
Key Points
-
Cerebral deposition of the amyloid-β peptide (Aβ) in the form of plaques is a defining feature of Alzheimer's disease (AD). The production and aggregation of Aβ does not seem to be simply an epiphenomenon, but is directly implicated in the aetiology of AD. Therefore, decreasing Aβ formation, aggregation and downstream toxic events are all reasonable therapeutic goals.
-
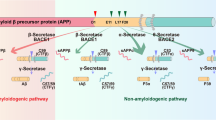
Aβ is produced from the amyloid-β precursor protein (APP) through the action of two proteases, β- and γ-secretase. Both of these proteases are considered important therapeutic targets. The gene that encodes β-secretase can be knocked out without any apparent effect, whereas γ-secretase is crucial for the normal development of the organism. Nevertheless, γ-secretase inhibitors have been reported to lower amyloid levels and reduce brain amyloid deposition in transgenic mice without signs of overt toxicity.
-
The toxic form of Aβ might be soluble oligomers. Therefore, agents that prevent Aβ nucleation could be more effective than those that merely block Aβ deposition. Peptide analogues have been identified that block Aβ nucleation in vitro, one of which prevents amyloid deposition and seems to reduce pathology in transgenic mice.
-
Anti-Aβ immunization, either active or passive, also prevents Aβ plaque formation and the associated pathology in transgenic mice. The first clinical trial for active immunization was cut short because of unacceptable central nervous system (CNS) inflammatory reactions in a small fraction of the subjects. However, modification of this protocol might circumvent this problem.
-
Epidemiological studies indicate that cholesterol-lowering drugs might prevent AD. These agents apparently reduce Aβ production by altering the ability of the secretases to cleave APP. Such findings offer great hope, as these drugs are known to be safe over long periods of time.
-
Other potential strategies for lowering Aβ levels include reducing APP expression at the promoter level and increasing Aβ clearance through the upregulation of Aβ-degrading proteases. Other efforts include developing agents that affect mediators that are downstream of Aβ, such as inhibition of TAU phosphorylation and treatment with antioxidants or anti-inflammatory agents.
Abstract
Alzheimer's disease is a progressive and ultimately fatal neurological disorder for which there is no effective treatment at present. The disease is characterized pathologically by cerebral plaques that contain the amyloid-β peptide and thread-like neuronal structures composed of the microtubule-associated protein TAU. Both amyloid-β and TAU are thought to be crucial to pathogenesis, but compelling evidence supports amyloid-β as the 'prime mover'. The main efforts for developing therapeutics are therefore focused on preventing amyloid-β production, aggregation or downstream neurotoxic events. The progress of these and other approaches raises the hope that effective agents for the prevention and treatment of Alzheimer's disease will be available in the near future.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others
References
Hardy, J. & Selkoe, D. J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 (2002). A recent review that highlights the considerable body of evidence in favour of the amyloid hypothesis.
Esler, W. P. & Wolfe, M. S. A portrait of Alzheimer secretases — new features and familiar faces. Science 293, 1449–1454 (2001). A detailed discussion of the biology of α-, β- and γ-secretase.
Goate, A. et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature 349, 704–706 (1991).
Chartier-Harlin, M. C. et al. Early-onset Alzheimer's disease caused by mutations at codon 717 of the β-amyloid precursor protein gene. Nature 353, 844–846 (1991).
Murrell, J., Farlow, M., Ghetti, B. & Benson, M. D. A mutation in the amyloid precursor protein associated with hereditary Alzheimer's disease. Science 254, 97–99 (1991). References 3–5 show that missense mutations in APP near the Aβ region cause AD in certain families. This was the first genetic evidence in favour of the amyloid hypothesis.
Selkoe, D. J. Cell biology of the amyloid-β protein precursor and the mechanism of Alzheimer's disease. Annu. Rev. Cell Biol. 10, 373–403 (1994).
Suzuki, N. et al. An increased percentage of long amyloid-β protein secreted by familial amyloid-β protein precursor (β-APP717) mutants. Science 264, 1336–1340 (1994).
Eckman, C. B. et al. A new pathogenic mutation in the APP gene (I716V) increases the relative proportion of Aβ42(43). Hum. Mol. Genet. 6, 2087–2089 (1997).
Sherrington, R. et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature 375, 754–760 (1995).
Levy-Lahad, E. et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science 269, 973–977 (1995).
Rogaev, E. I. et al. Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature 376, 775–778 (1995). References 9–11 report the seminal observation that missense mutations in the presenilins can cause familial AD.
Scheuner, D. et al. Secreted amyloid-β protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nature Med. 2, 864–870 (1996).
Duff, K. et al. Increased amyloid-β42(43) in brains of mice expressing mutant presenilin 1. Nature 383, 710–713 (1996).
Lemere, C. A. et al. The E280A presenilin 1 Alzheimer mutation produces increased Aβ42 deposition and severe cerebellar pathology. Nature Med. 2, 1146–1150 (1996).
Citron, M. et al. Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid-β protein in both transfected cells and transgenic mice. Nature Med. 3, 67–72 (1997).
Tomita, T. et al. The presenilin 2 mutation (N141I) linked to familial Alzheimer disease (Volga German families) increases the secretion of amyloid-β protein ending at the 42nd (or 43rd) residue. Proc. Natl Acad. Sci. USA 94, 2025–2030 (1997).
Borchelt, D. R. et al. Familial Alzheimer's disease-linked presenilin 1 variants elevate Aβ1–42/1–40 ratio in vitro and in vivo. Neuron 17, 1005–1013 (1996).
Wolfe, M. S. Secretase targets for Alzheimer's disease: identification and therapeutic potential. J. Med. Chem. 44, 2039–2060 (2001). A comprehensive review of the biology of the secretases and the small molecules that modulate their activities.
Vassar, R. et al. β-Secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741 (1999).
Sinha, S. et al. Purification and cloning of amyloid precursor protein β-secretase from human brain. Nature 402, 537–540 (1999).
Yan, R. et al. Membrane-anchored aspartyl protease with Alzheimer's disease β-secretase activity. Nature 402, 533–537 (1999).
Hussain, I. et al. Identification of a novel aspartic protease (ASP2) as β-secretase. Mol. Cell. Neurosci. 14, 419–427 (1999). References 19–22 are seminal papers that describe the discovery of β-secretase.
Hong, L. et al. Structure of the protease domain of memapsin 2 (β-secretase) complexed with inhibitor. Science 290, 150–153 (2000). A description of the crystal structure of β-secretase, paving the way for structure-based design of inhibitors.
Ghosh, A. K. et al. Structure-based design: potent inhibitors of human brain memapsin 2 (β-secretase). J. Med. Chem. 44, 2865–2868 (2001).
Kitazume, S. et al. Alzheimer's β-secretase, β-site amyloid precursor protein-cleaving enzyme, is responsible for cleavage secretion of a Golgi-resident sialyltransferase. Proc. Natl Acad. Sci. USA 98, 13554–13559 (2001).
Luo, Y. et al. Mice deficient in BACE1, the Alzheimer's β-secretase, have normal phenotype and abolished β-amyloid generation. Nature Neurosci. 4, 231–232 (2001).
Cai, H. et al. BACE1 is the major β-secretase for generation of Aβ peptides by neurons. Nature Neurosci. 4, 233–234 (2001).
Wolfe, M. S. et al. Peptidomimetic probes and molecular modeling suggest Alzheimer's γ-secretases are intramembrane-cleaving aspartyl proteases. Biochemistry 38, 4720–4727 (1999).
Shearman, M. S. et al. L-685,458, an aspartyl protease transition state mimic, is a potent inhibitor of amyloid-β protein precursor γ-secretase activity. Biochemistry 39, 8698–8704 (2000).
Herreman, A. et al. Total inactivation of γ-secretase activity in presenilin-deficient embryonic stem cells. Nature Cell Biol. 2, 461–462 (2000).
Zhang, Z. et al. Presenilins are required for γ-secretase cleavage of β-APP and transmembrane cleavage of Notch-1. Nature Cell Biol. 2, 463–465 (2000).
Wolfe, M. S. et al. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 398, 513–517 (1999). The first suggestion that presenilin could be a novel aspartyl protease.
Yu, G. et al. The presenilin 1 protein is a component of a high molecular weight intracellular complex that contains β-catenin. J. Biol. Chem. 273, 16470–16475 (1998).
Thinakaran, G. et al. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 17, 181–190 (1996). The seminal observation that presenilin is cleaved into two metabolically stable pieces.
Capell, A. et al. The proteolytic fragments of the Alzheimer's disease-associated presenilin-1 form heterodimers and occur as a 100–150-kDa molecular mass complex. J. Biol. Chem. 273, 3205–3211 (1998).
Ratovitski, T. et al. Endoproteolytic processing and stabilization of wild-type and mutant presenilin. J. Biol. Chem. 272, 24536–24541 (1997).
Steiner, H. et al. Expression of Alzheimer's disease-associated presenilin-1 is controlled by proteolytic degradation and complex formation. J. Biol. Chem. 273, 32322–32331 (1998).
Thinakaran, G. et al. Evidence that levels of presenilins (PS1 and PS2) are coordinately regulated by competition for limiting cellular factors. J. Biol. Chem. 272, 28415–28422 (1997).
Li, Y. M. et al. Photoactivated γ-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 405, 689–694 (2000).
Esler, W. P. et al. Transition-state analogue inhibitors of γ-secretase bind directly to presenilin-1. Nature Cell Biol. 2, 428–434 (2000). References 39 and 40 provided the first direct biochemical evidence that the active site of γ-secretase resides in presenilin.
Li, Y. M. et al. Presenilin 1 is linked with γ-secretase activity in the detergent solubilized state. Proc. Natl Acad. Sci. USA 97, 6138–6143 (2000).
Esler, W. P. et al. Activity-dependent isolation of the presenilin/γ-secretase complex reveals nicastrin and a γ substrate. Proc. Natl Acad. Sci. USA 99, 2720–2725 (2002).
Yu, G. et al. Nicastrin modulates presenilin-mediated Notch/Glp-1 signal transduction and β-APP processing. Nature 407, 48–54 (2000). The discovery of the presenilin-associated protein nicastrin and its role in γ-secretase activity.
Goutte, C., Tsunozaki, M., Hale, V. A. & Priess, J. R. APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc. Natl Acad. Sci. USA 99, 775–779 (2002).
Francis, R. et al. APH-1 and PEN-2 are required for Notch pathway signaling, γ-secretase cleavage of β-APP, and presenilin protein accumulation. Dev. Cell 3, 85–97 (2002). References 44 and 45 describe two new proteins that affect γ-secretase activity.
Seiffert, D. et al. Presenilin-1 and -2 are molecular targets for γ-secretase inhibitors. J. Biol. Chem. 275, 34086–34091 (2000).
Dovey, H. F. et al. Functional γ-secretase inhibitors reduce β-amyloid peptide levels in brain. J. Neurochem. 76, 173–181 (2001).
Hadland, B. K. et al. γ-Secretase inhibitors repress thymocyte development. Proc. Natl Acad. Sci. USA 98, 7487–7491 (2001).
Doerfler, P., Shearman, M. S. & Perlmutter, R. M. Presenilin-dependent γ-secretase activity modulates thymocyte development. Proc. Natl Acad. Sci. USA 98, 9312–9317 (2001).
Weggen, S. et al. A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 414, 212–216 (2001).
Hung, A. Y. et al. Activation of protein kinase C inhibits cellular production of the amyloid β-protein. J. Biol. Chem. 268, 22959–22962 (1993).
Buxbaum, J. D., Koo, E. H. & Greengard, P. Protein phosphorylation inhibits production of Alzheimer amyloid β/A4 peptide. Proc. Natl Acad. Sci. USA 90, 9195–9198 (1993).
Felsenstein, K. M., Ingalls, K. M., Hunihan, L. W. & Roberts, S. B. Reversal of the Swedish familial Alzheimer's disease mutant phenotype in cultured cells treated with phorbol 12,13-dibutyrate. Neurosci. Lett. 174, 173–176 (1994).
Jacobsen, J. S. et al. The release of Alzheimer's disease β-amyloid peptide is reduced by phorbol treatment. J. Biol. Chem. 269, 8376–8382 (1994).
Nitsch, R. M., Slack, B. E., Wurtman, R. J. & Growdon, J. H. Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science 258, 304–307 (1992). The first demonstration that muscarinic agents can modulate APP processing.
Haring, R. et al. Amyloid precursor protein secretion via muscarinic receptors: reduced desensitization using the M1-selective agonist AF102B. Biochem. Biophys. Res. Commun. 203, 652–658 (1994).
Wolf, B. A. et al. Muscarinic regulation of Alzheimer's disease amyloid precursor protein secretion and amyloid β-protein production in human neuronal NT2N cells. J. Biol. Chem. 270, 4916–4922 (1995).
Lin, L., Georgievska, B., Mattsson, A. & Isacson, O. Cognitive changes and modified processing of amyloid precursor protein in the cortical and hippocampal system after cholinergic synapse loss and muscarinic receptor activation. Proc. Natl Acad. Sci. USA 96, 12108–12113 (1999).
Corder, E. H. et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261, 921–923 (1993). An influential paper that reports the first major genetic risk factor for AD.
Corder, E. H. et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nature Genet. 7, 180–184 (1994).
Kivipelto, M. et al. Midlife vascular risk factors and Alzheimer's disease in later life: longitudinal, population based study. BMJ. 322, 1447–1451 (2001).
Wolozin, B., Kellman, W., Ruosseau, P., Celesia, G. G. & Siegel, G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch. Neurol. 57, 1439–1443 (2000).
Jick, H., Zornberg, G. L., Jick, S. S., Seshadri, S. & Drachman, D. A. Statins and the risk of dementia. Lancet 356, 1627–1631 (2000).
Sparks, D. L. et al. Induction of Alzheimer-like β-amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Exp. Neurol. 126, 88–94 (1994).
Simons, M. et al. Cholesterol depletion inhibits the generation of β-amyloid in hippocampal neurons. Proc. Natl Acad. Sci. USA 95, 6460–6464 (1998).
Fassbender, K. et al. Simvastatin strongly reduces levels of Alzheimer's disease β-amyloid peptides Aβ42 and Aβ40 in vitro and in vivo. Proc. Natl Acad. Sci. USA 98, 5856–5861 (2001).
Kojro, E., Gimpl, G., Lammich, S., Marz, W. & Fahrenholz, F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the α-secretase ADAM10. Proc. Natl Acad. Sci. USA 98, 5815–5820 (2001).
Puglielli, L. et al. Acyl-coenzyme A:cholesterol acyltransferase modulates the generation of the amyloid β-peptide. Nature Cell Biol. 3, 905–912 (2001).
Pike, C. J., Burdick, D., Walencewicz, A. J., Glabe, C. G. & Cotman, C. W. Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J. Neurosci. 13, 1676–1687 (1993).
Lorenzo, A. & Yankner, B. A. β-Amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc. Natl Acad. Sci. USA 91, 12243–12247 (1994).
Hartley, D. M. et al. Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J. Neurosci. 19, 8876–8884 (1999).
Jarrett, J. T., Berger, E. P. & Lansbury, P. T. Jr. The carboxy terminus of the β-amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry 32, 4693–4697 (1993). A seminal paper that provides biochemical evidence that Aβ 42 is highly prone to fibril formation, complementary to genetic and pathological evidence that this Aβ variant is particularly implicated in AD.
Harper, J. D., Wong, S. S., Lieber, C. M. & Lansbury, P. T. Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chem. Biol. 4, 119–125 (1997).
Walsh, D. M., Lomakin, A., Benedek, G. B., Condron, M. M. & Teplow, D. B. Amyloid-β protein fibrillogenesis. Detection of a protofibrillar intermediate. J. Biol. Chem. 272, 22364–22372 (1997).
Walsh, D. M. et al. Naturally secreted oligomers of amyloid-β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539 (2002). The first evidence that the toxic species of Aβ might be soluble oligomers and not necessarily fibrils.
Findeis, M. A. Peptide inhibitors of β-amyloid aggregation. Curr. Top. Med. Chem. 2, 417–423 (2002).
Permanne, B. et al. Reduction of amyloid load and cerebral damage in a transgenic mouse model of Alzheimer's disease by treatment with a β-sheet breaker peptide. FASEB J. 16, 860–862 (2002).
Bush, A. I. & Tanzi, R. E. The galvanization of β-amyloid in Alzheimer's disease. Proc. Natl Acad. Sci. USA 99, 7317–7319 (2002). A recent commentary on the role of zinc in Aβ deposition and toxicity and potential therapeutic implications.
Cherny, R. A. et al. Treatment with a copper–zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron 30, 665–676 (2001).
Lee, J. Y., Cole, T. B., Palmiter, R. D., Suh, S. W. & Koh, J. Y. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc. Natl Acad. Sci. USA 99, 7705–7710 (2002).
Schenk, D. et al. Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400, 173–177 (1999). The first report to show that immunization with Aβ reduced brain Aβ plaque formation in transgenic mice.
Weiner, H. L. et al. Nasal administration of amyloid-β peptide decreases cerebral amyloid burden in a mouse model of Alzheimer's disease. Ann. Neurol. 48, 567–579 (2000).
Morgan, D. et al. Aβ-peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature 408, 982–985 (2000).
Janus, C. et al. Aβ-peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature 408, 979–982 (2000).
Bard, F. et al. Peripherally administered antibodies against amyloid-β peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nature Med. 6, 916–919 (2000).
DeMattos, R. B. et al. Peripheral anti-Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer's disease. Proc. Natl Acad. Sci. USA 98, 8850–8855 (2001).
Dodart, J. C. et al. Immunization reverses memory deficits without reducing brain Aβ burden in Alzheimer's disease model. Nature Neurosci. 5, 452–427 (2002).
Rogers, J. T. et al. Alzheimer's disease drug discovery targeted to the APP mRNA 5′ untranslated region. J. Mol. Neurosci. 19, 77–82 (2002).
Rogers, J. T. et al. An iron-responsive element type II in the 5′ untranslated region of the Alzheimer's amyloid precursor protein transcript. J. Biol. Chem. 2002 Aug 26 (doi: 10.1074/jbc.M207435200).
Selkoe, D. J. Clearing the brain's amyloid cobwebs. Neuron 32, 177–180 (2001).
Pepys, M. B. et al. Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nature 417, 254–259 (2002).
Edbauer, D., Winkler, E., Haass, C. & Steiner, H. Presenilin and nicastrin regulate each other and determine amyloid beta-peptide production via complex formation. Proc. Natl Acad Sci USA 99, 8666–8671 (2002).
Yang, D. S. et al. Mature glycosylation and trafficking of nicastrin modulate its binding to presinilins. J. Biol. Chem. 277, 28135–28142 (2002).
Leem, J. Y. et al. Presenilin 1 is required for maturation and cell surface accumulation of nicastrin. J. Biol. Chem. 277, 19236–19240 (2002).
Kimberly, W. T. et al. Complex N-linked glycosylated nicastrin associates with active g-secretase and undergoes tight cellular regulation. J. Biol. Chem. 277, 35113–35117 (2002).
Wahrle, S. et al. Cholesterol-dependent γ-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol. Dis. 9, 11–23 (2002).
Acknowledgements
M. S. W. is supported by grants from the National Institutes of Health and the Alzheimer's Association.
Author information
Authors and Affiliations
Related links
Related links
DATABASES
LocusLink
OMIM
FURTHER INFORMATION
ELS
Glossary
- HYPERPHOSPHORYLATION
-
The addition of more phosphate groups to a protein than is typically observed under normal physiological conditions.
- AGGREGATION
-
The association between large biomolecules that leads to clumping and precipitation.
- MISSENSE MUTATION
-
An alteration in the nucleotide sequence of a gene that changes a single amino-acid residue for another in the encoded protein.
- TRANSITION-STATE MIMIC
-
A small, organic molecule that resembles an intermediate of enzyme catalysis, endowing the compound with inhibitory effects on the enzyme.
- NON-STEROIDAL ANTI-INFLAMMATORY DRUGS
-
(NSAIDs). Drugs that prevent or reduce inflammation, but which work by different mechanisms from steroids, such as hydrocortisone.
- PHORBOL ESTERS
-
Natural products that have a distinct, complex carbon skeleton, many of which activate protein kinase C.
- COGNITIVE DYSFUNCTION
-
An impaired ability to remember and learn.
Rights and permissions
About this article
Cite this article
Wolfe, M. Therapeutic strategies for Alzheimer's disease. Nat Rev Drug Discov 1, 859–866 (2002). https://doi.org/10.1038/nrd938
Issue Date:
DOI: https://doi.org/10.1038/nrd938
This article is cited by
-
Lipid-Based Nanocarriers via Nose-to-Brain Pathway for Central Nervous System Disorders
Neurochemical Research (2022)
-
Cryptotanshinione Inhibits β-Amyloid Aggregation and Protects Damage from β-Amyloid in SH-SY5Y Cells
Neurochemical Research (2012)
-
Reduction of β-Amyloid Deposits by γ-Secretase Inhibitor is Associated with the Attenuation of Secondary Damage in the Ipsilateral Thalamus and Sensory Functional Improvement after Focal Cortical Infarction in Hypertensive Rats
Journal of Cerebral Blood Flow & Metabolism (2011)
-
Development of cognitive enhancers based on inhibition of insulin-regulated aminopeptidase
BMC Neuroscience (2008)
-
Present and future drug treatments for chronic kidney diseases: evolving targets in renoprotection
Nature Reviews Drug Discovery (2008)
