
Abstract
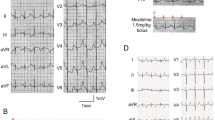
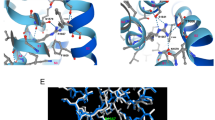
Deafness is genetically very heterogeneous and forms part of several syndromes. So far, delayed rectifier potassium channels have been linked to human deafness associated with prolongation of the QT interval on electrocardiograms and ventricular arrhythmia in Jervell and Lange-Nielsen syndrome. Cav1.3 voltage-gated L-type calcium channels (LTCCs) translate sound-induced depolarization into neurotransmitter release in auditory hair cells and control diastolic depolarization in the mouse sinoatrial node (SAN). Human deafness has not previously been linked to defects in LTCCs. We used positional cloning to identify a mutation in CACNA1D, which encodes the pore-forming α1 subunit of Cav1.3 LTCCs, in two consanguineous families with deafness. All deaf subjects showed pronounced SAN dysfunction at rest. The insertion of a glycine residue in a highly conserved, alternatively spliced region near the channel pore resulted in nonconducting calcium channels that had abnormal voltage-dependent gating. We describe a human channelopathy (termed SANDD syndrome, sinoatrial node dysfunction and deafness) with a cardiac and auditory phenotype that closely resembles that of Cacna1d−/− mice.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others

References
Hilgert, N., Smith, R.J. & Van Camp, G. Forty-six genes causing nonsyndromic hearing impairment: which ones should be analyzed in DNA diagnostics? Mutat. Res. 681, 189–196 (2009).
Cohen, M., Bitner-Glindzicz, M. & Luxon, L. The changing face of Usher syndrome: clinical implications. Int. J. Audiol. 46, 82–93 (2007).
Bitner-Glindzicz, M. & Tranebjaerg, L. The Jervell and Lange-Nielsen syndrome. Adv. Otorhinolaryngol. 56, 45–52 (2000).
Dou, H. et al. Null mutation of alpha1D Ca2+ channel gene results in deafness but no vestibular defect in mice. J. Assoc. Res. Otolaryngol. 5, 215–226 (2004).
Platzer, J. et al. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell 102, 89–97 (2000).
Fukushima, K. et al. An autosomal recessive nonsyndromic form of sensorineural hearing loss maps to 3p-DFNB6. Genome Res. 5, 305–308 (1995).
Woods, C.G. et al. Quantification of homozygosity in consanguineous individuals with autosomal recessive disease. Am. J. Hum. Genet. 78, 889–896 (2006).
Naz, S. et al. Mutations in a novel gene, TMIE, are associated with hearing loss linked to the DFNB6 locus. Am. J. Hum. Genet. 71, 632–636 (2002).
Sinnegger-Brauns, M.J. et al. Isoform-specific regulation of mood behavior and pancreatic beta cell and cardiovascular function by L-type Ca2+ channels. J. Clin. Invest. 113, 1430–1439 (2004).
Striessnig, J. & Koschak, A. Exploring the function and pharmacotherapeutic potential of voltage-gated Ca2+ channels with gene knockout models. Channels (Austin) 2, 233–251 (2008).
Seino, S. et al. Cloning of the alpha 1 subunit of a voltage-dependent calcium channel expressed in pancreatic beta cells. Proc. Natl. Acad. Sci. USA 89, 584–588 (1992).
Striessnig, J., Bolz, H.J. & Koschak, A. Channelopathies in Cav1.1, Cav1.3, and Cav1.4 voltage-gated L-type Ca2+ channels. Pflugers Arch. 460, 361–374 (2010).
Hoda, J.C., Zaghetto, F., Koschak, A. & Striessnig, J. Congenital stationary night blindness type 2 mutations S229P, G369D, L1068P, and W1440X alter channel gating or functional expression of Cav1.4 L-type Ca2+ channels. J. Neurosci. 25, 252–259 (2005).
Splawski, I. et al. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc. Natl. Acad. Sci. USA 102, 8089–8096, discussion 8086–8088 (2005).
McDonough, S.I., Mori, Y. & Bean, B.P. FPL 64176 modification of Cav1.2 L-type calcium channels: dissociation of effects on ionic current and gating current. Biophys. J. 88, 211–223 (2005).
Jones, L.P., Patil, P.G., Snutch, T.P. & Yue, D.T. G-protein modulation of N-type calcium channel gating current in human embryonic kidney cells (HEK 293). J. Physiol. (Lond.) 498, 601–610 (1997).
Bangalore, R., Mehrke, G., Gingrich, K., Hofmann, F. & Kass, R.S. Influence of L-type Ca channel alpha 2/delta-subunit on ionic and gating current in transiently transfected HEK 293 cells. Am. J. Physiol. 270, H1521–H1528 (1996).
Kamp, T.J., Perez-Garcia, M.T. & Marban, E. Enhancement of ionic current and charge movement by coexpression of calcium channel beta 1A subunit with alpha 1C subunit in a human embryonic kidney cell line. J. Physiol. (Lond.) 492, 89–96 (1996).
Koschak, A. et al. Alpha 1D Cav1.3 subunits can form l-type Ca2+ channels activating at negative voltages. J. Biol. Chem. 276, 22100–22106 (2001).
Mangoni, M.E. et al. Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc. Natl. Acad. Sci. USA 100, 5543–5548 (2003).
Neyroud, N. et al. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat. Genet. 15, 186–189 (1997).
Splawski, I., Timothy, K.W., Vincent, G.M., Atkinson, D.L. & Keating, M.T. Molecular basis of the long-QT syndrome associated with deafness. N. Engl. J. Med. 336, 1562–1567 (1997).
Tyson, J. et al. IsK and KvLQT1: mutation in either of the two subunits of the slow component of the delayed rectifier potassium channel can cause Jervell and Lange-Nielsen syndrome. Hum. Mol. Genet. 6, 2179–2185 (1997).
Busquet, P. et al. CaV1.3 L-type Ca2+ channels modulate depression-like behaviour in mice independent of deaf phenotype. Int. J. Neuropsychopharmacol. 13, 499–513 (2010).
Chan, C.S. et al. 'Rejuvenation' protects neurons in mouse models of Parkinson's disease. Nature 447, 1081–1086 (2007).
Guzman, J.N., Sanchez-Padilla, J., Chan, C.S. & Surmeier, D.J. Robust pacemaking in substantia nigra dopaminergic neurons. J. Neurosci. 29, 11011–11019 (2009).
McKinney, B.C. & Murphy, G.G. The L-type voltage-gated calcium channel Cav1.3 mediates consolidation, but not extinction, of contextually conditioned fear in mice. Learn. Mem. 13, 584–589 (2006).
Singh, A. et al. Modulation of voltage- and Ca2+-dependent gating of Cav1.3 L-type calcium channels by alternative splicing of a C-terminal regulatory domain. J. Biol. Chem. 283, 20733–20744 (2008).
Olson, P.A. et al. G protein–coupled receptor modulation of striatal Cav1.3 L-type Ca2+ channels is dependent on a Shank-binding domain. J. Neurosci. 25, 1050–1062 (2005).
Mangoni, M.E. & Nargeot, J. Genesis and regulation of the heart automaticity. Physiol. Rev. 88, 919–982 (2008).
Lakatta, E.G., Maltsev, V.A. & Vinogradova, T.M. A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart's pacemaker. Circ. Res. 106, 659–673 (2010).
Splawski, I. et al. Cav1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31 (2004).
Raybaud, A. et al. The role of the GX9GX3G motif in the gating of high voltage-activated Ca2+ channels. J. Biol. Chem. 281, 39424–39436 (2006).
Long, S.B., Campbell, E.B. & Mackinnon, R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 309, 897–903 (2005).
Stary, A., Shafrir, Y., Hering, S., Wolschann, P. & Guy, H.R. Structural model of the Cav1.2 pore. Channels (Austin) 2, 210–215 (2008).
Abecasis, G.R., Cherny, S.S., Cookson, W.O. & Cardon, L.R. GRR: graphical representation of relationship errors. Bioinformatics 17, 742–743 (2001).
O'Connell, J.R. & Weeks, D.E. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am. J. Hum. Genet. 63, 259–266 (1998).
Abecasis, G.R., Cherny, S.S., Cookson, W.O. & Cardon, L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 30, 97–101 (2002).
Gudbjartsson, D.F., Jonasson, K., Frigge, M.L. & Kong, A. Allegro, a new computer program for multipoint linkage analysis. Nat. Genet. 25, 12–13 (2000).
Rüschendorf, F. & Nurnberg, P. ALOHOMORA: a tool for linkage analysis using 10K SNP array data. Bioinformatics 21, 2123–2125 (2005).
Thiele, H. & Nurnberg, P. HaploPainter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics 21, 1730–1732 (2005).
Singh, A. et al. C-terminal modulator controls Ca2+-dependent gating of Cav1.4 L-type Ca2+ channels. Nat. Neurosci. 9, 1108–1116 (2006).
Safayhi, H. et al. L-type calcium channels in insulin-secreting cells: biochemical characterization and phosphorylation in RINm5F cells. Mol. Endocrinol. 11, 619–629 (1997).
Brandt, N. et al. Thyroid hormone deficiency affects postnatal spiking activity and expression of Ca2+ and K+ channels in rodent inner hair cells. J. Neurosci. 27, 3174–3186 (2007).
Knirsch, M. et al. Persistence of Cav1.3 Ca2+ channels in mature outer hair cells supports outer hair cell afferent signaling. J. Neurosci. 27, 6442–6451 (2007).
Brandt, A., Khimich, D. & Moser, T. Few Cav1.3 channels regulate the exocytosis of a synaptic vesicle at the hair cell ribbon synapse. J. Neurosci. 25, 11577–11585 (2005).
Acknowledgements
We thank the families who have participated in this study, K. Beam and W. Sandtner for discussion on gating currents, M. Thoenes, K. Zimmermann, S. Blick, G. Gajic and S. Kasperek for technical assistance, B. Ali for help with ECG recordings, K. Boss for discussion of the manuscript and C. Striessnig for artwork. This work was supported by the Geers-Stiftung, Bonn; Imhoff-Stiftung, Köln; Köln Fortune, University Hospital of Cologne, Deutsche Forschungsgemeinschaft (BO2954/1‐2); Forschung contra Blindheit: Initiative Usher‐Syndrom e.V. (to H.J.B.); the Austrian Science Fund (P-20670); the Agence Nationale pour la Recherche (ANR-06-PHYSIO-004-01 to M.E.M.); the Fondation de France; the Marie Curie Research Training Network CavNET (MRTN-CT-2006-035367); and the University of Innsbruck (to J.S. and A.K.).
Author information
Authors and Affiliations
Contributions
S.M.B. coordinated and conducted the identification of families with deafness and collection of DNA samples and clinical data. A.A., I.A. and M.F. arranged clinical investigations. H.U.K. facilitated and conducted most of the clinical investigations at Khyber Teaching Hospital, Peshawar. G.N. and P.N. performed genetic mapping. C.D. performed molecular genetic analyses. A.K. and A.L. performed electrophysiological analyses. M.G. cloned wild-type and mutant channels and performed PCR and western blot analysis. M.J.S.-B. performed PCR analysis. N.B., J.E. and M.E.M. provided IHC and SAN tissues. A.K. and J.S. coordinated experiments and wrote the manuscript. H.J.B. initiated, planned and coordinated the study and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–3 (PDF 1198 kb)
Rights and permissions
About this article
Cite this article
Baig, S., Koschak, A., Lieb, A. et al. Loss of Cav1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat Neurosci 14, 77–84 (2011). https://doi.org/10.1038/nn.2694
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nn.2694
This article is cited by
-
Pathogenicity of de novo CACNA1D Ca2+ channel variants predicted from sequence co-variation
European Journal of Human Genetics (2024)
-
Characterization of sinoatrial automaticity in Microcebus murinus to study the effect of aging on cardiac activity and the correlation with longevity
Scientific Reports (2023)
-
Role of Ca2+ in healthy and pathologic cardiac function: from normal excitation–contraction coupling to mutations that cause inherited arrhythmia
Archives of Toxicology (2023)
-
Candidate Key Proteins in Tinnitus: A Bioinformatic Study of Synaptic Transmission in Spiral Ganglion Neurons
Cellular and Molecular Neurobiology (2023)
-
Slowing down as we age: aging of the cardiac pacemaker’s neural control
GeroScience (2022)
