression of the int-1 gene in transgenic mice is associated with mammary gland hyperplasia and adenocarcinomas in male and female mice. Cell 55, 619–625 (1988).
Sharpless, M.E. et al. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 413, 86–91 (2001).
Donehower, L.A. et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356, 215–221 (1992).
Herman, J.G., Graff, J.R., Myöhänen, S., Nelkin, B.D. & Baylin, S.B. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA 93, 9821–9826 (1996).
Bulavin, D.V., Tararova, N.D., Aksenov, N.D., Pospelov, V.A. & Pospelova, T.V. Deregulation of p53/p21Cip1/Waf1 pathway contributes to polyploidy and apoptosis of E1A+cHa-ras transformed cells after gamma-irradiation. Oncogene 18, 5611–5619 (1999).
Zhang, S. et al. p16INK4a gene promoter variation and differential binding of a repressor, the ras-responsive zinc-finger transcription factor, RREB. Oncogene 22, 2285–2295 (2003).
Robertson, K.D. & Jones, P.A. The human ARF cell cycle regulatory gene promoter is a CpG island which can be silenced by DNA methylation and down-regulated by wild-type p53. Mol. Cell. Biol. 18, 6457–6473 (1998).
Koch-Paiz, C.A., Momenan, R., Amundson, S.A., Lamoreaux, E. & Fornace, A.J. Jr. Estimation of relative mRNA content by filter hybridization to a polyuridylic probe. Biotechniques 29, 706, 708, 712, 714 (2000).
Acknowledgements
We thank C. Deng for discussions and comments, D. Medina for help with whole–mammary gland staining, P. Sicinski for pBabe-puro-NeuT, H. Varmus for MMTV-Wnt-1 transgenic mice and M. Roussel for p16-null and p16/p19-null MEFs. O.T. is a Ph.D. student at the Institute of Cytology, St. Petersburg, Russia; his fellowship was sponsored by the US National Institutes of Health Exchange Program. C.W.A. was supported in part by a US Army Breast Cancer Idea Award at the Brookhaven National Laboratory under contract with the US Department of Energy. B.N. was supported by a US Army Breast Cancer Research Training Grant.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
About this article
Cite this article
Bulavin, D., Phillips, C., Nannenga, B. et al. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK–mediated activation of the p16Ink4a-p19Arf pathway. Nat Genet 36, 343–350 (2004). https://doi.org/10.1038/ng1317
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng1317
This article is cited by
-
Ppm1d truncating mutations promote the development of genotoxic stress-induced AML
Leukemia (2023)
-
PGC-1α Affects Epileptic Seizures by Regulating Mitochondrial Fusion in Epileptic Rats
Neurochemical Research (2023)
-
Co-targeting WIP1 and PARP induces synthetic lethality in hepatocellular carcinoma
Cell Communication and Signaling (2022)
-
Demethyleneberberine, a potential therapeutic agent in neurodegenerative disorders: a proposed mechanistic insight
Molecular Biology Reports (2022)
-
Inhibition of the DNA damage response phosphatase PPM1D reprograms neutrophils to enhance anti-tumor immune responses
Nature Communications (2021)
