Article Text

Abstract
Alzheimer’s disease is one of the most common causes of mental deterioration in elderly people, accounting for around 50%-60% of the overall cases of dementia among persons over 65 years of age. The past two decades have witnessed a considerable research effort directed towards discovering the cause of Alzheimer’s disease with the ultimate hope of developing safe and effective pharmacological treatments. This article examines the existing scientific applicability of the original cholinergic hypothesis of Alzheimer’s disease by describing the biochemical and histopathological changes of neurotransmitter markers that occur in the brains of patients with Alzheimer’s disease both at postmortem and neurosurgical cerebral biopsy and the behavioural consequences of cholinomimetic drugs and cholinergic lesions. Such studies have resulted in the discovery of an association between a decline in learning and memory, and a deficit in excitatory amino acid (EAA) neurotransmission, together with important roles for the cholinergic system in attentional processing and as a modulator of EAA neurotransmission. Accordingly, although there is presently no “cure” for Alzheimer’s disease, a large number of potential therapeutic interventions have emerged that are designed to correct loss of presynaptic cholinergic function. A few of these compounds have confirmed efficacy in delaying the deterioration of symptoms of Alzheimer’s disease, a valuable treatment target considering the progressive nature of the disease. Indeed, three compounds have received European approval for the treatment of the cognitive symptoms of Alzheimer’s disease, first tacrine and more recently, donepezil and rivastigmine, all of which are cholinesterase inhibitors.
Statistics from Altmetric.com
Alzheimer’s disease affects an estimated 15 million people worldwide and is the leading cause of dementia in elderly people. With the proportion of elderly people in the population increasing steadily, the burden of the disease, both to carers and national economies, is expected to become substantially greater over the next 2 to 3 decades.
Alzheimer’s disease is a progressive neurodegenerative disorder with a mean duration of around 8.5 years between onset of clinical symptoms and death. Brain regions that are associated with higher mental functions, particularly the neocortex and hippocampus, are those most affected by the characteristic pathology of Alzheimer’s disease. This includes the extracellular deposits of β-amyloid (derived from amyloid precursor protein; APP) in senile plaques, intracellular formation of neurofibrillary tangles (containing an abnormally phosphorylated form of a microtubule associated protein, tau), and the loss of neuronal synapses and pyramidal neurons. These changes result in the development of the typical symptomology of Alzheimer’s disease characterised by gross and progressive impairments of cognitive function and often accompanied by behavioural disturbances such as aggression, depression, and wandering. Carers find these features the most difficult to cope with and they often lead to the need for institutionalisation of the patient.1
The systematic biochemical investigation of the brains of patients with Alzheimer’s disease began in the late 1960s and early 1970s. The hope was that a clearly defined neurochemical abnormality would be identified, providing the basis for the development of rational therapeutic interventions analogous to levodopa treatment of Parkinson’s disease. Support for this perspective came in the mid-1970s with reports of substantial neocortical deficits in the enzyme responsible for the synthesis of acetylcholine (ACh), choline acetyltransferase (ChAT).2-4 Subsequent discoveries of reduced choline uptake,5 ACh release6 and loss of cholinergic perikarya from the nucleus basalis of Meynert7 confirmed a substantial presynaptic cholinergic deficit.
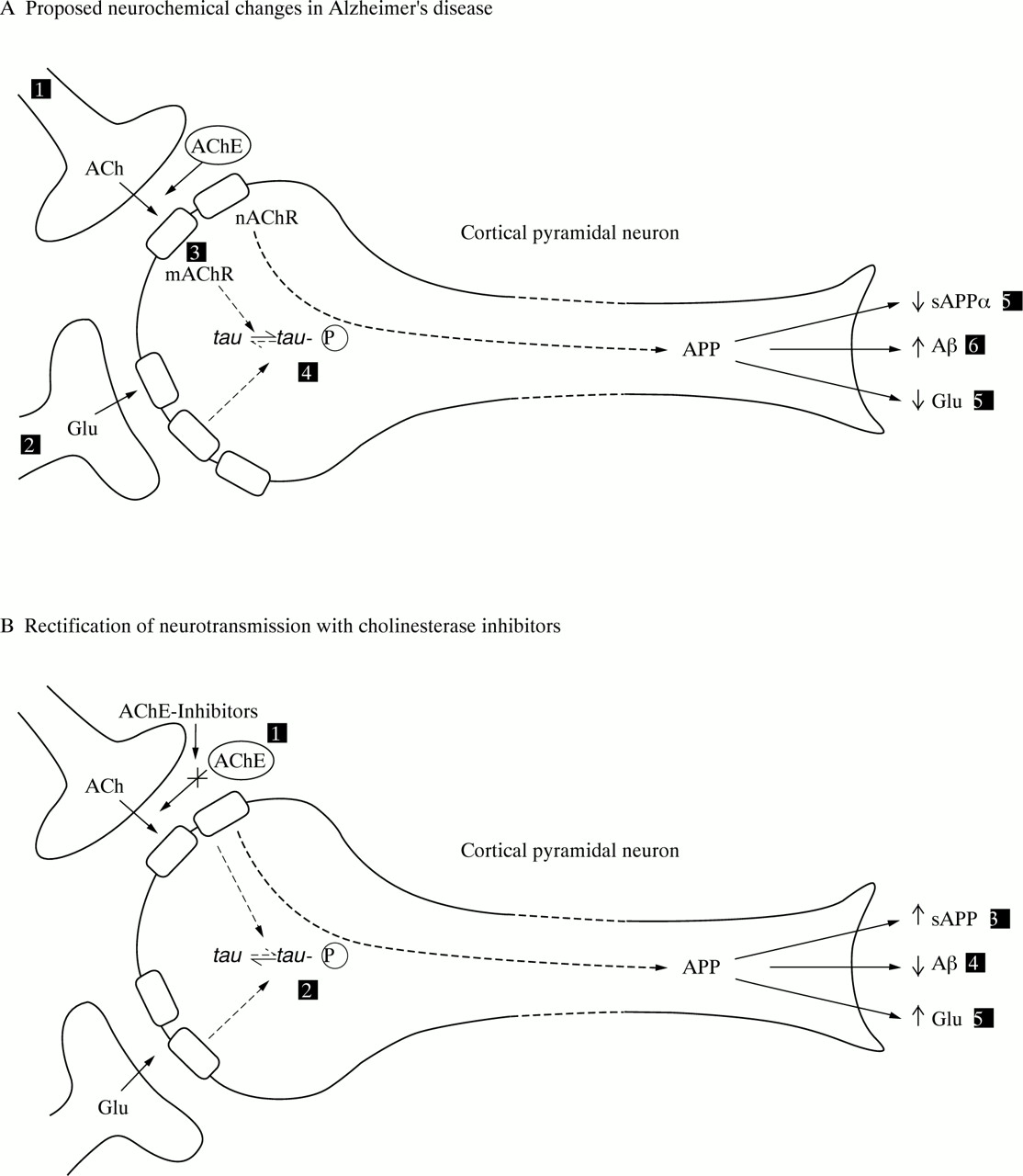
These studies, together with the emerging role of ACh in learning and memory,8 led to the “cholinergic hypothesis of Alzheimers disease” (figure A). Thus it was proposed that degeneration of cholinergic neurons in the basal forebrain and the associated loss of cholinergic neurotransmission in the cerebral cortex and other areas contributed significantly to the deterioration in cognitive function seen in patients with Alzheimer’s disease.9

{kind=link}
Schematic diagram of a neuron representing (A) alterations in neurotransmission in Alzheimer’s disease and (B) the hypothetical mode of action of AChE inhibitors. Key to figure (A): (1) reduced cortical cholinergic innervation; (2) reduced corticocortical glutamatergic neurotransmission due to neuron or synapse loss; (3) reduced coupling of muscarinic M1 receptors to second messenger system?; (4) shift of tau to the hyperphosphoryalted state—precursor of neurofibrillary tangles; (5) reduced secretion of soluble APP; (6) increased production of β-amyloid protein; (7) decreased glutamate production. *It is hypothesised that these changes give rise to the clinical symptoms of Alzheimer’s disease and contribute to the spread of pathology.12 49 54 Key to figure B: (1) AChE inhibitors reduce the breakdown of endogenously released ACh, resulting in greater activation of postsynaptic ACh receptors; hypothesised consequences: (2) reduced phosphorylation of tau; (3) secretion of sAPP returned towards normal; (4) reduced β-amyloid production; (5) glutamatergic neurotransmission returns towards normal, possibly due to activation of muscarinic and nicotinic receptors. ACh=acetylcholine; mAChR=ACh muscarinic receptor; APP=a myloid precursor protein; AChE=acetylcholinesterase; nAChR=ACh nicotinic receptor; Glu=glutamate.
Over the 20 years since the origins of the cholinergic hypothesis, data from numerous studies have challenged its veracity as an explanation for the syndrome of dementia in Alzheimer’s disease. Thus, this review attempts to re-evaluate the cholinergic hypothesis in the following ways:
(1) Setting the original findings of reduced cholinergic neurotransmission in the context of changes in other neurotransmitter systems, a clear understanding of the behavioural role of the cholinergic system, and a more detailed understanding of the molecular pathology of the disease.
(2) Charting the preclinical and clinical development of cholinomimetic drugs for the symptomatic treatment of Alzheimer’s disease, focusing on the first generation and second generation cholinesterase inhibitors currently available.
Neurochemical and histopathological changes in cholinergic and non-cholinergic neurons in Alzheimer’s disease
At postmortem, Alzheimer’s disease is characterised by neuronal loss and neurofibrillary tangle formation in circumscribed regions of the neocortex and hippocampus, primarily affecting pyramidal neurons and their synapses.10 11 Neurotransmitter specific subcortical nuclei that project to the cortex are also affected by neurodegenerative processes, including the cholinergic nucleus basalis of Meynert and medial septum, the serotonergic raphe nuclei, and the noradrenergic locus coeruleus.
Biochemical investigations of biopsy tissue taken from patients with Alzheimer’s disease 3.5 years (on average) after the onset of symptoms indicate that a selective neurotransmitter pathology occurs early in the course of the disease.12 Specifically, presynaptic markers of the cholinergic system appear uniformly reduced. This is exemplified by reductions in ChAT activity and ACh synthesis which are strongly correlated with the degree of cognitive impairment in patients with Alzheimer’s disease.12-15 Whereas serotonergic and some noradrenergic markers are affected, markers for dopamine, γ-aminobutyric acid (GABA), or somatostatin are not altered.12 When postmortem studies of Alzheimer’s disease brain are considered (typically representing a later stage of the disease) many more neurotransmitter systems are involved or are affected to a greater extent. These include GABA16 17 and somatostatin18 19 and may indicate that cortical interneurons, for which these are neurochemical markers, are affected later in the disease process. Based on postmortem studies, however, changes in serotonergic neurotransmission may be linked to the behavioural disturbances of Alzheimer’s disease such as depression, rather than cognitive dysfunction.1 20 21
On the basis of the above evidence, neocortical cholinergic innervation is probably lost at an early stage of the disease, a conclusion substantiated by evidence for similar changes in patients that have displayed clinical symptoms for less than 1 year.22However, although the loss of cholinergic function is correlated with the cognitive impairment in Alzheimer’s disease, an association between two such indices does not necessarily indicate a causal relation. Other indices also correlate with measures of cognitive decline in Alzheimer’s disease, such as loss of synapses and pyramidal cell perikarya.23 Moreover, a few patients with Alzheimer’s disease do not show large decreases in ChAT activity, albeit that a small reduction is found in the amygdala.24In addition, patients with inherited olivopontocerebellar atrophy have diminished ChAT activity of a magnitude similar to that seen in Alzheimer’s disease in the absence of cognitive deficits.25 Thus, although diminished ChAT activity is a necessary correlate of Alzheimer’s disease, additional factors other than impaired cholinergic function are likely to participate in the decline in cognitive function. Other studies have demonstrated a reduction in the number of nicotinic26 and muscarinic (M2) ACh receptors in Alzheimer’s disease brains, most of which are considered to be located on presynaptic cholinergic terminals, but a relative preservation of postsynaptic muscarinic (M1, M3) receptors.27 However, there is some evidence for a disruption of the coupling between the muscarinic M1 receptors, their G-proteins, and second messenger systems.28
In addition to cholinergic dysfunction, other strong correlates of dementia are the chemical and histopathological markers of excitatory amino acid (EAA) releasing cortical pyramidal neurons. These neurons, considered to contribute to normal cognitive function in their own right, also seem to have a pivotal role in cholinergic function as they are cholinoceptive.29-32 Although neurochemical studies of EAA neurotransmission have failed to show profound or extensive alterations in EAA neuronal indices,12 this may be related to the difficulty in distinguishing the transmitter pool of aspartate and glutamate from the metabolic pool. Nevertheless, glutamate concentration was reduced by 14% in temporal lobe biopsy samples of patients with Alzheimer’s disease. Greater reductions were evident at postmortem in regions enriched with EAA nerve terminals.33Uptake of d-aspartate, a putative marker of EAA nerve endings, is also reduced in many cortical areas in Alzheimer’s disease brains.34-36
Arguably, in vivo imaging studies of patients with Alzheimer’s disease also support the involvement of pyramidal neurons in the disease as the pattern of regional hypometabolism parallels neuronal loss/atrophy, tangle formation, and synapse loss.10 37-39 Loss of cortical pyramidal neurons,23 40 41 synapse loss,40 and reduced glutamate concentration,17 together with the formation of neurofibrillary tangles,42 all correlate with the severity of dementia. These findings indicate that pyramidal neurons and their transmitter glutamate (and/or aspartate) play a part in the cognitive symptoms of Alzheimer’s disease and may therefore represent an additional therapeutic target. However, these neurons are cholinoceptive and it is reasonable to propose that one of the actions of cholinomimetic drugs for the treatment of Alzheimer’s disease is to increase the activity of EAA neurons through muscarinic and nicotinic receptors that are present on such cells.29 This is supported by electrophysiological studies showing the excitatory actions of cholinomimetic drugs in cortical pyramidal neurons from both rats and humans30 31 and microdialysis studies in rats.32 Clearly, as a result of cholinergic and other pyramidal neuronal loss, the profound reduction in EAA neurotransmission will lead to pyramidal hypoactivity compounded by maintained levels of inhibition by GABAergic neurons. Consequently, it may be hypothesised that in addition to the deleterious effects of neuronal loss and tangle formation, there is a change in the balance of neurotransmission in the Alzheimer’s disease brain favouring lower neuronal activity.12 This may be reflected in the hypometabolism in patients with Alzheimer’s disease seen with imaging techniques, although a component of this is also likely to be due to neuronal atrophy.43 Likewise, it is of interest that regional cerebral blood flow may be increased in patients with Alzheimer’s disease by cholinesterase (ChE) inhibitors such as physostigmine.44 45
CHOLINERGIC AND NON-CHOLINERGIC NEURONS AND ALZHEIMER’S DISEASE NEUROPATHOLOGY
The discovery that rare mutations in the gene encoding for APP always led to Alzheimer’s disease in family members carrying the defect resulted in the proposal of the “amyloid cascade hypothesis” of Alzheimer’s disease.46 Thus, the mismetabolism of APP leading to increased production of β-amyloid was proposed as the critical event in both familial and sporadic Alzheimer’s disease with other changes, tangles, neuron loss, synapse loss, and neurotransmission dysfunction, following as a consequence. Cholinergic neurotransmission may be a specific target for β-amyloid, as it has been shown to reduce both choline uptake and ACh release in vitro.47 48 It is of interest here that disease related changes in the Alzheimer’s disease brain are focused on pyramidal neurons in that these cells are lost in the disease, subject to tangle formation, represent a major source of APP (and hence, a site for its mismetabolism leading to increased β-amyloid production) and are regulated by a neurotransmitter (ACh), affected early in the disease. These neurons therefore seem to have a central role in the clinical symptoms as well as in the pathophysiology of the disease. Observations in cell lines and primary neuronal cultures that the activation of muscarinic, metabotropic glutamate, and other phospholipase C-linked receptors favours the non-amyloidogenic processing of APP49 suggests that compounds being developed for symptomatic treatment may have a serendipitous effect on the continuing emergence of pathology by reducing the production of β-amyloid. Furthermore, β-amyloid neurotoxicity is attenuated by muscarinic agonists.50 No data have yet been reported regarding the potential beneficial effects of cholinomimetic drugs on either increasing APP or reducing β-amyloid production in patients with Alzheimer’s disease. There is, however, some evidence for reductions in CSF fluid APP in depressed patients receiving drugs with anticholinergic side effects.51 Clearly, long term studies are called for to test this hypothesis in the patient population. However, this may raise ethical problems—for example, the need for serial lumbar puncture and the justification for groups of patients to act as placebo controls.
Other studies have shown that the phosphorylation of tau, thought to be an important step in the formation of tangles (which occur predominately in EAA cortical pyramidal neurons), may also be influenced by the phospholipase C second messenger system.52 Thus, after muscarinic cholinergic receptor stimulation, activation of protein kinase C may lead to the inactivation of a protein kinase (GSK-3) which phosphorylates tau, in vitro, in a similar manner to that found in Alzheimer’s disease.52 In support of this tenet, neuronal cells in culture transfected with M1 muscarinic receptors show reduced phosphorylation of tau after treatment with cholinergic agonists.53 Therefore, as a consequence of reduced cholinergic activity, reduced activation of protein kinase C may lead to a higher level of activity of GSK-3 and hence hyperphosphorylation of tau. Thus, if these neurotransmitter-protein interactions occur in the Alzheimer’s disease brain, it is not inconceivable that the changes in the balance of neurotransmission in the Alzheimer’s disease brain may contribute to increased tau phosphorylation and β-amyloid production and hence neurodegeneration in selectively vulnerable regions. Furthermore, it is possible that ChE inhibitors may reduce the histopathological features of disease progression.
On the basis of recent studies of Alzheimer’s disease, a glutamatergic hypothesis of Alzheimer’s disease has been proposed as an auxiliary to the cholinergic hypothesis.12 54 Thus, the cholinergic hypothesis may be refined to include the idea that a major target of cholinomimetic action is EAA pyramidal neurons, and that cholinergic hypofunction compounds the loss of EAA function. Together these systems may be largely responsible for the neuropsychological deficits and may contribute to the continuing emergence of pathology in patients with Alzheimer’s disease. This revised cholinergic hypothesis provides a stronger case for the continued development of cholinomimetic drugs for the symptomatic treatment of Alzheimer’s disease.
Behavioural consequences of cholinomimetic drugs and cholinergic lesions
Many pharmacological studies have examined the effect of cholinomimetic drugs and cholinergic receptor antagonists on learning and memory tasks. The most commonly used model is based on the finding that scopolamine, a muscarinic receptor antagonist, induces amnesia in young healthy subjects comparable with that in old, untreated subjects.8 These deficits may be reversed by ChE inhibitors. Compounds that reverse these scopolamine induced deficits in experimental animals may be considered as potential drugs to treat cognitive impairment.
It is, however, difficult to separate reliably the effects on learning and memory processes from effects on other behavioural domains. For example, methylscopolamine (which does not cross the blood-brain barrier) is as active as scopolamine in several models of cognitive function,55 56 indicating that peripheral changes induced by these compounds indirectly influence performance in cognitive tasks. It is, therefore, very important to distinguish central versus peripheral effects of cholinminetic agents. Scopolamine induced impairment of performance may also be mediated by direct effects on sensorimotor function or motivation deficits.56 57Further, it is likely that the scopolamine induced impairment in the performance of both experimental animals and humans in the delayed matching to position task (a commonly used test of cognitive function) is secondary to attentional deficits that are induced by the drug.58 59
Both hippocampal and cortical areas of the brain receive major cholinergic input from basal forebrain nuclei. Thus, the lesioning of these nuclei has been used to model cholinergic denervation in Alzheimer’s disease and to establish the behavioural consequences of cholinergic deafferentation. The most significant and consistent effects of such cholinergic lesioning on learning and memory follow lesioning of cholinergic pathways that lead to the hippocampus.60 61 Initial studies used stereotaxic injection of ibotenic acid to lesion cholinergic nuclei, and caused profound deficits in discrimination learning and memory. However, injection of the toxins quisqualic acid and α-amino-3-hydroxy-5-methyl-4-isoxazole (AMPA) into the same site causes a greater loss of ChAT activity than ibotenate but only marginal impairments in the same range of cognitive tasks.62 Thus, in addition to the established role for ACh in learning and memory, there are data to suggest that ACh also plays a critical part in attentional processing.63-65 This is supported by a study showing that both tacrine and nicotine improve attentional functions in patients with Alzheimer’s disease.66
Cholinomimetic therapy in Alzheimer’s disease
A prediction of the cholinergic hypothesis is that drugs that potentiate central cholinergic function should improve cognition and perhaps even some of the behavioural problems experienced with Alzheimer’s disease. There are a number of approaches to the treatment of the cholinergic deficit in Alzheimer’s disease, most of which have initially focused on the replacement of ACh precursors (choline or lecithin) but these agents failed to increase central cholinergic activity. Other studies have investigated the use of ChE inhibitors that reduce the hydrolysis of ACh (figure, B)—for example, physostigmine. More recent investigational compounds include specific M1 muscarinic or nicotinic agonists, M2 muscarinic antagonists, or improved “second generation” ChE inhibitors (table).
Cholinomimetic drugs in clinical development for Alzheimer’s disease including European registrations
Additional potential symptomatic therapeutic avenues relevant to the cholinergic hypothesis of Alzheimer’s disease have resulted from the rapid development in the understanding of the molecular pathology of the disease. For example, during the development of cholinergic neurons in the basal forebrain, they express functional nerve growth factor (NGF) receptors. In adult life, these neurons seem to remain responsive to NGF. Consequently, intraventricular administration of NGF has been shown to prevent the lesion induced loss of cholinergic neuronal cell bodies and to accelerate the recovery of behavioural deficits in learning.67 Another approach is the transplantation of ACh rich foetal tissue grafts, which has been shown to improve the cognitive performance of primates after excitotoxic lesions of cholinergic nuclei.68 Thus, although such approaches may provide additional future possibilities for the palliative treatment for Alzheimer’s disease, the use of ChE inhibitors is the most well developed approach to treatment to date.
PRECLINICAL STUDIES OF CHOLINESTERASE INHIBITORS
Although a variety of ChE inhibitors have been developed as potential treatments for Alzheimer’s disease, their pharmacological activities differ. One of the most fundamental differences between them is in the mechanism of ChE inhibition. For example, enzyme kinetic studies have shown that tacrine, an acridine compound, and donepezil, a novel piperidine class agent, are “mixed type” reversible inhibitors of ChE. That is, these compounds inhibit ChE via both non-competitive (by blockade of the deacetylation process) and ACh competitive mechanisms.69 Thus, these compounds reversibly bind to the hydrophobic region of the enzyme to “allosterically” modulate catalytic activity. Further, the inhibition produced by these compounds is mutually exclusive, suggesting that both compounds act at similar sites within the enzyme, although donepezil is more potent and selective.70 71
This type of inhibition differs from that produced by the carbamates— for example, rivastigmine and physostigmine derivatives such as heptylphysostigmine. This class of compounds have been termed “pseudoirreversible” ChE inhibitors, in that they are actually cleaved by the enzyme, resulting in a covalent modification of the enzyme. Such inhibition is non-competitive with ACh and is irreversible. However, the association of the carbamate with the esteric site is transient (taking several minutes) due to both rapid metabolism and the relative rapid rate of decarbamylation which regenerates ChE.72-74 A further compound, metrifonate, inhibits ChE irreversibly. Metrifonate is a prodrug that is converted into dichlorvos, an organophosphorus ChE inhibitor with a very long duration of inhibition (the half life is 52 days).75
The principal influence of the mechanism of action of enzyme inhibitors in the clinic relates to their duration of action. A more theoretical issue is the effect of pronounced non-competitive inhibition on the rate of enzyme synthesis. Non-competitive inhibitors may produce only slowly reversible ChE inhibition. The rate at which this inhibition is reversed may be of the same order as the rate of enzyme synthesis.76 Thus, the long term effects of administration of slowly reversible, or irreversible, inhibitors on the overall cholinergic function are difficult to predict.
The selectivity of enzyme inhibition also plays a crucial part in determining the therapeutic profile of any ChE inhibitor. In this regard, several factors should be taken into account. All compounds will possess a greater or lesser degree of selectivity, and many of the differences between compounds may be influenced by the actions of the compound other than its intended ChE inhibition. Not surprisingly, therapeutic agents developed as inhibitors of AChE, which is found primarily in neural tissue, may also inhibit butyrylcholinesterase (BuChE), which acts mainly in the periphery. Although the function of BuChE remains unknown,77 clinical data with selective and non-selective AChE inhibitors suggest the BuChE inhibition may be associated with unwanted peripheral side effects,78 79although to date, this remains an unproved empirical finding. However, compared with tacrine, less peripheral cholinergic-related side effects have been found with donepezil, as it is over 1000-fold more selective for AChE than BuChE.70 74 79 80 Thus, greater brain AChE inhibition may be achieved with donepezil at the therapeutically effective dose compared with tacrine, increasing donepezil’s potential clinical efficacy.71
A further factor associated with the in vivo pharmacology of mixed type ChE inhibitors is that such compounds may interact with the site at which ACh is “captured” within the AChE enzyme, and may also act at other sites that bind or recognise Ach.81 82 Both tacrine and donepezil displace the binding of selective ligands from muscarinic and nicotinic ACh receptors,57 71 83-85 although neither compound has significant activity at other neurotransmitter receptors. At muscarinic receptors, both compounds act as antagonists.71 However, these effects only occur at concentrations of the compounds significantly greater than those needed to produce the required degree of ChE inhibition and are not therefore likely to have relevance in the clinic.86
Donepezil, like tacrine, has been reported to have effects on other neurotransmitter systems other than via receptors. For instance, donepezil is only 10-fold less potent than imipramine at inhibiting the uptake of serotonin.71 However, unlike tacrine, some second generation ChE inhibitors have been shown, using in vivo microdialysis techniques to measure the extracellular concentration of neurotransmitters and their metabolites, to increase monoamine concentrations in the cortex after administration of therapeutic doses.87 88 These type of effects might be expected to influence affective states—for example, mood—in a positive manner. Given that depression and aggression are important determinants of quality of life for patients with Alzheimer’s disease and their carers, such effects may have clinical relevance.1
CLINICAL TRIALS
First generation cholinesterase inhibitors
During the late 1980s and early 1990s, the first cholinomimetic compound, tacrine, underwent a large number of clinical studies using various doses and treatment periods ranging from a few days to 30 weeks. Tacrine was subsequently approved for use in some, but not all, countries. Evidence from three pivotal studies of tacrine has established clearly the benefits of ChE treatment in patients with a diagnosis of probable Alzheimer’s disease.89-91Statistically significant, dose related improvements on objective performance based tests of cognition, clinician and caregiver rated global evaluations of patient wellbeing, and also quality of life measures have been reported.89-92
Unfortunately, potentially serious adverse side effects have limited the use of this compound. Both tacrine, and the carbamate physostigmine, possess detrimental effects on hepatic and cardiovascular function. Indeed, perhaps the most often documented reason for withdrawal of tacrine is its potential hepatotoxicity. However, this effect seems to be unrelated to dose, and alanine aminotransferase (ALT) concentrations usually return to normal after drug withdrawal. In addition, many patients can be successfully rechallenged with tacrine after the enzymes return to normal.93
Among the other unwanted side effects of tacrine, the most often occurring are those caused by overstimulation of the peripheral cholinergic system at or below 30% ChE inhibition reflecting its dose related tolerability.94 These side effects are manifested predominantly by gastrointestinal tract discomfort and overactivity, resulting in nausea, vomiting, abdominal pain, and diarrhoea. Doses of tacrine within the therapeutic range elicit such side effects in about 20% of tacrine treated patients.95 In one 30 week clinical trial, over 50% of patients treated with tacrine discontinued treatment because of cholinergic related side effects. In addition, over 70% of patients titrated to the highest dosage of tacrine (160 mg/day) failed to complete this 30 week study.91 These incidental effects limit the compound’s maximum tolerated dose that may be administered to patients, and therefore the extent of brain AChE inhibition that can be achieved.
Despite these limitations, a substantial number of patients, some 250 000–300 000 worldwide, have been exposed to tacrine. Consequently, although tacrine produces a meaningful benefit in a significant proportion of patients with Alzheimer’s disease, the question has been raised as to whether this approach represents a fair test of the cholinergic hypothesis. This issue has been considered in the development of “second generation” ChE inhibitors. Such ChE inhibitors have been designed to limit side effect problems, and the maximum tolerated dose that can be achieved may be determined more by the effects of ChE inhibition itself.
Second generation cholinesterase inhibitors
At least an equivalent level of benefit is likely to be produced by the newer second generation ChE inhibitors including donepezil,96-98 rivastigmine,99metrifonate,100-102 galantamine103 and several other compounds. Such compounds show an effect and magnitude of benefit of at least that reported for tacrine, but with a more favourable clinical profile. For example, donepezil has a once daily dosage schedule and produces dose related significant improvements in cognition and global function, with over 80% of patients experiencing an improvement or no deterioration in cognition. Such responses should be viewed positively, considering the progressive, degenerative nature of the disease. In one 30 week randomised, double blind study of donepezil (5 or 10 mg/day) versus placebo (n=150/group, 450 total), statistically significant improvements were obtained with both 5 and 10 mg/day of donepezil for the intent to treat analysis of Alzheimer’s disease assessment scale (ADAS-cog104; p⩽0.001) and the clinician’s interview based impression of change (CIBIC plus105; p⩽0.005).97 This clinical improvement (as determined by the ADAS-cog) was correlated with both donepezil plasma concentrations and AChE inhibition.96Further, a retrospective subanalysis of the 30 week trial clinical dementia rating scale domains that reflect activities of daily living (ADLs): community affairs, home and hobbies, and personal care, suggests that donepezil (10 mg/day) delays the loss of ADLs by about 1 year.106 Preliminary evidence from open label studies showed that the treatment effect of donepezil is maintained over long periods (at least 2 years).107 This general thesis that ChE inhibitors will delay the progression of symptoms of Alzheimer’s disease and improve patients, on average, by the equivalent of 6–12 months deterioration, is now receiving further support with the publication of results from the trials of rivastigmine and metrifonate.99-102
Substantially more patients were able to tolerate and achieve therapeutic concentrations of donepezil than was possible with tacrine. Donepezil (5 and 10 mg/day) is well tolerated with no evidence of hepatotoxicity.70 96-98 108 and an incidence of side effects (5 mg/day) similar to that of placebo.96-98 The mainly cholinergic side effects that do occur are usually mild, transient, and resolve with continued treatment. As with the other available ChE inhibitors including tacrine, the incidence of side effects has been reported to be slightly increased in patients treated with the higher dosages of ChE inhibitors. This is likely due to the rapid, forced dose titration schedule used in clinical trials. Indeed, a lower incidence of side effects was found when a longer titration schedule was employed; for example, escalation to 10 mg/day donepezil after 4–6 weeks allowing achievement of a steady state at 5 mg/day donepezil.97
Diagnostic inaccuracy, which may be as high as 20%, can produce a strong bias in favour of non-response when the results in different treatment arms are “averaged out”. The heterogeneity of Alzheimer’s disease at a genetic, clinical, neurochemical, and neuropathological level may also contribute to differing response rates. Thus, whereas some patients, of course, respond considerably more than the mean reported in clinical trials, others similarly, will respond less. It may prove difficult to develop one therapy with an equivalent effect across the disease range of stage and severity. However, it may be possible to define patients by genotype, or by other markers, and tailor treatment to specific clinical subtypes, and work is underway to explore this possibility.
Response rates of ChE inhibitors do not detract from their clinical importance, as ChE inhibitors provide meaningful and important benefits for some patients with Alzheimer’s disease and their families. There is no doubt now that significant proportions of patients with probable mild to moderate Alzheimer’s disease gain some benefit from ChE inhibitors. In qualitative terms, a delay in symptomatic decline by about 6–12 months is valuable to those with Alzheimer’s disease, and also to those that care for and about them. At an anecdotal level, individual patients that have been treated successfully, and their relatives have reported improved awareness and attention, greater motivation and independence, improved language and communication abilities, and an improvement in the ability to undertake previously impaired or abandoned ADLs and hobbies. For example, a good response can include such features as the patient being able, once again, to manage their own day to day activities, being able to make and take telephone calls spontaneously, undertake their hobbies and pastimes and, in some cases, even to go shopping and successfully return with the required goods without having become lost.
Finally, evidence is emerging from clinical trials of cholinomimetic drugs that such drugs may improve the abnormal non-cognitive, behavioural symptoms of Alzheimer’s disease. Thus, ChE inhibitors have been reported to significantly improve many manifestations of behavioural disturbance including agitation, apathy, hallucinations, and aberrant motor behaviour101 109 and xanomeline, a selective muscarinic agonist, has been shown to improve vocal outbursts and psychotic symptoms.110 Further, such long term treatment with ChE inhibitors may delay or reduce the need for nursing home placement, and even reduce mortality (a trend for reduced mortality has been noted with tacrine).111 These findings require confirmation, and it will be important to establish whether this reduction in mortality increases the duration of the subsequent, more dependent stages of the disease. In addition, although nursing home placement may be delayed, the effect on the actual duration of nursing home care should be ascertained.
CLINICAL USE
Choosing the right patient
Cholinomimetic treatment is targeted specifically at patients with Alzheimer’s disease, albeit that such treatment may be beneficial in other dementias where a cholinergic deficit also exists—for example, Lewy body disease. Trials are currently underway to explore this possibility. The severity of the dementia is another important factor to be considered as currently these drugs have been assessed adequately in patients with mild to moderately severe Alzheimer’s disease only, but again this is subject to further evaluation and current practice may change as clinical experience increases. In addition, it is essential to make a careful assessment of the patients’ illness to ensure that they are likely to have Alzheimer’s disease. Primary care physicians may screen for and recognise patients with suspected Alzheimer’s disease within the community, but often referral to a specialist service is required. As there is no definitive diagnostic test for Alzheimer’s disease, it is important to base a diagnosis of “probable Alzheimer’s disease” on careful consideration of the patients’ symptoms and signs, preferably using the Diagnostic and Statistical Manual of Mental Disorders, fourth edition (DSM IV)112 or National Institute of Neurological and Communicative Disorders and Stroke - Alzheimer’s Disease and Related Disorders Association work group (NINCDS-ADRDA) criteria,113 or an equivalent protocol. If properly applied, the accuracy rate of diagnosis using such criteria, confirmed at necropsy, probably varies between 85% and 95%,114 115 depending on the experience of the centre in which the patient is assessed.
Decisions on continuing long term cholinesterase inhibitor treatment
Less than half of the patients receiving ChE inhibitors achieve a clinically significant response, although no further deterioration or even a slowing of deterioration are desirable outcomes, given the progressive, degenerative nature of the disease. Nevertheless, all patients with Alzheimer’s disease should have the opportunity of a treatment trial of at least 3 months in duration. Unfortunately, however, it has not yet been possible to predict or distinguish responders from non-responders.
In clinical trials, various psychometric outcome measures are used to assess the efficacy of drug treatment for Alzheimer’s disease. The cognitive subscale of the ADAS-cog and the CIBIC are used often to determine the efficacy of pharmacological agents. The ADAS-cog measures memory, orientation, attention, language, function, and praxis. CIBIC measures patients’ global function in terms of general, cognitive, behavioural, and ADL domains (for example, personal care and hobbies), and is determined by experienced clinicians, but may incorporate input from the primary caregiver of the patient with Alzheimer’s disease (for example, the CIBIC plus). A further assessment, the mini mental state examination (MMSE),116 is a short collection of cognitive tests that examines several areas of cognition. It is widely used to measure the onset, progression, and severity of Alzheimer’s disease in the clinical setting. The test is easy to administer and score, and can be used readily in a primary care setting, both at the office and in the patient’s home.
In clinical practice, it is rarely possible to undertake such a comprehensive assessment, but some degree of objectivity concerning treatment effect is, nevertheless, essential. Many physicians will rely on a simple general test such as the MMSE, coupled with a global measure formed from the relative or other carer’s opinion about the response, and their own assessment based on notes made at the first assessment. Those patients that clearly benefit from treatment should continue. However, treatment should be terminated in those patients who show deterioration, albeit that such patients must be closely monitored during such periods due to reports of precipitous decline after abrupt discontinuation. In addition, drug free periods, between 3–6 months for example, may be useful in evaluating the response to ChE inhibitors; deterioration during such periods would indicate that continued treatment is appropriate.117
Conclusion
The cholinergic hypothesis of Alzheimer’s disease is based on the presynaptic deficits found in the brains of patients with Alzheimer’s disease and studies of the role of ACh in animal and human behaviour. Although it is now clear that cholinergic dysfunction may not cause cognitive impairment directly, but rather indirectly, by interfering with attentional processing, the hypothesis predicted that cholinomimetic drugs would improve cognitive function. This prediction was not fully realised with compounds such as physostigmine and tacrine, probably because the emergence of side effects that may have constrained the dosing regimen to sub-efficacious doses. Poor tolerability seems to be less of an issue for the second generation compounds of the type now being licensed for the treatment of Alzheimer’s disease. With improved diagnosis, careful patient selection, and fewer side effects, such compounds will establish if cholinomimetic therapy provides effective and long lasting palliative therapy. Moreover, the emerging relation between neurotransmission and metabolism of two key proteins involved in Alzheimer’s disease, APP and tau, raises the possibility that second generation ChE inhibitors may alter disease pathology and progression.
Acknowledgments
Work on this manuscript was supported by an educational grant from Eisai Inc and Pfizer Pharmaceuticals Group, Pfizer Inc. We thank PPS International, Worthing, UK, for their assistance in the development of this manuscript and Drs K Stanhope and M Sheardown for helpful discussion.