Article Text

Abstract
Background/aims: Familial exudative vitreoretinopathy (FEVR) is an inherited blinding condition characterised by abnormal development of the retinal vasculature. The aim of this study was to perform linkage analysis in a large family affected with FEVR to determine whether the mutation involved was in one of the three known autosomal dominant FEVR loci or in another as yet unidentified gene.
Methods: Genomic DNA samples from family members were polymerase chain reaction (PCR) amplified with fluorescently tagged microsatellite markers spanning the EVR1/EVR4 locus (11q13-14) and the EVR3 locus (11p12-13). The resulting PCR products were resolved using an automated DNA sequencer and the alleles sized. These data were used to construct haplotypes across each locus and linkage analysis was performed to prove or exclude linkage.
Results: The clinical evaluation in this family suggested features typical of FEVR, with deficient peripheral retinal vascularisation being the common phenotype in all affected individuals. However, linkage analysis proved that this family has a form of FEVR genetically distinct from the EVR1, EVR3 and EVR4 loci.
Conclusion: The exclusion of linkage in this family to any of the known FEVR loci proves the existence of a fourth locus for autosomal dominant FEVR and shows that this rare disorder is far more heterogeneous than previously thought.
- FEVR, familial exudative vitreoretinopathy
- familial exudative vitreoretinopathy
- heterogeneity
- FEVR, familial exudative vitreoretinopathy
- familial exudative vitreoretinopathy
- heterogeneity
Statistics from Altmetric.com
Familial exudative vitreoretinopathy (FEVR) is a rare but well defined inherited disorder of retinal vessel development.1 The primary pathological process in FEVR is believed to be a premature arrest of retinal vasculogenesis/angiogenesis or retinal vascular differentiation, leading to incomplete vascularisation of the peripheral retina.2 All affected individuals have an avascular peripheral retina, so when this is used as the diagnostic criterion, FEVR has a penetrance of 100%. However, this feature alone usually causes no clinical symptoms and can often be diagnosed only by molecular testing or fluorescein angiography.3 Therefore, it is not uncommon for affected individuals to be unaware that they have the disorder until other more severely affected family members are diagnosed, prompting them to seek a diagnosis. The visual problems and variable phenotype present in FEVR are thought to be the result of secondary complications caused by the resulting retinal ischaemia. Neovasularisation around the region of demarcation near the avascular retina is thought to produce a fibrotic response with patients frequently demonstrating peripheral vitreoretinal opacities suggestive of fibrosis. As a consequence, retinal traction may be manifest as macular ectopia or peripheral retinal folds and a proportion of individuals develop a falciform retinal fold. Vascular exudation is a variable feature and occasionally is severe enough to produce a non-rhegmatogenous retinal detachment. Rhegmatogenous retinal detachment is more frequently seen and is often thought to be a consequence of traction although inherent abnormalities of the peripheral retina such as atrophic retinal holes may play a part. These features cause a reduction in visual acuity and in 20% of FEVR cases lead to partial or total blindness.2
FEVR is genetically heterogeneous and can be inherited as an autosomal dominant,4,5 autosomal recessive,6,7 or X linked trait.8,9 To date four loci have been mapped, one X linked (EVR2 on chromosome Xp)10 and three autosomal dominant (EVR111 and EVR412 on chromosome 11q and EVR313 on chromosome 11p). No recessive locus has yet been identified.
The first FEVR locus, EVR1, was localised to chromosome 11q13-q23, between D11S527/D11S533 and D11S35, by genetic mapping in two large autosomal dominant pedigrees.11 Subsequent analysis in further families confirmed linkage to this interval,14–17 and in 2002 the EVR1 gene was identified when FEVR patients were found to have mutations in the gene FZD4, which encodes the Wnt receptor Frizzled-4.18
The second FEVR locus, EVR2, was mapped in X linked pedigrees to chromosome Xp11.4.10 The mutated gene within this locus was almost immediately identified and shown to be the same as that mutated in Norrie disease, NDP.19,20 Norrie disease is a rare X linked recessive neurodevelopmental disorder characterised by congenital blindness, with a proportion of patients (30%) also having sensorineural deafness and mental disturbances.21NDP encodes a secreted protein containing a cystine knot motif22 which has recently been shown to function as a high affinity ligand for Frizzled-4.23
The third locus for FEVR, EVR3, was identified on chromosome 11p12-p13 in a large Scottish family segregating FEVR in an autosomal dominant fashion.13EVR3 spans 14 cM and is flanked by the markers GATA34E08 (telomeric) and D11S4102 (centromeric). To date no other families have been reported to map to EVR3 and the mutated gene is still unidentified.13
The fourth locus, EVR4, was only recently identified on chromosome 11q13 in a family originally reported as linking to the EVR1 locus.15 After the identification of FZD4 as the EVR1 gene, it was screened in this family and excluded on the basis of both mutation analysis and haplotype data.12 The causative gene in this family was mapped to a region within the original EVR1 locus, but 10 cM centromeric to FZD4. Subsequently, in this and other families mutations were identified in LRP5, a gene which also encodes a Wnt receptor, low density lipoprotein receptor related protein-5.24
In the current study we test and exclude linkage to the three known autosomal dominant FEVR loci in a new FEVR pedigree originating from Mexico. These data provide evidence for a fourth autosomal dominant locus and imply the existence of at least five genes responsible for this phenotype.
PATIENTS AND METHODS
Patients
The family originated from Mexico. Detailed clinical examination of 18 family members was undertaken and blood samples were collected after informed consent was obtained. Ethical approval was provided by the Leeds Teaching Hospitals Trust research ethics committee. All patients were examined in their homes except for IV:6 and V:6 who were examined at the University of Texas-Houston Medical School. The examinations of all patients included ascertainment of best corrected visual acuity, external examination, motility evaluation, and examination with the indirect ophthalmoscope. The Kowa RC-2 fundus camera was used to photograph the anterior segments of all patients and the posterior segments of all patients except for II:4 who had dense cataracts. The two affected patients examined in the medical centre (father and son) had slit lamp evaluations, indocyanine green angiography of the irides, and fluorescein angiography of the fundi.
Linkage analysis
Genotyping was performed using fluorescently tagged microsatellite markers. Polymerase chain reactions (PCR) were carried out in a 25 μl volume with 50 ng of genomic DNA, 20 pmol of each primer (a fluorescent and an unlabelled primer in each pair), 200 μM each dATP, dCTP, dGTP, dTTP, 10 mM TRIS-HCl pH 8.3, 50 mM KCl, 1.5 mM MgCl2, 0.01% gelatin, and 1 unit Taq DNA polymerase (Invitrogen). After the initial denaturation step at 96°C for 3 minutes, the samples were processed through 35 cycles of 92°C for 30 seconds, 50–60°C for 30 seconds, and 72°C for 30 seconds. A final extension step was performed at 72°C for 10 minutes. Following amplification, PCR products were resolved using an ABI 377 sequencer and were analysed using Genscan 2.0.2 and Genotper 1.1.1 software (Applied Biosystems). Linkage analysis was performed under the assumption of a dominant model with 100% penetrance (with no normal siblings included in the analysis or individuals II:4, V:4, and V:5 who had an uncertain diagnosis) and 0.001 frequency of the disease allele. The phenocopy rate was set at 0.1%. The data were checked for Mendelian inheritance using the UNKNOWN program and two point linkage analyses were performed using the MLINK program from the LINKAGE package, version 5.1.23 Multipoint lod scores were computed by the LINKMAP program from the LINKAGE package, version 5.1.25 Marker allele frequencies were estimated on the basis of data from the founders and spouses of the family used in this study. The Marshfield, Généthon, and DeCODE maps were used for selection of markers and to determine the genetic distances.26–28
RESULTS
Clinical evaluation
The family studied is shown in figure 1. The phenotype in affected individuals concurred well with descriptions of FEVR in the literature. The proband (V:6) and two additional family members (III:1 and III:3) had bilateral tractional retinal detachments in childhood (fig 2A). Other affected family members were more mildly affected with a picture predominantly of retinal exudation. Seven adults (II:3, IV:2, IV:4, IV:5, IV:6, IV:8, and IV:9) had clusters of exudates throughout the retina (fig 2B), whereas three young patients (V:1, V:2, and V:3) had only isolated exudates. We were unable to identify the clinical status in three individuals. Individual II:4 had cataracts which obscured an adequate evaluation of his retina. Two young patients (V:4 and V:5) had minimal abnormalities in the far retinal periphery which could not be adequately photographed with the Kowa RC-2 camera. The proband had hand motion vision in his right eye and 20/60 vision in his left eye following surgery to halt the progression of its peripheral retinal detachment and lens irrigation and aspiration to treat its shallow anterior chamber. III:1 and III:3 were totally blind. All other family members were 20/20 best corrected except II:4 who had 20/400 vision primarily because of his cataracts. The slit lamp examinations on two affected patients revealed nasally displaced pupils without well defined collarettes and some shallowing of the anterior chambers (more pronounced in V:6 than in IV:6). Indocyanine green angiography failed to demonstrate normal vascularisation of the iris. Fluorescein angiography demonstrated the typical exudates of FEVR and the termination of peripheral vessels before reaching the ora serrata in IV:6, and macular dragging with peripheral retinal atrophic areas in V:6.
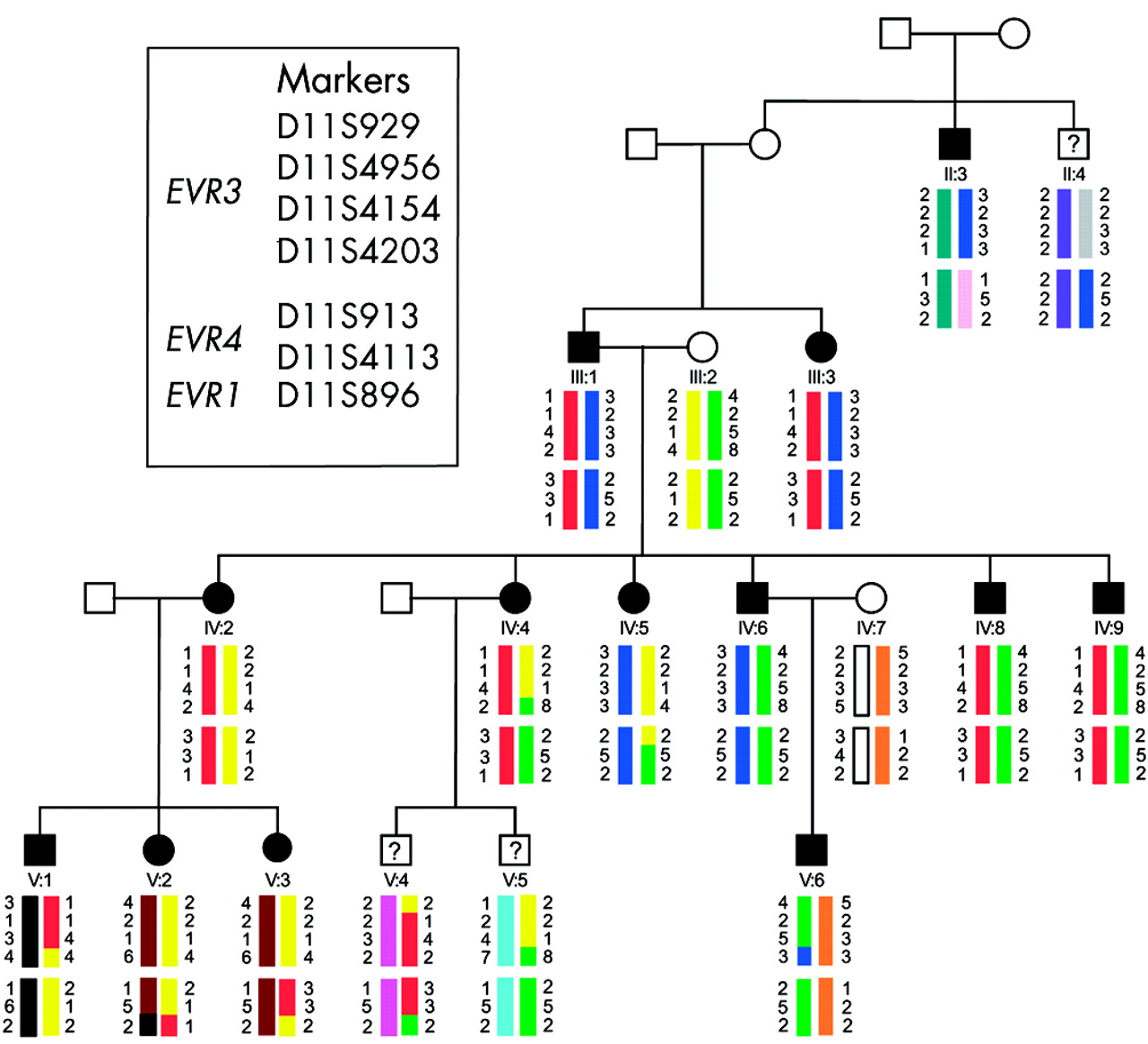
Pedigree of the autosomal dominant FEVR family analysed in this study. Individuals represented by solid symbols are confirmed as affected by clinical examination. Individuals represented by symbols with a question mark have an uncertain diagnosis and were excluded from linkage analysis studies. Haplotypes spanning the EVR1, EVR3, and EVR4 loci are shown below each symbol. Each different haplotype block is represented by a different colour. No single haplotype is present in affected individuals for any of these loci.

Clinical pictures of family members showing classic features of FEVR. (A) Fundus photograph of the proband’s (V:6) optic nerve and macula in the right eye taken at 7 months. Virtually all of the retinal vessels are drawn up into a “comet”-shaped retinal fold which extends from the optic nerve to a large fibrovascular mass in the temporal retinal periphery. (B) Fundus photograph of the proband’s father’s (IV:6) temporal peripheral retina. The optic nerve and macula in this individual appeared normal but examination of the peripheral retina revealed the presence of multiple exudates (yellow dots).
Exclusion of the FEVR loci
The family clearly shows an autosomal dominant inheritance pattern (fig 1), with male to male transmission ruling out the possibility that this family harbours a mutation in the X linked EVR2 gene, NDP. Analysis therefore focused on the three known FEVR autosomal dominant loci and family members were genotyped with markers from chromosome 11 spanning EVR1, EVR3, and EVR4.
To test for linkage to the EVR1 gene FZD4, genotyping was performed with marker D11S896, which according to the July 2003 version of the UCSC genome browser (genome.cse.ucsc.edu/index) is contained within the same genomic clone as FZD4 (RP11-736K20-AP001528). To test for linkage to the EVR4 gene LRP5, genotyping was performed with markers D11S913 and D11S4113. These markers flank LRP5 and, according to the Marshfield genetic map, are separated by 1 cM.27 The genotyping data for these markers is shown in figure 1 and the results of two point linkage analysis obtained with these markers are shown in table 1. The results show convincing exclusion of linkage to all three markers and therefore rule out both FZD4 and LRP5 as the mutated gene in this family.
Two point exclusion data for the EVR1 (FZD4) and EVR4 (LRP5) genes and the EVR3 locus (11p12-13)
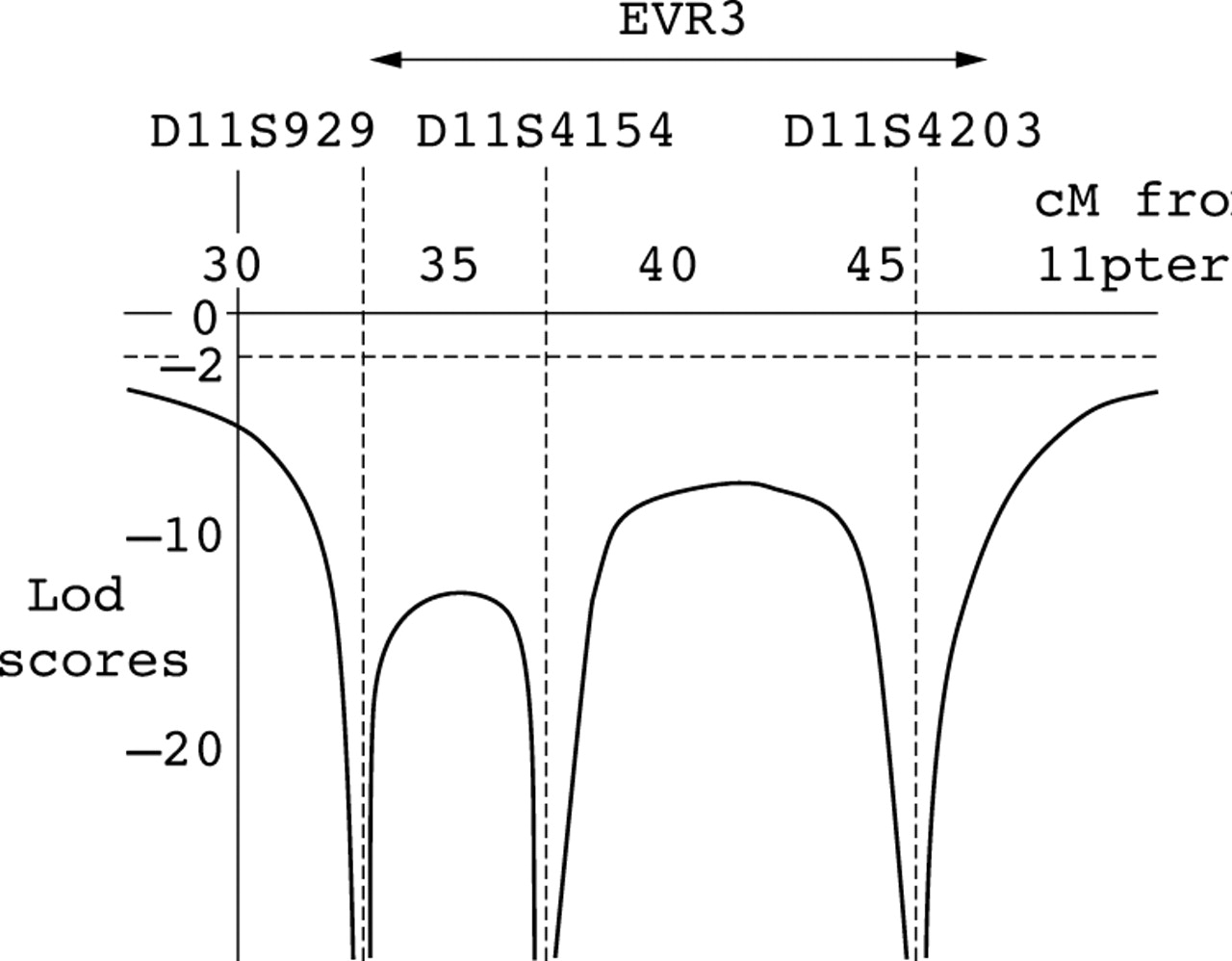
As the FEVR gene at the EVR3 locus is still unknown, linkage analysis was performed using four markers spanning this locus. Markers and distances used are as follows; 11ptel - 33 cM - D11S929 - 1 cM - D11S4956 - 4 cM - D11S4154 - 8 cM - D11S4203 - 102 cM - 11qtel. Each family member’s haplotype obtained from these markers is shown in figure 1. Two point lod scores for these markers show convincing exclusion of linkage for EVR3 (table 1) and multipoint linkage analysis with a subset of these markers (D11S929, D11S4154, and D11S4203) also show statistically significant exclusion across EVR3 (lod score <−8) (fig 3). These data provide convincing exclusion of all three known autosomal dominant FEVR loci in this family.

{kind=link}
{kind=link}
{kind=link}
Multipoint graph showing exclusion of the EVR3 locus. The approximate position of EVR3 is shown above the graph. Lod scores lower than −2 are statistically significant for exclusion.
DISCUSSION
We have provided evidence for the existence of a fourth autosomal dominant FEVR locus by excluding the three known dominant loci in a large Mexican family. This result implies that there are now at least five genes responsible for this phenotype, four autosomal dominant13,18,24 and one X linked.19 This number of genes is likely to increase further as recessive FEVR families have been described in the literature but a locus has yet to be reported.6,7 The increasing heterogeneity observed in FEVR is also evident from recent FZD4 and LRP5 mutation screens. Although EVR1 has historically been reported as the major autosomal dominant locus for FEVR, mutations in FZD4 and LRP5 have been shown to account for only 40% of patients,24,29,30 suggesting that other unidentified genes play a significant part in this condition.
The phenotype described in this family fits well with the classic description of FEVR1,4 and with the phenotype seen in families linking to other FEVR loci.15,27–29 This pedigree shows a majority of individuals with retinal exudates which corresponds well with the original classification of the disease.4 Other pedigrees published showed less evidence of this feature31 but as exudation is a variable aspect of the phenotype this may not be significant. The only atypical features observed in this family were the anterior segment anomalies seen in individuals IV:6 and V:6. Several other affected family members were thought to have shallow anterior chambers and abnormal irides but because these individuals were examined in their own homes during a field trip to Mexico, they were examined without slit lamp biomicroscopy. It is therefore not possible to say whether these features invariably co-segregated with the retinal phenotype in this family. Lenticular anomalies in FEVR patients have been described previously1 but whether anterior segment anomalies are specific to mutations in particular FEVR genes remains to be determined. To date it is difficult to distinguish between the phenotypes seen at all four dominant FEVR loci without molecular testing. This fact is likely to hinder the development of a diagnostic screening service for FEVR as screening multiple genes for a rare disorder is unlikely to be cost effective.
By continuing to identify the genes responsible for FEVR and dissecting the genetic pathways involved in normal and abnormal retinal vessel formation, it is hoped that we can improve the understanding of this disease and facilitate improved diagnosis and treatment for people who have it.
Note added on proof
Since this paper was accepted recessive mutations have been described in the autosomal dominant EVR4 gene, LRP5 (Jiao et al, Autosomal recessive familial exudative vitreoretinopathy is associated with mutations in LRP5. Am J Hum Genet 2004;75:878–84).
Acknowledgments
We thank the FEVR family for their help with this study. The financial support of the Wellcome Trust (grants 069718/Z/02 and 055145/Z/98) is gratefully acknowledged. HMH is supported by grants from the Hermann Eye Fund, Houston, TX; Research to Prevent Blindness, Inc, New York, NY, USA; and by Vision Core National Institutes of Health, Bethesda, MD, USA (grant EY10608).
REFERENCES
Footnotes
-
CT and LMD contributed equally to this work.