


Studies Using the AHR-Null Mice
Abstract
The aryl hydrocarbon receptor (AHR) is believed to mediate the toxic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), polychlorinated biphenyls, and polycyclic aromatic hydrocarbons. AHR is a member of the Per, ARNT, Sim/basic-helix-loop-helix superfamily of ligand-activated transcription factors that also harbors the transcription factors involved in the hypoxia response, development of the central nervous system, and day-night adaptations. To investigate the role of AHR in chemical toxicity and carcinogenesis and to determine any possible function in mammalian development and physiological homeostasis, AHR-null mice were developed. The AHR-null mice were resistant to the acute toxicity of TCDD and had an altered teratogenic response to this compound. These mice were found to have a number of abnormal phenotypes, thus confirming that AHR plays an important developmental and physiological role. Among the most consistent phenotypes was an altered liver pathology that was associated with accelerated rates of apoptosis. Evidence suggests that this may be related to an abnormal accumulation of levels of hepatic retinoic acid that cause an activation of transforming growth factor β, resulting in stimulation of apoptosis. AHR may directly or indirectly control levels of a cytochrome P450 that is responsible for catabolizing retinoic acid.
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD)1, polychlorinated biphenyls, and polycyclic aromatic hydrocarbons are notorious environmental hazards that cause acute and chronic toxicity and are believed to be nongenotoxic carcinogens. These chemicals all bind to a ligand-dependent transcription factor called the aryl hydrocarbon receptor (AHR), resulting in the activation of a battery of genes, including the cytochromes P450 CYP1A1, CYP1A2, and CYP1B1 (Rowlands and Gustafsson, 1997). To date, an endogenous ligand for this receptor has not been found, although several natural dietary molecules, such as diosmin and diosmetin, have been reported to be able to bind to the AHR as agonists inducing transcription of the CYP1A1 gene (Ciolino et al.,1998). The AHR is ubiquitously expressed in most organs and cells in the body and requires a dimerization partner called AH receptor nuclear translocator (ARNT) in order to translocate to the nucleus and bind to upstream DNA elements of target genes. Both proteins are members of a small superfamily of basic-helix-loop-helix (bHLH) transcription factors that also include the transcriptional regulators PER (Period) and SIM (Single minded) first identified in Drosophila as required for the development of the central nervous system. Thus this superfamily of transcription factors is typically called the PAS superfamily (Per, ARNT, Sim) (Hankinson, 1995). Another member of this superfamily of transcription factors is the protein that mediates gene activation by anoxia, called Hif1α (Wang et al.,1995). AHR, Hif1α, and Sim all require ARNT as a dimerization partner for gene activation. A second factor related to ARNT, called ARNT2, has also been described (Hirose et al.,1996), in addition to a series of proteins called MOPs (members of the PAS superfamily) (Hogenesch et al.,1997). The bHLH proteins have been found in mammals, insects, plants, fungi, flatworms, and cyanobacteria (Zhulin et al.,1997; Powell-Coffman et al.,1998). Some of these factors are associated with light reception and circadian rhythms associated with animal behavior (Hogenesch et al.,1998; Price et al.,1998).
The mechanism of action of AHR is shown in fig.1. In the absence of a ligand, AHR is found bound to two molecules of HSP90 in the cytoplasm. Upon ligand binding, the HSP90 molecules are displaced from the AHR, which now enters the nucleus and complexes with ARNT. The heterodimer then binds to its response elements (xenobiotic response element) to mediate increases in the rates of transcription of specific target genes (Rowlands and Gustafsson, 1997). HSP90 is a molecular chaperone that binds to the AHR and maintains the unliganded, cytosolic complex. In addition to HSP90, other proteins may interact with the receptor in the cytosol. For example, hepatitis B virus X-associated protein 2 was also found to be associated with the AHR/HSP90 trimer to form a cytosolic tetrameric 9S complex that may also include other proteins (Meyeret al.,1998). Transcriptional activation by this complex is mediated by the co-activator CBP/p300 that bridges the interaction between the AHR-ARNT heterodimer with the TATA box-associated factors (TAFs) (Kobayashi et al.,1997). This results in recruitment of RNA polymerase II. It has also been suggested that other mechanisms, such as protein kinase C–dependent phosphorylation, could participate in gene activation by the AHR or potentiation/inhibition of the induction response (Chen and Tukey, 1996).

Model of gene activation by AHR.
The model is described in the text.
AHR-Null Mice
In order to determine the physiological role of the AHR, gene knockout mice were prepared (Fernandez-Salguero et al.,1995). The basic domain of the basic-helix-loop-helix motif of the murine AHR gene was disrupted by insertion of a cassette encoding the phosphoribosyltransferase II gene, which encodes a bacterial protein that mediates resistance to the neomycin-based eukaryote cell toxin G418. This is traditionally used as a positive selection marker in pluripotent embryonic stem cells to generate the knockout mice. AHR knockout mice (AHR-null) do not express receptor protein, and typical AHR target genes, such as CYP1A1 andCYP1A2, are not induced upon the administration of TCDD (Fernandez-Salguero et al.,1995). In addition, basal expression of certain genes, including CYP1A2 and UDP-glucuronosyltransferase form 6 (UGT*06), is also markedly decreased. AHR-null mice were found to exhibit a large number of phenotypic abnormalities, including a peripheral immune system deficiency primarily affecting total numbers of B and T cells in the spleen. This is most notable in newborn pups and older animals and is believed to be responsible for premature deaths as a result of opportunistic bacterial infections, probably caused byHelicobacter pylori (Fernandez-Salguero et al.,1997). However, when AHR-null mice at 1, 2, 12, and 32 weeks of age were challenged with a standard immunization protocol, the titers of IgG, IgM and IgA obtained were in the same range to those found for AHR wild-type mice, indicating a normal response to an exogenous antigen in the experimental conditions used (D. M. Hilbert, P. Fernandez-Salguero, F. J. Gonzalez and S. Rudikoff, unpublished results, 1996). Numerous other phenotypic changes, many of which are manifested only as the animal ages (usually at over 8 months of age) are noted (Fernandez-Salguero et al.,1997). These include heart hypertrophy with the presence of fibrosis in the heart muscle. In the skin, severe localized epidermal hyperplasia with hyperkeratosis and an altered expression of cytokines, keratins, and integrins was observed; marked dermal fibrosis and hyperproliferation of hair follicles that exhibit an abnormal orientation pattern were also evident (Fernandez-Salguero et al.,1997). AHR-null mice develop a pronounced rectal prolapse associated with the presence ofH pylori and probably reflecting the inability of their defective immune systems to control opportunistic bacteria. Hyperplasia is found in the gastric pylorus and develops into polyps in animals at approximately 10 months of age. Hyperproliferation of blood vessels in the portal areas of the liver, with the presence of fibrosis, and massive calcifications in the uterus are also found in older AHR-null mice. Tumors in the liver and lung, identified as adenocarcinomas, were also observed in some of the older (11 to 12 months of age) AHR-null mice. These observations suggest that the AHR plays a fundamental role in cell and organ physiology and homeostasis and lend further support for the existence of an endogenous ligand.
Role of the AHR in the Acute Toxicity of TCDD
Exposure of mammals to the environmental pollutant TCDD results in a diverse set of toxicologic and pathologic effects. The mechanisms of some of these effects have been studied extensively in vitro, and correlative studies have indicated the involvement of AHR. These studies have been used as a basis to assign human risk to exposure to low doses of TCDD (Sewall and Lucier, 1995). However, a definitive association of the AHR with TCDD-mediated toxicity has been difficult to establish because of the diversity of its effects and the ubiquitous expression of this receptor. In an effort to distinguish AHR-mediated TCDD toxicity from those resulting from alternative pathways, the AHR-null mice were compared with wild-type mice regarding their susceptibility to acute TCDD-induced toxicity (Fernandez-Salgueroet al.,1996). The results demonstrate that AHR-null mice are relatively unaffected by doses of TCDD (2000 μg/kg) that are tenfold higher than those found to induce severe toxic and pathologic effects in wild-type mice and even heterozygous littermates expressing a functional AHR; these effects included wasting syndrome, lipid accumulation within hepatocytes, and thymic atrophy. Analyses of liver, thymus, heart, kidney, pancreas, spleen, lymph nodes and uterus from AHR-null mice identified no significant TCDD-induced pathology. The resistance of AHR-null mice to TCDD-induced thymic atrophy appears to be restricted to processes involving the AHR since the corticosteroid dexamethasone rapidly and efficiently induced cortical depletion in both AHR-null and normal littermate control mice. Furthermore, by constructing chimeric mice, using TCDD-responsive (from control mice having a functional AHR) stromal components and TCDD-unresponsive (from AHR-null mice) hematopoietic components, or the reverse, it was demonstrated that the targets for TCDD-induced thymic atrophy are located exclusively in the hematopoietic compartment and that TCDD activation of epithelial cells in the stroma is not required for the induction of thymic alterations (Staples et al.,1998). Taken together, these results suggest that the pathological changes induced by TCDD in the liver and thymus are mediated entirely by the AHR. However, it is important to note that at the highest doses of TCDD administered, AHR-deficient mice displayed limited vasculitis and scattered single-cell necrosis in their lungs and livers, respectively. The mechanism(s) responsible for these apparently receptor-independent processes remain unclear but may involve novel, alternative pathways for TCDD-induced toxicity. However, this was only observed at doses that are an order of magnitude higher than those that are lethal in wild-type mice. Interestingly, a bHLH protein, called MET, that seems to be responsible for mediating toxicity induced by juvenile hormone analog insecticides was cloned from Drosophila; indeed, Met mutants were shown to be resistant to juvenile hormone analog–induced toxicity, thus suggesting the existence of a common mechanism among different organisms for certain xenobiotics to exert toxicity (Ashoket al.,1998).
Role of the AHR in Teratogenesis
TCDD is a notorious teratogen, causing cleft palate in rodents when administered to pregnant rats and mice at mid-gestation (Coutureet al.,1990). To determine whether the AHR is involved in mediating the embryotoxicity of TCDD and related compounds, AHR-null mice were analyzed for susceptibility to TCDD (Mimura et al.,1997; Peters et al.,1998). Pregnant dams were administered TCDD at gestation days10 and 12.5, and fetuses were examined at gestation day 18. Whereas mice lacking expression of the receptor exhibited no fetal abnormalities, both heterozygotes having only one functional copy of the AHR and wild-type mice exhibited cleft palate, hydronephrosis, small kidneys, and several other abnormalities (Peters et al.,1998). Paradoxically, AHR-null dams exposed to TCDD did show a marked increase in resorption rates, indicating that AHR-independent pathways may contribute to some developmental abnormalities triggered by dioxin. The mechanism by which the AHR mediates cleft palate is believed to involve the effects of abnormal expression of growth factors. In this regard, it is noteworthy that TGFβ3-null mice exhibited a cleft palate with the same mechanistic origin and histology as that induced by TCDD during embryonic development (Kaartinen et al.,1995). TCDD is known to cause decreased expression of TGFα, TGFβ, and EGF in the developing embryo (Abbott and Birnbaum, 1990; Bryant et al.,1997). However, it is not known whether the AHR has a direct or indirect role in determining levels of expression of cytokines since there exists no evidence that the AHR directly mediates thetrans-activation of genes encoding growth factors. TCDD is also known to affect the levels of expression of growth factors (Leeet al.,1996; Abbott et al.,1992) and growth factor receptors, including the one for EGF (Sewall et al.,1995). Recent studies in the adult AHR-null mice suggest that the AHR may cause an increase in TGFβ through a post-transcriptional mechanism (Andreola et al.,1997; Zaher et al.,1998).
Role of the AHR in Liver Development
Among the earliest phenotypes that emerge from the AHR-null mice is a marked liver pathology. Livers in AHR-null mice are approximately half the size per gram of body weight of those in normal mice and show fibrosis that is most prominent around the portal triads, with some scattered foci in the parenchyma (Fernandez-Salguero et al.,1995; Fernandez-Salguero et al.,1997; Schmidtet al.,1996). A proliferation of small blood vessels is also noted in the portal areas and in some areas within the parenchyma. The extent of fibrosis increases with age, and by 11–13 months adenomas and carcinomas are sometimes found in the AHR-null mice. Liver tumors are never found in heterozygotes or wild-type mice of similar age.
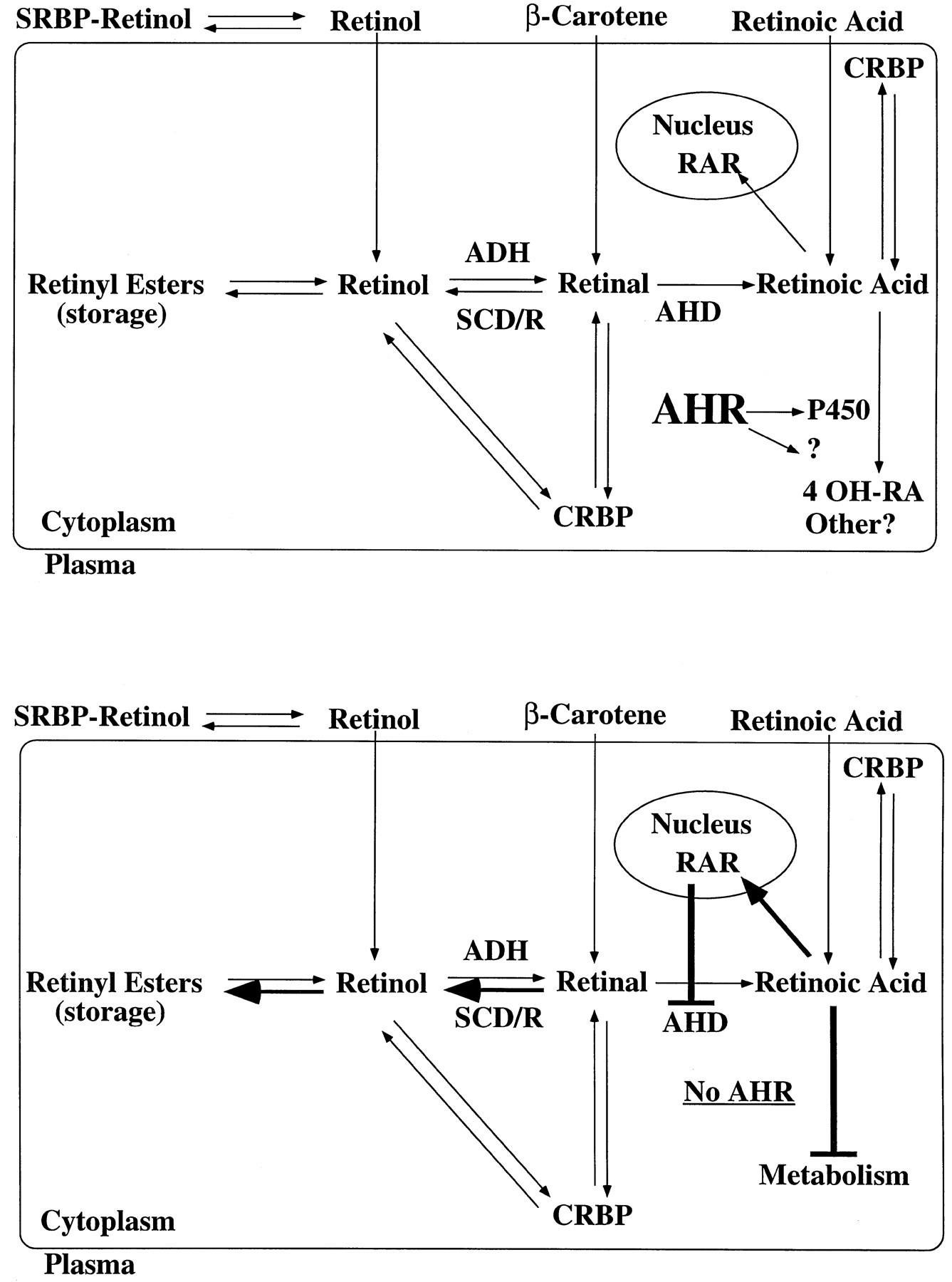
By serendipity, the levels of retinoic acid and its various derivatives were found to be elevated in AHR-null mice (Andreola et al.,1997). Retinoic acid (RA), retinol, and retinyl palmitate were elevated from two- to threefold in the livers of AHR-null, compared with those of wild-type mice (fig. 2). This was because of a deficiency in the ability of the AHR-null mice to catabolize retinoic acid. In the liver, retinoic acid is most probably degraded by oxidation via a P450. Although the identity of the P450 responsible for RA metabolism in mouse liver is not yet known, a potential role for the recently identified P450RAI (White et al.,1997; Fujii et al.,1997) in retinoic acid metabolism has been ruled out (Andreola et al.,1997). Additional biochemical evidence supporting the increase in the content of RA in the livers of AHR-null mice was produced by a parallel increase in tissue type II transglutaminase (TGase-2), a protein that is inducible by RA through the retinoic acid receptor (Nagyet al.,1996) and that is highly induced in cells undergoing apoptosis (Singh et al.,1998). AHR-null mice also exhibited decreased (less than 15% to 20%) levels of hepatic aldehyde dehydrogenase 2 (AHD) an enzyme that catalyzes conversion of retinal to RA. Analysis of liver RNA from wild-type and AHR-null mice indicated that hepatic aldehyde dehydrogenase 2 is not inducible by TCDD in either mouse strain, thus suggesting that the observed downregulation is not an AHR-mediated process (Andreola et al.,1997). Therefore, these results suggest a novel feedback mechanism by which, as the levels of production of RA from retinal decreases, the excess of RA precursors are converted through reversible steps to retinyl esters instead of RA. A scheme by which the AHR could control levels of hepatic RA is shown in fig.3. The AHR controls—either directly or indirectly—levels of expression of one or more P450s that catabolize retinoic acid. In the AHR-null mouse, this P450 is present at low levels, resulting in decreased catabolism and increased retinoic acid content. This results in feedback inhibition of AHD with a concomitant increase in retinyl esters. The increase in retinoic acid could also result in the activation of target genes through RAR.

Levels of retinyl palmitate and retinoic acid in the livers of wild-type and AHR-null mice.
The data were taken from (Andreola et al.,1997).

Model depicting the involvement of AHR in controlling the levels of retinoic acid in liver.
The description of the model is in the text. SRBP, serum retinol-binding protein; CRBP, cellular retinol-binding protein; ADH, alcohol dehydrogenase; SCD/R, short-chain dehydrogenase/reductase.
The abnormal liver phenotype in the AHR-null mice suggested that hepatocyte growth was altered by receptor deficiency. Since TGFβ is known to affect cell growth and proliferation, the levels of TGFβ1 and TGFβ3 were examined and found to be elevated in the liver, particularly in areas that are coincident with fibrosis (Zaher et al.,1998). Since the TGFβ family of cytokines are known to stimulate programmed cell death (Glick et al.,1989; Letterio and Roberts, 1996), apoptosis was examined by analyzing chromosomal DNA integrity and nuclear fragmentation and found to be accelerated in primary cultures of hepatocytes derived from the AHR-null mice. These hepatocytes also had elevated levels of secreted TGFβ1, as observed by proliferation inhibition assays using mink-lung epithelial cells. AHR-null hepatocytes in primary culture secreted not only latent but also active TGFβ in such an amount that when conditioned media from AHR-null cultures was added to hepatocyte cultures from control mice, apoptosis was significantly stimulated. Thus abnormally elevated TGFβ levels in the livers of developing AHR-null mice could result in an increase in apoptosis that ultimately results in small liver size and fibrosis. Interaction between TGFβ and AHR signaling pathways has also been pointed out by studies in cell lines that showed AHR downregulation by this cytokine (Dohr et al.,1997; Dohr and Abel, 1997).
The increase in TGFβ appears to be due to a post-transcriptional modification since TGFβ mRNA is not elevated in AHR-null mouse livers (Zaher et al.,1998). The increase in active TGFβ1 and TGFβ3 in the AHR-null mice may be mediated by the elevated levels of tissue TGase-2, which is thought to convert pre-TGFβ to the biologically active cytokine (Nunes et al.,1997; Gleizeset al.,1997). Therefore, these results indicate that the increase in TGFβ could be a consequence of the elevated levels of tissue TGase-2 that is induced by RA in AHR-null livers. The increase in TGFβ-induced apoptosis could result in the small size and pockets of fibrosis found in AHR-null mice livers.
Taken together, these results clearly indicate that the AHR, in addition to mediating TCDD-induced toxicity, has a significant role in cell and tissue homeostasis in vivo. AHR-dependent signaling pathways seem to mediate the activity or participate with growth factors and hormone signaling mechanisms in controlling the cell cycle. Crossregulation between AHR and TGFβ appears to be an attractive possibility that deserves further study.
Footnotes
-
Send reprint requests to: Dr. Frank J. Gonzalez, Building 37, Room 3E-24, National Cancer Institute, Bethesda, MD 20892. e-mail: fjgonz{at}helix.nih.gov
- Abbreviations used are::
- TCDD
- 2,3,7,8-tetrachlorodibenzo-p-dioxin
- AHR
- aryl hydrocarbon receptor
- ARNT
- AH receptor nuclear translocator
- bHLH
- basic-helix-loop-helix
- PAS
- Per AHRSim
- HSP90
- heat shock protein 90
- Ig
- immunoglobulin
- TGF
- transforming growth factor
- RA
- retinoic acid
- P450
- cytochrome P450
- EGF
- epidermal growth factor
- TGase-2
- type II transglutaminase
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}