
Key Points
-
Physiological hypertrophy is an adaptive form of cardiac hypertrophy that does not lead to heart disease in healthy individuals.
-
This cardiac growth maintains or augments cardiac function and increases angiogenesis and metabolism. Physiological hypertrophy does not promote fibrotic remodelling or cardiomyocyte death.
-
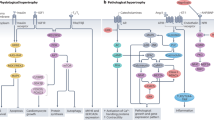
Physiological hypertrophy is initiated by specific hormones (triiodothyronine, insulin, insulin-like growth factor 1 and vascular endothelial growth factor) or stretch (loading), which activate a restricted number of intracellular signalling pathways (PI3K, AKT, mTOR and ERK1/2).
-
Metabolic reprogramming governed by AMP-activated protein kinase (AMPK) is essential for adaptive cardiac hypertrophy.
-
Exercise induced hypertrophy causes a downregulation of CCAAT/enhancer binding protein-β and the transcription of an exercise-specific gene set.
-
Intermittent PI3K, AKT, ERK1/2 or AMPK activation promotes the activation of a physiological hypertrophic programme to maintain or augment function, thus antagonizing pathological conditions.
Abstract
The heart hypertrophies in response to developmental signals as well as increased workload. Although adult-onset hypertrophy can ultimately lead to disease, cardiac hypertrophy is not necessarily maladaptive and can even be beneficial. Progress has been made in our understanding of the structural and molecular characteristics of physiological cardiac hypertrophy, as well as of the endocrine effectors and associated signalling pathways that regulate it. Physiological hypertrophy is initiated by finite signals, which include growth hormones (such as thyroid hormone, insulin, insulin-like growth factor 1 and vascular endothelial growth factor) and mechanical forces that converge on a limited number of intracellular signalling pathways (such as PI3K, AKT, AMP-activated protein kinase and mTOR) to affect gene transcription, protein translation and metabolism. Harnessing adaptive signalling mediators to reinvigorate the diseased heart could have important medical ramifications.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$189.00 per year
only $15.75 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others
References
Bernardo, B. C., Weeks, K. L., Pretorius, L. & McMullen, J. R. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol. Ther. 128, 191–227 (2010).
Heineke, J. & Molkentin, J. D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nature Rev. Mol. Cell Biol. 7, 589–600 (2006).
Van Berlo, J. H., Maillet, M. & Molkentin, J. D. Signaling effectors underlying pathologic growth and remodeling of the heart. J. Clin. Invest. (in the press).
Bergmann, O. et al. Evidence for cardiomyocyte renewal in humans. Science 324, 98–102 (2009).
Kajstura, J. et al. Cardiomyogenesis in the aging and failing human heart. Circulation 126, 1869–1881 (2012).
Dorn, G. W. The fuzzy logic of physiological cardiac hypertrophy. Hypertension 49, 962–970 (2007).
Porrello, E. R., Widdop, R. E. & Delbridge, L. M. Early origins of cardiac hypertrophy: does cardiomyocyte attrition programme for pathological 'catch-up' growth of the heart? Clin. Exp. Pharmacol. Physiol. 35, 1358–1364 (2008).
Hopkins, S. F. Jr, McCutcheon, E. P. & Wekstein, D. R. Postnatal changes in rat ventricular function. Circ. Res. 32, 685–691 (1973).
Clubb, F. J. Jr & Bishop, S. P. Formation of binucleated myocardial cells in the neonatal rat. An index for growth hypertrophy. Lab. Invest. 50, 571–577 (1984).
Eghbali, M., Wang, Y., Toro, L. & Stefani, E. Heart hypertrophy during pregnancy: a better functioning heart? Trends Cardiovasc. Med. 16, 285–291 (2006).
Winsor, T. & Beckner, G. Hypertrophy of the heart; electrocardiographic distinction between physiologic and pathologic enlargement. Calif. Med. 82, 151–158 (1955).
Henschen, S. Skilanglauf and skiwettlauf: ein medizinische sportstudie. Mitt. Med. Klin. Uppsala 2, 15–18 (1899).
Nishimura, T., Yamada, Y. & Kawai, C. Echocardiographic evaluation of long-term effects of exercise on left ventricular hypertrophy and function in professional bicyclists. Circulation 61, 832–840 (1980).
Sugishita, Y., Koseki, S., Matsuda, M., Yamaguchi, T. & Ito, I. Myocardial mechanics of athletic hearts in comparison with diseased hearts. Am. Heart J. 105, 273–280 (1983).
Dickhuth, H. H., Reindell, H., Lehmann, M. & Keul, J. [Capacity for regression of the athletic heart]. Z. Kardiol. 74 (Suppl. 7), 135–143 (1985).
Schannwell, C. M. et al. Left ventricular hypertrophy and diastolic dysfunction in healthy pregnant women. Cardiology 97, 73–78 (2002).
Janz, K. F., Dawson, J. D. & Mahoney, L. T. Predicting heart growth during puberty: The Muscatine Study. Pediatrics 105, e63 (2000).
Hew, K. W. & Keller, K. A. Postnatal anatomical and functional development of the heart: a species comparison. Birth Defects Res. B. Dev. Reprod. Toxicol. 68, 309–320 (2003).
Pluim, B. M., Zwinderman, A. H., van der Laarse, A. & van der Wall, E. E. The athlete's heart. A meta-analysis of cardiac structure and function. Circulation 101, 336–344 (2000).
Ehsani, A. A., Hagberg, J. M. & Hickson, R. C. Rapid changes in left ventricular dimensions and mass in response to physical conditioning and deconditioning. Am. J. Cardiol. 42, 52–56 (1978).
Maron, B. J., Pelliccia, A., Spataro, A. & Granata, M. Reduction in left ventricular wall thickness after deconditioning in highly trained Olympic athletes. Br. Heart J. 69, 125–128 (1993).
Sarquella-Brugada, G. et al. Genetics of sudden cardiac death in children and young athletes. Cardiol. Young 24, 1–15 (2012).
Patten, I. S. et al. Cardiac angiogenic imbalance leads to peripartum cardiomyopathy. Nature 485, 333–338 (2012).
Heiss, H. W. et al. Studies on the regulation of myocardial blood flow in man. I.: Training effects on blood flow and metabolism of the healthy heart at rest and during standardized heavy exercise. Bas. Res. Cardiol. 71, 658–675 (1976).
Pelliccia, A. et al. Coronary arteries in physiological hypertrophy: echocardiographic evidence of increased proximal size in elite athletes. Int. J. Sports Med. 11, 120–126 (1990).
Laughlin, M. H., Bowles, D. K. & Duncker, D. J. The coronary circulation in exercise training. Am. J. Physiol. Heart Circ. Physiol. 302, H10–H23 (2012).
Lopaschuk, G. D. & Jaswal, J. S. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J. Cardiovasc. Pharmacol. 56, 130–140 (2010).
Gertz, E. W., Wisneski, J. A., Stanley, W. C. & Neese, R. A. Myocardial substrate utilization during exercise in humans. Dual carbon-labeled carbohydrate isotope experiments. J. Clin. Invest. 82, 2017–2025 (1988).
Abel, E. D. & Doenst, T. Mitochondrial adaptations to physiological versus pathological cardiac hypertrophy. Cardiovasc. Res. 90, 234–242 (2011). In-depth review of the mitochondrial adaptations to physiological or pathological cardiac hypertrophic signals. Describes how the distinct cardiac metabolic profiles associated with physiological and pathological hypertrophy are initiated by specific signalling pathways: PI3K, AMPK and PGC1α.
Wilkins, B. J. et al. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ. Res. 94, 110–118 (2004).
Burgess, M. L. et al. Exercise- and hypertension-induced collagen changes are related to left ventricular function in rat hearts. Am. J. Physiol. 270, H151–H159 (1996).
Jin, H. et al. Effects of exercise training on cardiac function, gene expression, and apoptosis in rats. Am. J. Physiol. Heart Circ. Physiol. 279, H2994–H3002 (2000).
Neri Serneri, G. G. et al. Increased cardiac sympathetic activity and insulin-like growth factor-I formation are associated with physiological hypertrophy in athletes. Circ. Res. 89, 977–982 (2001).
Bellomo, D. et al. Mice lacking the vascular endothelial growth factor-B gene (Vegfb) have smaller hearts, dysfunctional coronary vasculature, and impaired recovery from cardiac ischemia. Circ. Res. 86, e29–e35 (2000).
Karpanen, T. et al. Overexpression of vascular endothelial growth factor-B in mouse heart alters cardiac lipid metabolism and induces myocardial hypertrophy. Circ. Res. 103, 1018–1026 (2008).
Bry, M. et al. Vascular endothelial growth factor-B acts as a coronary growth factor in transgenic rats without inducing angiogenesis, vascular leak, or inflammation. Circulation 122, 1725–1733 (2010).
Stubbe, P., Gatz, J., Heidemann, P., Muhlen, A. & Hesch, R. Thyroxine-binding globulin, triiodothyronine, thyroxine and thyrotropin in newborn infants and children. Horm. Metab. Res. 10, 58–61 (1978).
Hadj-Sahraoui, N., Seugnet, I., Ghorbel, M. T. & Demeneix, B. Hypothyroidism prolongs mitotic activity in the post-natal mouse brain. Neurosci. Lett. 280, 79–82 (2000).
Morkin, E. Regulation of myosin heavy chain genes in the heart. Circulation 87, 1451–1460 (1993).
Arsanjani, R., McCarren, M., Bahl, J. J. & Goldman, S. Translational potential of thyroid hormone and its analogs. J. Mol. Cell. Cardiol. 51, 506–511 (2011).
Kenessey, A. & Ojamaa, K. Thyroid hormone stimulates protein synthesis in the cardiomyocyte by activating the Akt-mTOR and p70S6K pathways. J. Biol. Chem. 281, 20666–20672 (2006).
Kenessey, A., Sullivan, E. A. & Ojamaa, K. Nuclear localization of protein kinase C-α induces thyroid hormone receptor-α1 expression in the cardiomyocyte. Am. J. Physiol. Heart Circ. Physiol. 290, H381–H389 (2006).
Belakavadi, M., Saunders, J., Weisleder, N., Raghava, P. S. & Fondell, J. D. Repression of cardiac phospholamban gene expression is mediated by thyroid hormone receptor-α1 and involves targeted covalent histone modifications. Endocrinology 151, 2946–2956 (2010).
Brownsey, R. W., Boone, A. N. & Allard, M. F. Actions of insulin on the mammalian heart: metabolism, pathology and biochemical mechanisms. Cardiovasc. Res. 34, 3–24 (1997).
Shiojima, I. & Walsh, K. Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev. 20, 3347–3365 (2006). Comprehensive review on the role of PI3K–AKT signalling pathways in regulating physiological cardiac hypertrophy, cardiac contractile function and coronary angiogenesis.
Araki, E. et al. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature 372, 186–190 (1994).
Burks, D. J. et al. IRS-2 pathways integrate female reproduction and energy homeostasis. Nature 407, 377–382 (2000).
Belke, D. D. et al. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J. Clin. Invest. 109, 629–639 (2002). Shows that insulin signalling controls postnatal cardiac growth. Cardiomyocyte-specific IR knockout was shown to result in cardiomyocytes with a reduced volume, and postnatal contractile and metabolic switches were altered.
Hu, P. et al. Minimally invasive aortic banding in mice: effects of altered cardiomyocyte insulin signaling during pressure overload. Am. J. Physiol. Heart Circ. Physiol. 285, H1261–H1269 (2003).
Sena, S. et al. Impaired insulin signaling accelerates cardiac mitochondrial dysfunction after myocardial infarction. J. Mol. Cell. Cardiol. 46, 910–918 (2009).
Boudina, S. et al. Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation 119, 1272–1283 (2009).
Efstratiadis, A. Genetics of mouse growth. Int. J. Dev. Biol. 42, 955–976 (1998).
Sutton, J. & Lazarus, L. A. Growth hormone in exercise: comparison of physiological and pharmacological stimuli. J. Appl. Physiol. 41, 523–527 (1976).
Baker, J., Liu, J. P., Robertson, E. J. & Efstratiadis, A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell 75, 73–82 (1993).
Liu, J. P., Baker, J., Perkins, A. S., Robertson, E. J. & Efstratiadis, A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 75, 59–72 (1993). References 54 and 55 show that IGF1 signalling is essential for postnatal growth.
Reiss, K. et al. Overexpression of insulin-like growth factor-1 in the heart is coupled with myocyte proliferation in transgenic mice. Proc. Natl Acad. Sci. USA 93, 8630–8635 (1996).
Delaughter, M. C., Taffet, G. E., Fiorotto, M. L., Entman, M. L. & Schwartz, R. J. Local insulin-like growth factor I expression induces physiologic, then pathologic, cardiac hypertrophy in transgenic mice. FASEB J. 13, 1923–1929 (1999).
McMullen, J. R. et al. The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase(p110α) pathway. J. Biol. Chem. 279, 4782–4793 (2004).
Kim, J. et al. Insulin-like growth factor I receptor signaling is required for exercise-induced cardiac hypertrophy. Mol. Endocrinol. 22, 2531–2543 (2008). In this study, IGF1R targeted deletion in adult cardiomyocytes did not result in any baseline hypertrophic phenotype in young mice. IGF1R-targeted hearts were resistant to exercise induced hypertrophy.
Moellendorf, S. et al. IGF-IR signaling attenuates the age-related decline of diastolic cardiac function. Am. J. Physiol. Endocrinol. Metab. 303, e213–e222 (2012).
Patel, A. et al. Canonical TRP channels and mechanotransduction: from physiology to disease states. Pflugers Arch. 460, 571–581 (2010).
Musarò, A., McCullagh, K. J., Naya, F. J., Olson, E. N. & Rosenthal, N. IGF-1 induces skeletal myocyte hypertrophy through calcineurin in association with GATA-2 and NF-ATc1. Nature 400, 581–585 (1999).
Maroto, R. et al. TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nature Cell Biol. 7, 179–185 (2005).
Spassova, M. A., Hewavitharana, T., Xu, W., Soboloff, J. & Gill, D. L. A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc. Natl Acad. Sci. USA 103, 16586–16591 (2006).
Seth, M. et al. TRPC1 channels are critical for hypertrophic signaling in the heart. Circ. Res. 105, 1023–1030 (2009).
Wu, X., Eder, P., Chang, B. & Molkentin, J. D. TRPC channels are necessary mediators of pathologic cardiac hypertrophy. Proc. Natl Acad. Sci. USA 107, 7000–7005 (2010). This study, along with reference 65, showed that TRPC channels are necessary mediators of cardiac hypertrophy.
Shai, S. Y. et al. Cardiac myocyte-specific excision of the β1 integrin gene results in myocardial fibrosis and cardiac failure. Circ. Res. 90, 458–464 (2002).
Johnston, R. K. et al. β3 integrin-mediated ubiquitination activates survival signaling during myocardial hypertrophy. FASEB J. 23, 2759–2771 (2009).
Cox, L., Umans, L., Cornelis, F., Huylebroeck, D. & Zwijsen, A. A broken heart: a stretch too far: an overview of mouse models with mutations in stretch-sensor components. Int. J. Cardiol. 131, 33–44 (2008).
Linke, W. A. Sense and stretchability: the role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc. Res. 77, 637–648 (2008).
Herman, D. S. et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 366, 619–628 (2012).
Shioi, T. et al. The conserved phosphoinositide 3-kinase pathway determines heart size in mice. EMBO J. 19, 2537–2548 (2000).
McMullen, J. R. et al. Phosphoinositide 3-kinase(p110α) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc. Natl Acad. Sci. USA 100, 12355–12360 (2003). This study, along with reference 72, showed that PI3Kα regulates the physiological growth of the heart in gain and loss of function studies. PI3Kα activity controls exercise-induced physiological cardiac hypertrophy but not pathological hypertrophy.
Luo, J. et al. Class IA phosphoinositide 3-kinase regulates heart size and physiological cardiac hypertrophy. Mol. Cell. Biol. 25, 9491–9502 (2005).
Lu, Z. et al. Loss of cardiac phosphoinositide 3-kinase p110α results in contractile dysfunction. Circulation 120, 318–325 (2009).
Crackower, M. A. et al. Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell 110, 737–749 (2002). This study showed that PTEN deletion in cardiomyocytes promotes heart growth at the organ and cellular level. Overexpression of dominant negative p110α downstream of PTEN normalizes the phenotype.
McManus, E. J. et al. The in vivo role of PtdIns(3,4,5)P3 binding to PDK1 PH domain defined by knockin mutation. EMBO J. 23, 2071–2082 (2004).
Mora, A. et al. Deficiency of PDK1 in cardiac muscle results in heart failure and increased sensitivity to hypoxia. EMBO J. 22, 4666–4676 (2003).
Oudit, G. Y. et al. The role of phosphoinositide-3 kinase and PTEN in cardiovascular physiology and disease. J. Mol. Cell. Cardiol. 37, 449–471 (2004).
Cho, H. et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKBβ). Science 292, 1728–1731 (2001).
Chen, W. S. et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 15, 2203–2208 (2001).
Cho, H., Thorvaldsen, J. L., Chu, Q., Feng, F. & Birnbaum, M. J. Akt1/PKBα is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J. Biol. Chem. 276, 38349–38352 (2001).
DeBosch, B. et al. Akt1 is required for physiological cardiac growth. Circulation 113, 2097–2104 (2006).
Shioi, T. et al. Akt/protein kinase B promotes organ growth in transgenic mice. Mol. Cell. Biol. 22, 2799–2809 (2002).
Matsui, T. et al. Phenotypic spectrum caused by transgenic overexpression of activated Akt in the heart. J. Biol. Chem. 277, 22896–22901 (2002).
Condorelli, G. et al. Akt induces enhanced myocardial contractility and cell size in vivo in transgenic mice. Proc. Natl Acad. Sci. USA 99, 12333–12338 (2002).
Shiojima, I. et al. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J. Clin. Invest. 115, 2108–2118 (2005). Showed that conditional expression of AKT1 in the heart for 2 weeks induces a reversible physiological hypertrophy while sustained AKT1 expression for 6 weeks causes heart failure.
Shiraishi, I. et al. Nuclear targeting of Akt enhances kinase activity and survival of cardiomyocytes. Circ. Res. 94, 884–891 (2004).
Rota, M. et al. Nuclear targeting of Akt enhances ventricular function and myocyte contractility. Circ. Res. 97, 1332–1341 (2005).
Haq, S. et al. Glycogen synthase kinase-3β is a negative regulator of cardiomyocyte hypertrophy. J. Cell. Biol. 151, 117–130 (2000).
Antos, C. L. et al. Activated glycogen synthase-3β suppresses cardiac hypertrophy in vivo. Proc. Natl Acad. Sci. USA 99, 907–912 (2002).
Michael, A. et al. Glycogen synthase kinase-3β regulates growth, calcium homeostasis, and diastolic function in the heart. J. Biol. Chem. 279, 21383–21393 (2004).
Skurk, C. et al. The FOXO3a transcription factor regulates cardiac myocyte size downstream of AKT signaling. J. Biol. Chem. 280, 20814–20823 (2005).
Malhowski, A. J. et al. Smooth muscle protein-22-mediated deletion of Tsc1 results in cardiac hypertrophy that is mTORC1-mediated and reversed by rapamycin. Hum. Mol. Genet. 20, 1290–1305 (2011). Showed that Tsc1 deletion results in lethal developmental and postnatal cardiac hypertrophy that is reversed by rapamycin treatment.
Wang, Y. et al. Rheb activates protein synthesis and growth in adult rat ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 45, 812–820 (2008).
Shen, W. H. et al. Cardiac restricted overexpression of kinase-dead mammalian target of rapamycin (mTOR) mutant impairs the mTOR-mediated signaling and cardiac function. J. Biol. Chem. 283, 13842–13849 (2008).
Zhang, D. et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J. Clin. Invest. 120, 2805–2816 (2010).
Shende, P. et al. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation 123, 1073–1082 (2011). This study, along with reference 97, shows that cardiac-specific mTOR or RAPTOR ablation results in heart failure without an initial phase of hypertrophy.
McMullen, J. R. et al. Deletion of ribosomal S6 kinases does not attenuate pathological, physiological, or insulin-like growth factor 1 receptor-phosphoinositide 3-kinase-induced cardiac hypertrophy. Mol. Cell. Biol. 24, 6231–6240 (2004).
Tsukada, J., Yoshida, Y., Kominato, Y. & Auron, P. E. The CCAAT/enhancer (C/EBP) family of basic-leucine zipper (bZIP) transcription factors is a multifaceted highly-regulated system for gene regulation. Cytokine 54, 6–19 (2011).
Bostrom, P. et al. C/EBPβ controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell 143, 1072–1083 (2010). Shows that C/EBPβ is specifically downregulated by exercise training. C/EBPβ downregulation increases cardiomyocyte proliferation and leads to the transcription of genes specific to exercise.
Bueno, O. F. et al. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 19, 6341–6350 (2000). Demonstrates that activation of MEK1–ERK1/2 signalling in the mouse heart induces a non-pathological form of compensated cardiac hypertrophy.
Lips, D. J. et al. MEK1-ERK2 signaling pathway protects myocardium from ischemic injury in vivo. Circulation 109, 1938–1941 (2004).
Purcell, N. H. et al. Genetic inhibition of cardiac ERK1/2 promotes stress-induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc. Natl Acad. Sci. USA 104, 14074–14079 (2007).
Kehat, I. et al. Extracellular signal-regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth. Circ. Res. 108, 176–183 (2011).
Kehat, I. & Molkentin, J. D. Extracellular signal-regulated kinase 1/2 (ERK1/2) signaling in cardiac hypertrophy. Ann. NY Acad. Sci. 1188, 96–102 (2010).
Horman, S., Beauloye, C., Vanoverschelde, J. L. & Bertrand, L. AMP-activated protein kinase in the control of cardiac metabolism and remodeling. Curr. Heart Fail. Rep. 9, 164–173 (2012).
Shibata, R. et al. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nature Med. 10, 1384–1389 (2004).
Zarrinpashneh, E. et al. AMPKα2 counteracts the development of cardiac hypertrophy induced by isoproterenol. Biochem. Biophys. Res. Commun. 376, 677–681 (2008).
Zhang, P. et al. AMP activated protein kinase-α2 deficiency exacerbates pressure-overload-induced left ventricular hypertrophy and dysfunction in mice. Hypertension 52, 918–924 (2008).
Sakamoto, K. et al. Deficiency of LKB1 in heart prevents ischemia-mediated activation of AMPKα2 but not AMPKα1. Am. J. Physiol. Endocrinol. Metab. 290, e780–e788 (2006).
Ikeda, Y. et al. Cardiac-specific deletion of LKB1 leads to hypertrophy and dysfunction. J. Biol. Chem. 284, 35839–35849 (2009).
Gundewar, S. et al. Activation of AMP-activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ. Res. 104, 403–411 (2009). Shows that metformin exerts its cardioprotective effects through AMPK activation.
Maloyan, A. et al. Exercise reverses preamyloid oligomer and prolongs survival in αB-crystallin-based desmin-related cardiomyopathy. Proc. Natl Acad. Sci. USA 104, 5995–6000 (2007).
Konhilas, J. P. et al. Exercise can prevent and reverse the severity of hypertrophic cardiomyopathy. Circ. Res. 98, 540–548 (2006).
Care, A. et al. MicroRNA-133 controls cardiac hypertrophy. Nature Med. 13, 613–618 (2007).
Fernandes, T. et al. Exercise training prevents the microvascular rarefaction in hypertension balancing angiogenic and apoptotic factors: role of microRNAs-16, -21, and -126. Hypertension 59, 513–520 (2012).
Porrello, E. R. et al. MiR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ. Res. 109, 670–679 (2011).
van Rooij, E. et al. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 316, 575–579 (2007).
Callis, T. E. et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J. Clin. Invest. 119, 2772–2786 (2009).
Nishi, H. et al. MicroRNA-27a regulates β cardiac myosin heavy chain gene expression by targeting thyroid hormone receptor β1 in neonatal rat ventricular myocytes. Mol. Cell. Biol. 31, 744–755 (2011).
Acknowledgements
This work was supported by grants from the US National Institutes of Health (J.D.M., J.H.v.B. and M.M.), and the Howard Hughes Medical Institute (J.D.M).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
Glossary
- Myocardium
-
From the Greek mys (muscle) and kardia (heart). It is the thick middle muscular layer of the heart that contracts.
- Valvular stenosis
-
Also called heart valve disease. Valvular stenosis occurs in response to stiffening, thickening, fusion or blockage of one or more valves of the heart. The heart comprises four valves: the mitral, aortic, tricuspid and pulmonic valves.
- Transverse-tubule system
-
Also called the T-tubule system. A T-tubule is a deep invagination of the sarcolemma (cardiomyocyte plasma membrane) enriched in excitation–contraction coupling molecules. T-tubule system refers to the network of T-tubules within an adult cardiomyocyte.
- Systolic function
-
The performance of the left ventricle during systole, which is the contraction of the heart. The best index of left ventricle systolic function is ejection fraction, which is calculated as the difference between end-diastolic and end-systolic left ventricle volume, divided by the end-diastolic left ventricle volume.
- Diastolic function
-
The performance of the left ventricle during diastole, which is the relaxation of the heart and the filling of the ventricle.
- Arrhythmogenic channelopathies
-
Genetic or acquired cardiac ion channel diseases. Ion channels (sodium, potassium and calcium channels) control the electrical activity of the heart. Abnormal electrical activity can lead to cardiac arrhythmias (irregular cardiac rhythm) and sudden death.
- Myocarditis
-
Inflammation of the heart caused by a viral or bacterial infection or an autoimmune disease. Myocarditis sometimes induces eccentric hypertrophy and heart failure.
- Stretch–spring sensing
-
Translation of changes in the cardiomyocyte extracellular environment (stretch) and the sarcomeres elasticity (spring) into biochemical hypertrophic signals.
- Sarcolemma
-
Specialized plasma membrane of a myocyte.
Rights and permissions
About this article
Cite this article
Maillet, M., van Berlo, J. & Molkentin, J. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat Rev Mol Cell Biol 14, 38–48 (2013). https://doi.org/10.1038/nrm3495
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrm3495
This article is cited by
-
Blood volume contributes to the mechanical synchrony of the myocardium during moderate and high intensity exercise in women
European Journal of Applied Physiology (2024)
-
Ser14 phosphorylation of Bcl-xL mediates compensatory cardiac hypertrophy in male mice
Nature Communications (2023)
-
JMJD6 protects against isoproterenol-induced cardiac hypertrophy via inhibition of NF-κB activation by demethylating R149 of the p65 subunit
Acta Pharmacologica Sinica (2023)
-
The Role of Small Extracellular Vesicles Derived from Mesenchymal Stromal Cells on Myocardial Protection: a Review of Current Advances and Future Perspectives
Cardiovascular Drugs and Therapy (2023)
-
Coordinated Metabolic Responses Facilitate Cardiac Growth in Pregnancy and Exercise
Current Heart Failure Reports (2023)
