


Abstract
Human cytochrome P450 (P450) 2B6 plays an important role in the metabolism of many drugs used in the clinic, and it has been shown to be highly polymorphic and inducible by a variety of substrates. The metabolism of phencyclidine (PCP) by P450 2B6 results in mechanism-based inactivation of the enzyme. We investigated the effects of a naturally occurring mutation of P450 2B6 where a lysine 262 is changed to an arginine (K262R) on PCP metabolism and mechanism-based inactivation of 2B6 by PCP. The K262R mutant retained the 7-ethoxy-4-trifluoromethylcoumarin O-deethylation activity when it was incubated with PCP and NADPH in the reconstituted system, whereas the wild-type enzyme was readily inactivated by PCP. Spectral binding studies showed that PCP was reversibly bound in the active site of the K262R mutant with slightly higher affinity (156 μM) compared with the wild-type 2B6 (397 μM). In addition, all the metabolites of PCP (M1–M8) that were formed by the wild-type enzyme were also formed by the K262R mutant. Although the K262R mutant metabolized PCP to give similar metabolite profiles, the overall rate of metabolite formation was lower than the wild-type enzyme. A reactive intermediate of PCP was formed by wild-type P450 2B6 and trapped with glutathione (GSH). However, no GSH conjugates were detected from incubations with the K262R mutant. These data suggest that the lysine 262 residue plays an important role in the formation of a reactive intermediate of PCP that leads to the mechanism-based inactivation of P450 2B6.
The cytochrome P450 (P450) enzymes are a family of mixed-function oxidases that catalyze a wide variety of reactions. The mammalian P450s play a central role in drug metabolism and detoxification (Rendic, 2002). Although crystal structures of several mammalian P450s have been solved recently, it now appears that the active sites of the individual P450 may adopt multiple conformations in response to various ligands (Zhao et al., 2006). A variety of other methods have been used to determine the structure-function relationships of mammalian P450s to verify and provide further support for the crystallography data. Among these methods are mechanism-based inactivation (Kent et al., 2001) and studies with naturally occurring mutants (Biagini et al., 1999). These studies have increased our understanding of the active site structures of several P450s and the role of critical amino acid residues involved in substrate binding and/or catalysis. In studies that involve mechanism-based inactivation, a substrate of the P450 is converted during metabolism to a highly reactive intermediate that covalently and irreversibly modifies the P450 active site and renders the enzyme inactive (Kent et al., 2001). Therefore, mechanism-based inactivation is considered to be a promising tool for identifying critical structures in the active sites of P450s (Roberts et al., 1994; He et al., 1996; Lightning et al., 2000; Kent et al., 2006).
Human P450 2B6 plays an important role in the metabolism of a variety of substrates, including many drugs in clinical use. Miksys et al. (2003) have shown that P450 2B6 is inducible by nicotine, and its expression varies in certain areas of the human brain. In 2003, Jushchyshyn et al. (2003) characterized the inactivation of P450 2B6 by phencyclidine (PCP), a psychomimetic drug of abuse, and showed that the inactivation occurs in a mechanism-based manner via modification of the P450 apoprotein and not the heme moiety. The reactive intermediate responsible for the inactivation was subsequently identified by characterizing the thiol adducts of PCP (Shebley et al., 2006). 2B6 has also been shown to be highly polymorphic, and several single nucleotide polymorphisms have been identified, expressed in Escherichia coli systems, and characterized (Lang et al., 2001; Klein et al., 2005). The P450 2B6 K262R genetic polymorphism, also known as 2B6*4, has been shown to be associated with altered metabolism of several drugs, including the antidepressant bupropion (Kirchheiner et al., 2003). In addition, this mutant exhibits alterations in substrate metabolism, as well as inactivation in vitro as shown by Bumpus et al. (2005), in studies with a mechanism-based inactivator of the wild-type P450 2B6, 17-α-ethynylestradiol (17EE), which was not metabolized by the K262R mutant enzyme, nor did it cause inactivation.
In this study, we investigated the consequences of the K262R mutation of P450 2B6 on mechanism-based inactivation by PCP compared with the wild-type enzyme. The results reported here show that lysine 262 plays a central role in forming the reactive intermediate of PCP that is responsible for mechanism-based inactivation.
Materials and Methods
Materials. PCP hydrochloride, NADPH, glutathione (GSH), bovine serum albumin, and catalase were purchased from Sigma-Aldrich (St. Louis, MO). Trifluoroacetic acid (TFA) was purchased from Pierce Chemicals (Rockford, IL). 7-Ethoxy-4-trifluoromethylcoumarin (7-EFC) was from Molecular Probes (Eugene, OR). AccuBond II ODS-C18 solid-phase extraction (1-ml) cartridges were purchased from Agilent Technologies (Palo Alto, CA). PCP authentic standards cis-1-(1-phenyl-4-hydroxy-cyclohexyl)-piperidine HCl (M1), trans-1-(1-phenyl-4-hydroxy-cyclohexyl)-piperidine (M2), cis-1-(1-phenylcyclohexyl)-4-hydroxypiperidine (M3), trans-1-(1-phenylcyclohexyl)-4-hydroxypiperidine (M4), and 4-(4′-hydroxypiperidino)-4-phenylcyclohexanol (M6) were obtained from the National Institute on Drug Abuse (Research Triangle Park, NC). 1-(1-Phenylcyclohexyl)-2,3,4,5-tetrahydro-pyridinium perchlorate (M5) was a gift from Dr. Neal Castagnoli, Jr. (Virginia Polytechnic Institute and State University, Blacksburg, VA).
Protein Purification. P450s 2B6 and the K262R mutant were expressed in E. coli Topp3 cells and purified according to published protocols (Hanna et al., 2000) with some modifications. In the case of P450 2B6 K262R, the protein was purified from the supernatant following centrifugation of the lysozyme-treated and sonicated cells (Bumpus et al., 2005). After overnight treatment of the cell pellet with lysozyme (25 mg/l), the cells were centrifuged at 4000 rpm for 25 min at 4°C. The cell pellet was recovered and resuspended in 50 mM Tris-acetate, pH 8.0, buffer. The homogenized cells were sonicated for 12 min (6 cycles of 2 min on/2 min off). The lysed and sonicated cells were centrifuged at 10,000 rpm for 40 min at 4°C, and the P450-containing supernatant was kept for further purification. After adjusting to pH 8.0 by the addition of 2 mM imidazole, a nickel affinity column was used to purify the His-tagged protein.
Enzyme Activity Assays and Inactivation by PCP. P450 2B6 or the K262R mutant (1 μM) was reconstituted with 2 μM NADPH-P450 reductase (reductase) for 45 min at 4°C, after which 500 units of catalase was added in 50 mM potassium phosphate buffer (pH 7.4). The incubation mixture was divided into five 100-μl reaction tubes, and each reaction received 1 μlof dimethyl sulfoxide or PCP in dimethyl sulfoxide to final concentrations of 0, 10, 50, 80, and 100 μM. These primary reactions were preincubated at 30°C for 10 min, and the reactions were then initiated by adding NADPH to a final concentration of 1.3 mM while shaking in a water bath. At the indicated times, 10-μl samples (10 pmol of P450) of the primary reaction mixtures were transferred to 990 μl of a secondary reaction containing 7-EFC (100 μM), NADPH (0.2 mM), and bovine serum albumin (40 μg/ml) in 50 mM KPi (pH 7.4) and incubated at 30°C for 5 min. The enzyme activity was stopped by the addition of ice-cold acetonitrile to give a final concentration of 25%. The 7-EFC O-deethylation activity was measured spectrofluorometrically as described previously (Buters et al., 1993). The fluorescence of the O-dealkylated product was measured at room temperature on an RF5301 spectrofluorometer (Shimadzu Scientific Instruments, Inc., Columbia, MD) with excitation at 410 nm and emission at 510 nm.
Spectral Binding Properties. To determine the binding properties of PCP to P450 2B6 and the K262R mutant, spectral binding constants were measured using previously published methods (Estabrook and Werringloer, 1978). P450 2B6 or the K262R mutant was diluted in 50 mM KPi (pH 7.4) to a final concentration of 1 μM in a final volume of 2 ml, and equal volumes of each sample were placed in the reference and sample cuvettes. A difference spectrum between the two cuvettes was recorded from 350 to 500 nm on a DW2 UV-visible spectrophotometer (SLM Aminco, Urbana, IL) equipped with an OLIS spectroscopy operating system (On-Line Instrument Systems, Inc., Bogart, GA). Increasing concentrations of PCP (0.2–1.5 mM) in methanol were added to the sample cuvette, whereas the reference cuvette received equal volumes of methanol. The total volume of methanol added was 1% of the sample volume. The peak at 390 nm and the minimum at 420 nm were monitored for each scan.
Effects of Cytochromeb5 on the Inactivation of P450s by PCP. P450s 2B6 and the K262R mutant were reconstituted as described previously. Cytochrome b5 (1 μM) was then added 3 min after the addition of reductase, and the three enzymes were reconstituted for 45 min at 4°C. The activity assays and studies on the inactivation by PCP were performed as described above.
PCP Metabolism and Formation of GSH Conjugates. P450s 2B6 or the K262R mutant (1 μM) was reconstituted with reductase (2 μM) for 45 min at 4°C, after which catalase (1000 units/ml) and 0.2 mM PCP were added in 100 mM phosphate buffer, pH 7.4. The reaction mixture was preincubated for 10 min at 30°C, and the metabolism was initiated by the addition of NADPH to a final concentration of 1.2 mM. After 40 min, the reactions were stopped by the addition of TFA to a final concentration of 1%. The acidified samples were added to a 1-ml Accubond solid-phase extraction II ODS C-18 cartridge previously washed with 1 ml of methanol followed by 2 ml of water. After the samples were loaded, the cartridge was washed with 2 ml of water, and the metabolites were eluted sequentially with 2 ml of methanol, followed by 0.3 ml of acetonitrile. The organic samples were pooled and dried under a steady stream of nitrogen gas to approximately 50 μl. The dried samples were reconstituted with 100 μl of mobile phase A (10% methanol, 90% water, 0.05% TFA), and 50 μl was injected onto a Phenomenex (Torrance, CA) Luna C18 (3 μm, 4.6 × 100 mm) column (Agilent Technologies) and separated on a Hewlett Packard (Palo Alto, CA) 1100 series high-performance liquid chromatography system using a solvent system composed of solvent A and solvent B (90% acetonitrile, 10% methanol, 0.05% TFA). The column was equilibrated with 80% A and 20% B for 15 min before injecting the samples. A flow rate of 0.3 ml/min and a linear gradient of 20 to 50% B over 30 min were used to resolve the metabolites. The authentic standards (M1–M6) were resolved under the same conditions. Mass spectrometry was performed using a Finnigan MAT LCQ Classic mass spectrometer with an electrospray ionization (ESI) interface from the liquid chromatography system. The analytes were ionized in positive mode using a capillary voltage of 55 V, a spray voltage set at 4.5 kV, a tube lens offset voltage of 25 V, a capillary temperature of 170°C, and an auxiliary gas and sheath gas flow of 30 and 90, respectively. The data-dependent scanning mode was used to detect the PCP metabolites on the LCQ mass spectrometer.

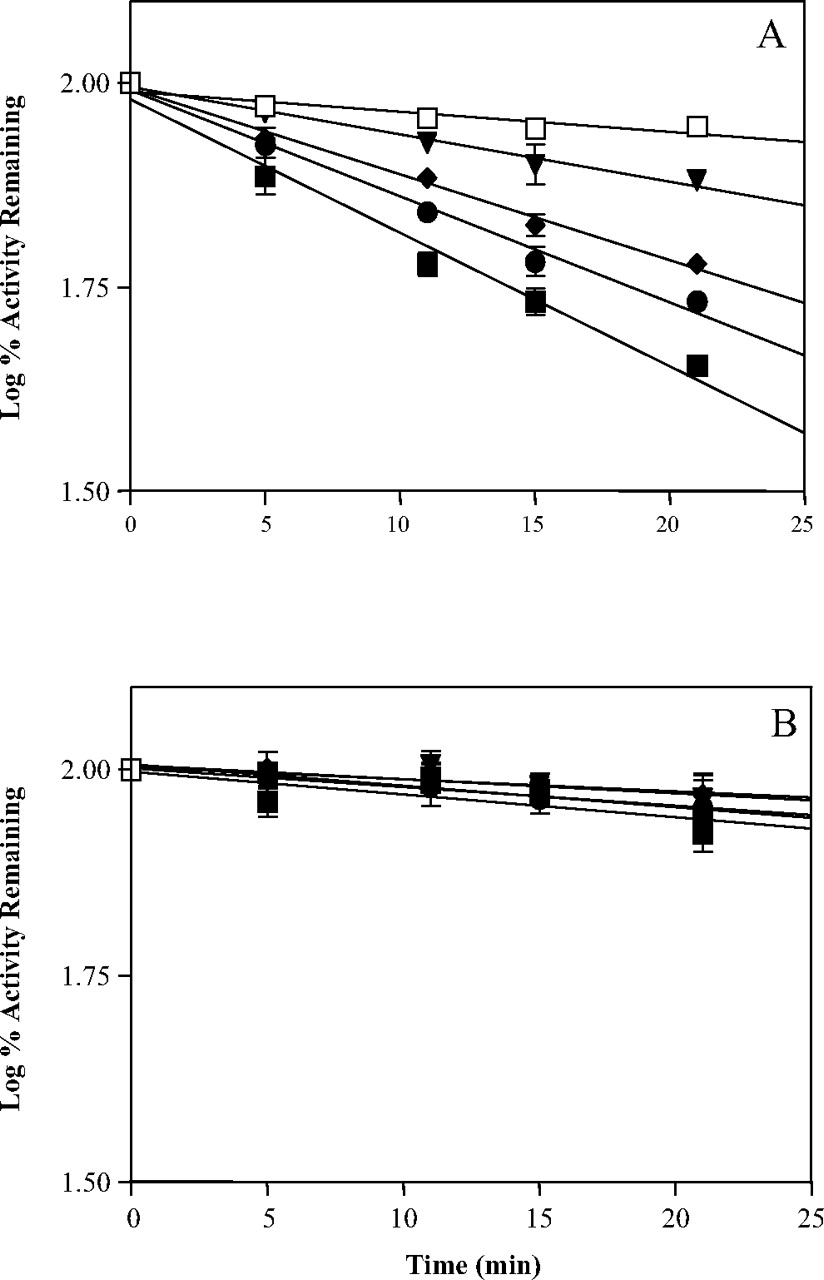
Time- and concentration-dependent inactivation of P450 2B6 by PCP. Incubation conditions were described under Materials and Methods. The concentrations of PCP used were as follows: 0 μM(□), 10 μM (▾), 50 μM (♦), 80 μM (•), and 100 μM (▪). Each point in A and B is reported as percent of control at time 0 and represents the mean and S.D. of three experiments performed in duplicates. A, incubation of PCP with P450 2B6 in the presence of 1.2 mM NADPH. B, incubation of PCP with P450 2B6 (K262R) in the presence of 1.2 mM NADPH.
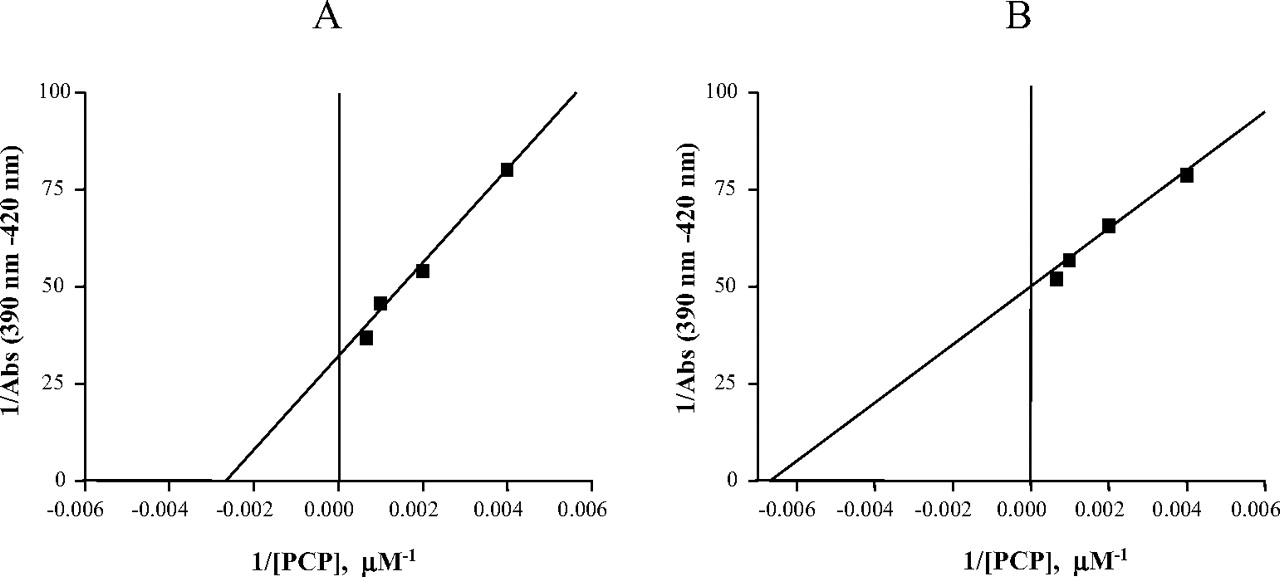
Determination of the binding constants for the binding of PCP to P450s 2B6. The conditions for the spectral titrations were described under Materials and Methods. The inverse of the changes in the P450 2B6 (A) and the K262R mutant (B) difference spectra (390–420 nm) were plotted versus the inverse of PCP concentrations. The binding constant (Ks) was calculated from the x-intercept (Estabrook and Werringloer, 1978).
Similar experiments were performed to trap and identify the GSH conjugates. For these experiments, the reaction mixtures were prepared as described above, and 10 mM GSH was added to the primary reaction mixtures. The GSH conjugates were extracted as described above for the PCP metabolites. Aliquots of each sample were injected onto a Zorbax Rx-C8 (5 μm, 4.6 × 250 mm) column (Agilent Technologies) and separated on a Surveyor high-performance liquid chromatography system (Thermo Electron, Waltham, MA) using a solvent system composed of solvents A and B. A flow rate of 1 ml/min and a linear gradient of 10 to 50% solvent B over 30 min were used to resolve the GSH conjugates. A Thermo Electron LTQ linear ion trap mass spectrometer equipped with an ESI source interfaced to the mass spectrometer was used to detect the GSH conjugates using the selected ion monitoring scanning mode. This scan mode allows the detection of specific parent ions of known m/z values that correspond to the GSH conjugates. Collision-induced dissociation (CID) with normalized collision energy of 22% was applied to the parent ions [MH]+. In addition, precursor ion scans at m/z 308 and constant neutral loss (–129 Da) were performed using the TSQ Quantum Ultra AM (Thermo Electron) to detect any GSH conjugates that had lost a molecule of GSH, or the pyroglutamate (–129 Da) moiety of GSH, on CID. In these experiments, a Phenomenex Luna C18 (3 μm, 4.6 × 100 mm) column and a solvent system composed of solvents A and B were used to resolve the GSH conjugates. A flow rate of 0.3 ml/min and a linear gradient of 20 to 50% B over 30 min were used. All the mass spectrometers and all the peripheral components were controlled by the Xcalibur software (Thermo Electron). The instrument parameters were optimized using the PCP standards.
Results
Lack of Inactivation of P450 2B6 K262R by PCP. Incubation of increasing concentrations of PCP with P450 2B6 caused significant losses in the 7-EFC O-deethylation activity compared with the control. As shown in Fig. 1A, the inactivation was time-, NADPH-, and concentration-dependent and exhibited pseudo–first-order kinetics. The KI was approximately 18 μM as calculated from the double-reciprocal plot generated from the slopes of the lines, and this is similar to what was previously reported (Jushchyshyn et al., 2003). As shown in Fig. 1B, incubations of the K262R mutant with up to 100 μM PCP did not result in any significant loss in activity, even when the reaction time was extended to 40 min (data not shown). These results show that lysine 262 is required for the inactivation of P450 2B6 by PCP.
Spectral Binding Analysis. Substrate interactions with P450 have been shown to result in changes in the Soret region of the UV-visible spectrum around 400 nm (Schenkman and Jansson, 2006). These spectral changes are classified as type I, where a peak appears at 385 to 390 nm and a trough at 420 nm, and type II, with a minimum at 390 to 405 nm and a maximum absorption peak at 425 to 435 nm in the difference spectra. Spectral titrations of P450s 2B6 and the K262R mutant were performed with PCP, and the resulting changes in the difference spectra were determined at each concentration of the substrate. The resulting changes in absorbance (the increase at 390 nm minus the decrease at 420 nm) were plotted as the inverse of the change in absorbance versus the inverse of PCP concentration. From these plots, the spectral binding constants (Ks) were determined. The Ks is defined as the concentration of the compound that results in 50% of the theoretical maximal spectral change (Estabrook and Werringloer, 1978). The binding of PCP to P450 2B6 and the K262R mutant produced spectra that are characteristic of type I compounds. Figure 2 shows the determination of the Ks values for the binding of PCP to P450 2B6 and the K262R mutant. Interestingly, the K262R mutant seems to bind PCP with slightly higher binding affinity (156 μM) than the wild-type enzyme (397 μM). These data suggest that the mutation of lysine 262 to arginine may have resulted in structural changes of the P450 active site that lead to tighter binding of PCP to the K262R mutant.
Investigation of the Potential Effects of Cytochromeb5 onInactivation. Bumpus et al. (2005) have previously shown that although 17EE readily inactivated wild-type 2B6 in the absence of cytochrome b5, it was not metabolized by the K262R mutant, and hence it had no effect on the enzymatic activity. The addition of cytochrome b5 to the reconstitution mixture supported the metabolism of 17EE by the K262R mutant and also led to inactivation of the enzyme (N. N. Bumpus et al., unpublished results). Therefore, we investigated the effects of this accessory protein on the inactivation of the K262R mutant by PCP. Although cytochrome b5 has been shown to be absolutely required for some reactions, its behavior has been shown to be dependent not only on the form of P450 involved but also on the substrate (Schenkman and Jansson, 2003). When P450 2B6 was incubated with an equimolar concentration of cytochrome b5 in the presence of PCP and NADPH, the enzyme was still inactivated by PCP and we observed no effect on the rate of inactivation. When the K262R mutant was reconstituted with cytochrome b5 under the same conditions, no apparent loss in activity was observed (Table 1). The K262R mutant was still able to catalyze the O-deethylation of 7-EFC, but because of the mutation, it was unable to exhibit inactivation by PCP, even when cytochrome b5 was added the reaction mixture. The data described so far raise the question as to whether the K262R mutant of 2B6 was able to metabolize PCP to the same products as the wild-type enzyme. Therefore, we tested the ability of the mutant enzyme to metabolize PCP and analyzed the metabolism by liquid chromatography (LC)/ESI/tandem mass spectrometry (MS/MS).
Effects of cytochrome b5 on the inactivation of P450s by PCP

LC/ESI/MS analysis of the metabolism of PCP by P450 2B6 (K262R). Incubations, extraction of metabolites, chromatography, and the LC/MS conditions were described under Materials and Methods. PCP was incubated for 40 min in a reconstituted incubation mixture containing P450 2B6 (K262R), 200 μM PCP, and 1.2 mM NADPH. The results shown in this panel represent the extracted ion chromatograms (XIC) of PCP metabolites. M1, cis-1-(1-phenyl-4-hydroxy-cyclohexyl)-piperidine; M2, trans-1-(1-phenyl-4-hydroxy-cyclohexyl)-piperidine; M3, cis-1-(1-phenylcyclohexyl)-4-hydroxy piperidine; M4, trans-1-(1-phenylcyclohexyl)-4-hydroxy piperidine; M5, the iminium ion of PCP; M5, the enamine of PCP; M6, 4-(4′-hydroxy piperidino)-4-phenylcyclo hexanol; M1 and M2 are positional isomers of M1 and M2, respectively; M7 and M8, trihydroxylated PCP.
Metabolism of PCP. As described previously (Shebley et al., 2006), several metabolites of PCP were detected by LC/ESI/MS of incubation mixtures with P450 2B6 in the reconstituted system. These metabolites are several monohydroxylated metabolites of PCP (M1–M4) with their corresponding cis/trans isomers where the hydroxylation occurs on either the piperidinyl or the cyclohexyl ring, and the iminium ion and its corresponding enamine of PCP (M5, M5), which are the products of oxidation on the piperidine ring. In addition, dihydroxylated (M6) and trihydroxylated (M7, M8) metabolites of PCP were detected from incubations with P450 2B6. The K262R mutant exhibited a similar metabolic profile (Fig. 3), in that all these PCP metabolites were still formed by the mutant and no new metabolites were observed. Quantitation of the metabolites formed by the K262R mutant showed that it produced PCP metabolites at a slightly decreased rate with the decrease ranging from 3 to 16%, with the magnitude of the decreases depending on the metabolite. The overall rate of metabolite formation was approximately 85% of the wild-type enzyme (data not shown). These data show that PCP binds to the active site of the K262R mutant and is metabolized by this variant to give the same products, albeit at a rate that is slightly decreased.
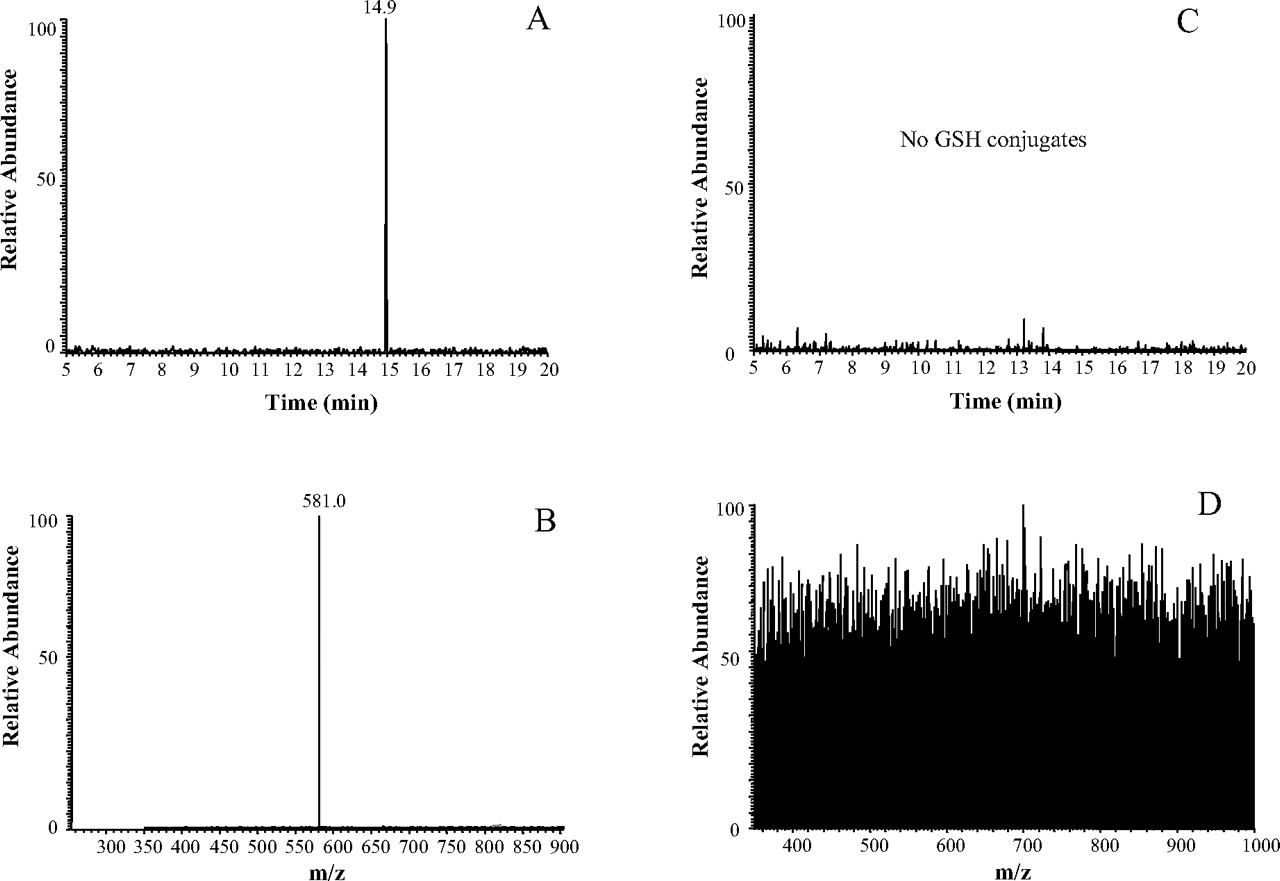
Trapping Reactive Intermediates of PCP. We have previously shown that the incubation of P450 2B6 with PCP in the presence of GSH resulted in the formation of a reactive intermediate-GSH conjugate that could be characterized by LC/ESI/MS/MS (Shebley et al., 2006). This PCP intermediate is believed to be a dihydroxylated iminium or a monohydroxylated epoxide species having an m/z of 275, and it exhibits an m/z of 581 on forming a conjugate with GSH. As shown in Fig. 4, this GSH conjugate with an m/z of 581 eluted at 17.5 min from incubation mixtures of P450 2B6 with PCP and GSH, and was detected using the selected ion monitoring scanning mode. In contrast, this GSH conjugate could not be detected in any of the samples that were analyzed from incubation mixtures containing the K262R mutant. These results strongly suggest that although PCP was metabolized by the K262R mutant to give the same stable products, the mutant enzyme was unable to form the specific reactive intermediate of PCP responsible for inactivating the wild-type enzyme and which reacted with GSH to give the conjugate shown in Fig. 4. In attempt to detect any other reactive intermediates that might have been formed during the metabolism of PCP by the K262R mutant and that could be trapped as their corresponding GSH conjugates, we analyzed the samples using a precursor ion scan mode that would selectively detect any parent ion that produces a product ion having an m/z 308 (protonated GSH), along with other experiments where we used constant neutral loss scanning (–129 Da) to detect the loss of the γ-glutamyl moiety of GSH. The same GSH conjugate was again detected from incubations with the wild-type enzyme (Fig. 5, A and B) using precursor ion scanning; however, we did not detect any GSH conjugates from incubations with the K262R mutant (Fig. 5, C and D) under the same conditions. These data indicate that the K262R mutant is able to bind and metabolize PCP. However, it appears to be unable to form a reactive intermediate that can modify the active site and lead to mechanism-based inactivation.

Analysis of GSH conjugates of PCP by LC/ESI/MS/MS. The sample was obtained from incubation of the reconstituted system containing P450 2B6 with 200 μM PCP, 1.2 mM NADPH, and 10 mM GSH. The sample was prepared as described under Materials and Methods. The top panel shows the XIC of a GSH conjugate with an m/z of 581, which eluted at approximately 17.5 min. Selected ion monitoring for the parent mass of 581 was used to scan for this conjugate. The bottom panel shows the MS/MS spectrum of the peak eluting at 17.5 min and the fragmentation assignments for this GSH conjugate.
Discussion
The identification of critical amino acid residues in P450 active sites has been approached by several methods. Site-directed mutagenesis (Fraser et al., 1999), analysis of naturally occurring polymorphisms (Biagini et al., 1999), and X-ray crystallography of mammalian P450s (Johnson and Stout, 2005) have all been shown to be useful for obtaining information concerning those residues that may be involved in substrate binding and/or catalysis. Although the crystal structures of several mammalian P450s have been solved (Johnson et al., 2002; Scott et al., 2003, 2004; Williams et al., 2003, 2004; Johnson and Stout, 2005; Yano et al., 2005; Rowland et al., 2006; Zhao et al., 2006), other approaches are still required to validate and support these structures using data from proteins in solution. Another valuable method is mechanism-based inactivation, where the P450 metabolizes a substrate molecule to generate a reactive intermediate that can covalently bind to amino acid residue(s) essential to the function of the enzyme, thereby inactivating it. This tool has been shown to be successful in providing important information on the active site architecture of several P450s (Roberts et al., 1994; He et al., 1996; Kent et al., 2006). In this study, we showed that a naturally occurring P450 2B6 K262R mutant retains its ability to bind and metabolize PCP but is not able to form a reactive intermediate that can form a covalent adduct with the protein or with GSH.
The lysine 262 in 2B6 is believed to lie outside of the active site in the loop connecting helices G and H based on an analysis of the crystal structure of the closely related rabbit P450 2B4 (Scott et al., 2004). Therefore, this residue may not be involved in direct interactions with substrates in the active site, but it may influence other regions of the active site to cause functional differences. Interestingly, the lysine 262 is unique to P450 2B6 because sequence alignments with other P450 2B isoforms, including the rabbit 2B4, reveal a conserved arginine at this position (data not shown). The crystal structure of the 4-(4-chlorophenyl)-imidazole-bound P450 2B4 indicates that arginine 262 is located in a region that results in the highest structural deviations between the substrate-free “open” structure of 2B4 and the inhibitor-bound “closed” form (Scott et al., 2004). Therefore, Scott et al. (2004) concluded that interactions on the proximal end of the protein between the G/H and C/D loops might provide a physical mechanism for coordinating ligand-induced conformational changes on the distal side of the protein to electron delivery on the proximal side.

LC/ESI/MS analysis using the precursor ion scanning mode at m/z of 308. A, total ion chromatogram (TIC) of a sample from incubation of P450 2B6 with 200 μM PCP, 1.2 mM NADPH, and 10 mM GSH. The peak at 14.9 min is from a precursor ion scan of a species that lost an m/z of 308 (GSH) on CID. B, full-scan MS data observed at 14.9 min showing that the major species has an m/z of 581. C, TIC of a sample from incubation of the K262R mutant with 200 μM PCP, 1.2 mM NADPH, and 10 mM GSH. D, full-scan MS data at 14.9 min, where no major ions were detected at this retention time.
Based on the observations made from the crystal structures of P450 2B4, and the high sequence homology between 2B4 and 2B6, we hypothesized that the K262R mutation might possibly affect the active site architecture of P450 2B6, leading to changes in substrate binding and/or metabolism. Our results show that the K262R mutant is catalytically active using the 7-EFC O-deethylation assay and that the metabolism of PCP by the mutant no longer affects the enzyme's activity. The binding or orientation of PCP within the active site of P450 2B6 may have changed somewhat as a result of the K262R mutation, as indicated by the change in the Ks of PCP with the mutant enzyme, which indicates a slightly tighter binding affinity of PCP to the K262R active site compared with binding to the wild-type enzyme. This suggests that lysine 262 may facilitate a more favorable binding orientation of PCP in the active site of the wild-type P450 2B6 via interactions with neighboring residues that leads to formation of the inactivating species; however, the enzyme is unable to create such a favorable binding orientation as a consequence of the K262R mutation, which saves the enzyme from inactivation.
It has previously been shown that the mechanism-based inactivator 17EE did not inactivate the K262R mutant because it was not metabolized by this enzyme (Bumpus et al., 2005). However, the addition of cytochrome b5 to the reconstitution mixture resulted in both the metabolism of 17EE and the inactivation of the K262R mutant (N. N. Bumpus and P. F. Hollenberg, unpublished observations). Our data show that cytochrome b5 has no effect on either the inactivation of the wild-type or mutant P450 2B6, suggesting that the mechanism of inactivation by PCP does not require the presence of cytochrome b5 in the reconstitution mixture with P450 2B6 and that it does not support the inactivation of the K262R mutant, as was seen with 17EE.
Our metabolism data indicate that the K262R mutant metabolizes PCP to give the same stable metabolites obtained with the wild-type enzyme, indicating that the mutant is catalytically capable of carrying out metabolism of PCP. In addition, the Michaelis-Menten kinetics for 7-EFC O-deethylation by the wild-type 2B6 show a Km = 0.6 μM and Vmax = 0.1 pmol 7-HFC/min/pmol P450, whereas the *4 variant has an increase in the Km (1.3 μM) and Vmax (0.3 pmol 7-HFC/min/pmol P450) and a slight increase (1.4-fold) in the Vmax/Km relative to the wild-type enzyme (M. Shebley and P. F. Hollenberg, unpublished observations). These results are similar to a previously published report by Jinno et al. (2003) and show that these two enzymes are not all that different catalytically. However, the data on the GSH conjugate strongly suggest that the reactive intermediate of PCP can only be generated during metabolism by the wild-type P450 2B6 and can be detected as the trapped GSH conjugate, whereas the K262R mutant could not form this reactive intermediate that can result in conjugate formation (Figs. 4 and 5). The structure of the GSH conjugate suggests that PCP must undergo multiple oxidations by the wild-type 2B6 to generate the reactive intermediate. The initial step may involve formation of an epoxide or a diene that could react with water to give the diol and then form the final iminium that then reacts with GSH. Support for multiple oxidation reactions is evident from the observation of dihydroxylated and trihydroxylated products of PCP. The fact that the K262R mutant was not able to form a reactive intermediate of PCP that could be trapped with GSH or lead to inactivation suggests that this enzyme is not capable of carrying a secondary oxidation step that could lead to the formation of the inactivating species or to conjugation with GSH. Additional characterization of the structures of the metabolites of PCP formed by the K262R mutant using NMR and improved separation of isomeric products may shed additional light on the differences in the metabolism and formation of reactive intermediates between the wild-type P450 2B6 and its K262R mutant.
It is possible that the substitution of an arginine for a lysine may lead to increased hydrogen bonding with neighboring residues, such as Lys 139, Arg 145, Gly 185, Thr 255, Asp 257, Asp 263, or Asp 266, which lie within 4Å from the Lys 262 of 2B6 and are located in the C/D loop, D helix, E/F loop, G helix, G/H loop (Asp 257 and 263), and H helix, respectively. These interactions may result in a slight modification of the active site architecture of the enzyme, leading to a binding orientation of PCP that prevents inactivation, while at the same time preserving its metabolism. However, at this time we cannot rule out the possibility that the K262R mutant is able to generate the inactivating species of PCP during metabolism, but that because of the mutation and subsequent structural changes to the site of adduct formation, the position or orientation of the target residue becomes modified such that the reactive intermediate can no longer interact with its target to cause covalent adduction that would lead to inactivation of the enzyme. To rule out the possibility that the lysine 262 residue is a direct target for adduct formation, we performed experiments using incubations of wild-type P450 2B6 with PCP in the presence of N-acetyl lysine as a trapping agent in attempt to form PCP adducts with the lysine. The results showed no adduct formation with the lysine (data not shown), suggesting that lysine 262 is not the residue that becomes modified directly by the reactive intermediate of PCP.
Taken together, the data presented here provide insights into the role of lysine 262 in the inactivation of P450 2B6 by PCP. Our studies with the P450 2B6 K262R mutant suggest that the lysine 262 of 2B6 may be critical for the formation of the PCP-reactive intermediate that leads to mechanism-based inactivation of the enzyme.
Acknowledgments
We thank Dr. Ute M. Kent for her helpful suggestions and advice during the course of this study. We also thank Hsia-lien Lin for the purification of reductase.
Footnotes
-
This work was supported in part by National Institutes of Health Grant CA-16954 (P.F.H.) and a predoctoral fellowship from Merck & Co. Inc. (M.S.).
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.107.014985.
-
ABBREVIATIONS: P450, cytochrome P450; PCP, phencyclidine; 17EE, 17-α-ethynylestradiol; GSH, glutathione; TFA, trifluoroacetic acid; 7-EFC, 7-ethoxy-4-trifluoromethylcoumarin; ESI, electrospray ionization; CID, collision-induced dissociation; LC, liquid chromatography; MS/MS, tandem mass spectrometry.
- Received January 29, 2007.
- Accepted April 24, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}