


Abstract
Voltage-gated sodium channels play a critical role in excitability of nociceptors (pain-sensing neurons). Several different sodium channels are thought to be potential targets for pain therapeutics, including Nav1.7, which is highly expressed in nociceptors and plays crucial roles in human pain and hereditary painful neuropathies, Nav1.3, which is up-regulated in sensory neurons following chronic inflammation and nerve injury, and Nav1.8, which has been implicated in inflammatory and neuropathic pain mechanisms. We compared the effects of lacosamide [(2R)-2-(acetylamino)-N-benzyl-3-methoxypropanamide], a new pain therapeutic, with those of lidocaine and carbamazepine on recombinant Nav1.7 and Nav1.3 currents and neuronal tetrodotoxin-resistant (Nav1.8-type) sodium currents using whole-cell patch-clamp electrophysiology. Lacosamide is able to substantially reduce all three current types. However, in contrast to lidocaine and carbamazepine, 1 mM lacosamide did not alter steady-state fast inactivation. Inhibition by lacosamide exhibited substantially slower kinetics, consistent with the proposal that lacosamide interacts with slow-inactivated sodium channels. The estimated IC50 values for inhibition by lacosamide of Nav1.7-, Nav1.3-, and Nav1.8-type channels following prolonged inactivation were 182, 415, and 16 μM, respectively. Nav1.7-, Nav1.3-, and Nav1.8-type channels in the resting state were 221-, 123-, and 257-fold less sensitive, respectively, to lacosamide than inactivated channels. Interestingly, the ratios of resting to inactivated IC50s for carbamazepine and lidocaine were much smaller (ranging from 3 to 16). This suggests that lacosamide should be more effective than carbamazepine and lidocaine at selectively blocking the electrical activity of neurons that are chronically depolarized compared with those at more normal resting potentials.
Neuropathic pain is a difficult type of pain to treat, and it is estimated that as many as half of all patients receive inadequate pain relief. More effective and better tolerated treatments are needed. Lacosamide is a functionalized amino acid that has shown effectiveness as an anticonvulsant and analgesic and therefore might help fill this need (Beyreuther et al., 2007b; Stöhr et al., 2007). Lacosamide has shown efficacy in several animal models of neuropathic pain (Beyreuther et al., 2006, 2007a) and is currently in late stages of clinical development for epilepsy and diabetic neuropathic pain (Doty et al., 2007). Two modes of action have been identified for lacosamide, interaction with collapsin-response mediator protein 2, and inhibition of voltage-gated sodium channel activity by selective interaction with the slow-inactivated conformation of the channel (Beyreuther et al., 2007b; Errington et al., 2008). In this study, we asked whether lacosamide could inhibit voltage-gated sodium channels expressed in peripheral sensory neurons and believed to be involved in pain mechanisms.
Voltage-gated sodium channels underlie the rapid action potentials characteristic of neurons and muscle. They are transmembrane proteins with gated pores whose opening and closing is controlled by changes in the voltage gradient across the membrane (Catterall, 2000). The primary functional unit of the voltage-gated sodium channel is a ∼240-kDa polypeptide α subunit. Nine distinct voltage-gated sodium channel α subunits (Nav1.1–1.9) have been cloned from mammals (Goldin, 2002). Adult dorsal root ganglion (DRG) sensory neurons can express multiple α subunits, including tetrodotoxin-sensitive (TTX-S) sodium channels (Nav1.1, Nav1.6, and Nav1.7) and tetrodotoxin-resistant (TTX-R) sodium channels (Nav1.8 and Nav1.9).
Several sensory neuronal channels have been implicated in pain mechanisms. A “nociceptor-specific” Nav1.7 knockout mouse showed significant deficits in response to inflammatory stimuli (Nassar et al., 2004), and humans that lack functional Nav1.7 channels exhibit a complete insensitivity to pain (Cox et al., 2006) with apparently no other deficits, indicating that the loss of Nav1.7 activity selectively affects pain. Furthermore, gain-of-function mutations in Nav1.7 cause two distinct inherited painful neuropathies (Cummins et al., 2004; Drenth et al., 2005; Fertleman et al., 2006) and cause hyperexcitability of DRG neurons (Rush et al., 2006; Sheets et al., 2007). These data clearly indicate that Nav1.7 plays a crucial role in nociception. Nav1.3 might also play a role in pain mechanisms. Nav1.3 is not normally expressed in adult sensory neurons but is expressed following either chronic inflammation or nerve injury (Waxman et al., 1994; Black et al., 2004) and has been connected to central neuropathic pain (Hains et al., 2003). Based on these data and because of its unique functional properties (Cummins et al., 2001), Nav1.3 is hypothesized to play an important role in some types of pain. Two distinct types of TTX-R currents (Nav1.8- and Nav1.9-type) have been identified in DRG neurons (Cummins et al., 1999; Dib-Hajj et al., 1999). Nav1.8 channels have been implicated in both inflammatory (Akopian et al., 1999) and neuropathic (Lai et al., 2002) pain, and selective block of Nav1.8 can alleviate both of these types of pain (Jarvis et al., 2007). Nav1.8 currents are therefore also clearly of interest when considering the mechanism of action of new pain therapeutics.
All mammalian voltage-gated sodium channel α subunits have four domains, each consisting of six transmembrane segments (S1–S6). The S6 segments are thought to be important in binding local anesthetics, such as lidocaine and anti-convulsants (Ragsdale and Avoli, 1998; Clare et al., 2000). Many of the clinically relevant modulators exhibit a pronounced state-dependent binding, where sodium channels that are rapidly and repeatedly activated and inactivated are more readily blocked. This state dependence is thought to be very important in limiting the activity of cells exhibiting abnormal activity. In a simplified scheme, voltage-gated sodium channels have four distinct states, open, closed, fast-inactivated, and slow-inactivated. The classic sodium channel modulators, such as lidocaine, are believed to exhibit the highest affinity for the fast-inactivated state. However, some studies have suggested that alteration of slow inactivation could be clinically relevant as well (Fukuda et al., 2005; Haeseler et al., 2006).
In this study, we used whole-cell patch-clamp electrophysiology to investigate the effects of lacosamide on Nav1.7 and Nav1.3 voltage-gated sodium channels expressed in human embryonic kidney (HEK) 293 cells. In addition, we studied the effects of lacosamide on Nav1.8-type TTX-R currents from DRG neurons. We also compared the effects of lacosamide with those of two common pain therapeutics, carbamazepine and lidocaine.
Materials and Methods
Cell Culture. HEK293 cells were grown under standard tissue culture conditions (5% CO2; 37°C) in high-glucose DMEM (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (HyClone Laboratories, Logan, UT), 100 U/ml penicillin, and 100 μg/ml streptomycin. Two HEK293 cells lines were used. One line stably expressed the rat Nav1.3 voltage-gated sodium channel (Cummins et al., 2001), and the other cell line stably expressed the human Nav1.7 voltage-gated sodium channel (Cummins et al., 1998). Stable cell lines were originally selected for using the antibiotic G418 (Geneticin; Cellgro, Herndon, VA). No β subunits were transfected with the channels. At least 16 h before electrophysiological recording experiments, the cells were harvested using mechanical trituration and replated on 12-mm glass coverslips.
DRG Culture. Adult male Sprague-Dawley rats (Harlan, Indianapolis, IN) were rendered unconscious by exposure to CO2 and decapitated. The L4 and L5 DRGs were removed and incubated with a mixture of protease (12.5 mg/ml) and collagenase (5 mg/ml) in bicarbonate-free DMEM for 40 min at 37°C. The enzyme-treated DRGs were then triturated using fire-polished glass pipettes, plated on poly-d-lysine-coated dishes, and maintained under standard tissue culture conditions (5% CO2; 37°C) in DMEM supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. TTX-R Nav1.8-type sodium currents were recorded from DRG neurons 12 to 36 h after plating.
Whole-Cell Patch-Clamp Recordings. Whole-cell patch-clamp recordings were conducted at room temperature (∼21°C) using a HEKA EPC-10 amplifier. Data were acquired on a Windows-based Pentium IV computer using the Pulse program (version 8.65; HEKA, Lambrecht/Pfalz, Germany). Fire-polished electrodes (0.9–1.3 MΩ) were fabricated from 1.7-mm capillary glass using a Sutter P-97 puller (Sutter Instrument Company, Novato, CA). The standard pipette solution contained: 140 mM CsF, 10 mM NaCl, 1.1 mM EGTA, 1 mM MgCl2, and 10 mM HEPES, pH 7.3. The standard bathing solution contained: 140 mM NaCl, 1 mM MgCl2, 3 mM KCl, 1 mM CaCl2, and 10 mM HEPES, pH 7.3 (adjusted with NaOH). Series resistance errors were compensated to be less than 3 mV by using 80 to 85% series resistance compensation and 1.0- to 1.5-MΩ pipettes. Cells on glass coverslips were transferred into a recording chamber containing 250 μl of standard bathing solution. The amplifier's offset potential was zeroed with the electrode almost touching the cell of interest. After obtaining a gigaseal, suction was used to establish the whole-cell recording configuration. Cells were held at –80 mV for 3.5 min before initiating the experimental protocols. Cells with less than 1 nA of peak sodium current were not used for this study. Compounds were added to the bath compartment by first withdrawing 25 μl of bathing solution, then adding 25 μl of 10-fold concentrated compound and mixing 10 to 15 times with a 25-μl pipetter. Only one compound was tested on each cell, although we typically tested two concentrations of a compound (without intervening washout) on each cell. Cells were held at –80 mV for 2 to 3 min during initial application of a compound before running the specific voltage protocols described under Results and illustrated in the figures.
Chemicals and Solutions. Lacosamide was obtained from Schwarz Pharma (Monheim, Germany). A 500 mM solution was made up in dimethyl sulfoxide (DMSO). A 25 mM stock solution was made from this by 20-fold dilution into standard bathing solution (see above). This stock solution was aliquoted and stored at –20°C. Lidocaine hydrochloride was purchased from Sigma-Aldrich (St. Louis, MO). Lidocaine stock solutions were prepared fresh daily. A 100 mM stock solution was prepared in standard bathing solution and stored on ice. Carbamazepine was purchased from Sigma-Aldrich. A 250 mM solution was made up in DMSO. Frozen aliquots of this stock solution were stored at –20°C. Each of the stock solutions were diluted into standard bathing solution to create 10-fold concentrated solutions of 30 mM, 10 mM, 3 mM, 1 mM, 300 μM, 100 μM, 30 μM, and 10 μM. The concentrated solutions were diluted 1:10 into the recording chamber and thoroughly mixed during the experiments to provide final concentrations of 3 mM, 1 mM, 300 μM, 100 μM, 30 μM, 10 μM, 3 μM, and 1 μM for each of the three compounds. The 10-fold concentrated solutions were prepared fresh daily and stored on ice. In a separate set of control experiments, DMSO at 0.5% (v/v) had negligible effects on the functional properties of Nav1.3 (n = 4) or Nav1.7 (n = 5) currents (see Supplemental Figs. 1 and 2).
Data Analysis. Voltage-clamp experimental data were analyzed using the Pulsefit (version 8.65; HEKA) and Origin (OriginLab Corp., Northampton, MA) software programs. Slope factors of steady-state inactivation curves were calculated using the general Boltzmann function: I(V) = offset + (amplitude/1 + exp(–(V – Vhalf)/slope). To estimate inhibition potencies, dose-response curves were fit using the Hill function: fi = 1 – (fmax)/(1 + ([drug]/IC50)nH), where fi is the fraction inhibited, fmax is the maximal fraction that can be inhibited, and nH is the Hill coefficient. Error bars in the figures represent the S.E.M. Data were assumed to be normally distributed, and unpaired Student's t test was used for statistical comparisons.
Results
Inhibition of Nav1.7 and Nav1.3 Currents by Lacosamide
In the initial set of experiments, the ability of 30 and 100 μM lacosamide to inhibit sodium currents carried by Nav1.7 and Nav1.3 channels was tested by holding the cells at –80 mV, running a current-voltage (IV) protocol, applying the test concentration of lacosamide, then running a second IV protocol to determine the effect of lacosamide on peak current amplitude and on the voltage dependence of current activation. The IV protocol consisted of giving 50-ms step depolarizations to voltages ranging from –80 to + 40 mV in 5-mV increments and recording the sodium currents elicited during each step depolarization. Figure 1A shows an IV family of Nav1.7 currents recorded before and after application of 30 μM lacosamide. The peak current elicited by each step depolarization is measured and can be plotted versus the voltage. Figure 1B shows the IV data from Nav1.7 cells (n = 3) before and after 30 μM lacosamide (normalized to the peak current measured before lacosamide). Lacosamide reduced the peak Nav1.7 currents obtained from a –80-mV holding potential by 30%. Lacosamide (100 μM) reduced peak Nav1.7 currents by almost 60% (Fig. 1C; n = 3). In contrast, Nav1.3 currents elicited from a holding potential of –80 mV were less sensitive to lacosamide, exhibiting only an 11% decrease with application of 30 μM lacosamide (Fig. 1D; n = 4) and 25% decrease with application of 100 μM lacosamide (Fig. 1E; n = 3). Lacosamide (100 μM) did not significantly alter the voltage dependence of activation for either Nav1.7 or Nav1.3 currents.
Comparison of Effects of Lacosamide with Lidocaine and Carbamazepine Effects
Inhibition of Resting (Closed) Channels. Lidocaine and carbamazepine exhibit differential binding to resting (or closed) channels and inactivated channels. We examined the ability of lacosamide, lidocaine, and carbamazepine to inhibit resting channels by holding cells expressing Nav1.7 and Nav1.3 currents at –120 mV for 10 s (to allow all channels to move to the resting state) and then eliciting currents with a 20-ms step depolarization to 0 mV. Peak sodium current amplitude was measured before and after application of lacosamide (1–3000 μM; n = 3–7), lidocaine (1–3000 μM; n = 3–4), and carbamazepine (1–3000 μM; n = 3–4). Fits to the concentration-response curves estimate that the IC50 of resting Nav1.7 channels is 0.7 mM for lidocaine, 1.6 mM for carbamazepine, and roughly 40 mM for lacosamide (Fig. 2A; Table 1). Fits to the concentration-response curves estimate that the IC50 of resting Nav1.3 channels is 1.5 mM for lidocaine, 2.5 mM for carbamazepine, and roughly 51 mM for lacosamide (Fig. 2B; Table 1). These data indicate that lacosamide has a much higher IC50 for resting Nav1.3 and Nav1.7 channels than either carbamazepine or lidocaine. Although the estimates of the affinity of resting Nav1.7 and Nav1.3 channels for lacosamide should only be considered as very rough estimates due to lack of data at higher concentrations, at 1 mM, lidocaine and carbamazepine inhibit Nav1.3 and Nav1.7 to a significantly (p < 0.0005) greater extent than 1 mM lacosamide.

A, IV family trace of an HEK293 cell stably expressing the human Nav1.7 channel before (left) and after (middle) application of 30 μM lacosamide (LCM). The voltage protocol is shown on the right. B and C, IV relationship of Nav1.7 current in the absence and presence of 30 (B) and 100 (C) μM lacosamide. D and E, IV relationship of Nav1.3 current in the absence and presence of 30 (D) and 100 (E) μM lacosamide.
Inhibition of Inactivated Channels. Next, we examined the ability of lacosamide, lidocaine, and carbamazepine to interact with inactivated channels by holding cells expressing Nav1.7 currents at –50 mV for 10 s (to allow all channels to move to inactivated states) and then stepping the cells to –120 mV for 100 ms (to allow channels without drug bound to recover from fast inactivation; Cummins and Waxman, 1997; Cummins et al., 2001) and finally eliciting currents with a 20-ms step depolarization to 0 mV to estimate channel availability (Fig. 3C). Peak sodium current amplitude was measured before and after application of lacosamide (1–3000 μM; n = 3–7), lidocaine (1–3000 μM; n = 3–4), and carbamazepine (1–3000 μM; n = 3–4). The concentration-response curve for inhibition of inactivated Nav1.7 channels is shown in Fig. 3A. The concentration-response curve for inactivated Nav1.3 channels (Fig. 3B) was similarly obtained, except that we used a 10-s step to –40 mV (due to differences in steady-state inactivation; see Fig. 4), with the cells expressing Nav1.3 currents. The different inactivation prepulse potentials were chosen to achieve comparable extents of inactivation (a 500-ms prepulse to –40 mV inactivates 97.5% Nav1.3 channels, and a 500-ms prepulse to –50 mV inactivates 96.7% Nav1.7 channels). Fits to the concentration-response curves estimate that the IC50 for inhibition of inactivated Nav1.7 channels is 44 μM for lidocaine, 406 μM for carbamazepine, and 182 μM for lacosamide. Fits to the concentration-response curves estimate that the IC50 for inhibition of inactivated Nav1.3 channels is 284 μM for lidocaine, 900 μM for carbamazepine, and 415 μM for lacosamide. Table 1 shows the parameters for the Hill equation fits. These data indicate that: 1) lacosamide has a much higher affinity for inactivated Nav1.7 and Nav1.3 channels than for resting Nav1.7 and Nav1.3 channels, and 2) inactivated Nav1.3 channels are less sensitive to all three compounds than inactivated Nav1.7 channels.

A, concentration-response curve for the inhibitory effects of LCM, lidocaine, and carbamazepine (CBZ) on resting Nav1.7 channels. B, concentration-response curve for the inhibitory effects of LCM, lidocaine, and CBZ on resting Nav1.3 channels. C, voltage protocol for testing current amplitude is shown.
Effects on Voltage Dependence of Steady-State Fast and Slow Inactivation
Our data indicated that lacosamide, like lidocaine and carbamazepine, exhibits preferential binding to inactivated channels. We asked whether lacosamide would enhance fast inactivation of Nav1.7 and Nav1.3 channels in the same manner as lidocaine and carbamazepine. Therefore, we measured the effect of lacosamide, lidocaine, and carbamazepine on the voltage dependence of steady-state fast and slow inactivation. We used 1 mM for each of the compounds to help maximize the binding of the compound to the channels. Higher concentrations were not used for this because solubility becomes a problem, especially with carbamazepine. Cells expressing Nav1.7 or Nav1.3 channels were held at –120 mV, stepped to inactivating prepulse potentials ranging from –150 to –30 mV (in 10-mV increments) for 500 ms, before stepping the cells to 0 mV for 20 ms to measure the available current (Fig. 4C). To eliminate the bias of time-dependent shifts in the voltage dependence of fast inactivation, we compared data recorded 4 min after establishing the whole-cell recording configuration from control cells (in the absence of test compound; n = 5) with data recorded 4 min after establishing the whole-cell recording configuration from cells exposed to one of the three test compounds (n = 5–8). Both lidocaine and carbamazepine (1 mM) shifted the voltage dependence of steady-state fast inactivation by roughly 20 mV in the negative direction for both Nav1.7 (Fig. 4A) and Nav1.3 (Fig. 4B) currents. The magnitude of the shift in the presence of both lidocaine and carbamazepine is significant for both Nav1.3 and Nav1.7 compared with control cells (p < 0.005). However, lacosamide (1 mM) did not affect the voltage dependence of steady-state fast inactivation for either Nav1.7 or Nav1.3 currents.
Our data demonstrate that although lacosamide seems to exhibit preferential binding to inactivated channels, this was not seen with 500-ms prepulses. Therefore, we next measured the effect of lacosamide, lidocaine, and carbamazepine (all at 1 mM) on the voltage dependence of steady-state slow inactivation. Cells expressing Nav1.7 or Nav1.3 channels were held at –120 mV, stepped to inactivating prepulse potentials ranging from –140 to 0 mV (in 10-mV increments) for 10 s, hyperpolarized to –160 mV for 100 ms, and then stepped to 0 mV for 20 ms to measure the available current (Fig. 4F). The brief hyperpolarization was used to allow channels (both with and without drug bound) to recover from fast inactivation while limiting the recovery from slow inactivation. To eliminate the bias of time-dependent shifts in the voltage dependence of fast inactivation, we compared data recorded 4 min after establishing the whole-cell recording configuration from control cells (in the absence of test compound; n = 5) to data recorded 4 min after establishing the whole-cell recording configuration from cells exposed to one of the three test compounds (n = 5–8). Carbamazepine did not alter the voltage dependence of steady-state slow inactivation for either Nav1.7 (Fig. 4D) or Nav1.3 (Fig. 4E) currents. However, both lacosamide and lidocaine shifted the apparent voltage dependence of steady-state slow inactivation for both Nav1.7 and Nav1.3 currents.

A, concentration-response curve for the inhibitory effects of LCM, lidocaine, and CBZ on inactivated Nav1.7 channels. B, concentration-response curve for the inhibitory effects of LCM, lidocaine, and CBZ on inactivated Nav1.3 channels. C, voltage protocol for testing current amplitude is shown.
Rate of Development of Inhibition
To better understand the interaction between lacosamide and Nav1.7 and Nav1.3 sodium channels, we investigated the time course for development of block by 1 mM lacosamide, lidocaine, and carbamazepine. Development of block was examined by holding the cells (n = 5–7 for each of the four conditions) expressing Nav1.7 (Fig. 5A) or Nav1.3 (Fig. 5B) at –120 mV in the presence of one of the three compounds, prepulsing the cells to 0 mV for varying amounts of time to allow block to develop, hyperpolarizing the cells to –120 mV for 20 ms to allow unbound channels to recover from fast inactivation, then stepping the cells to 0 mV for 20 ms to determine the fraction of channels available for activation (Fig. 5C). In control cells the reduction in the fraction of available current represents the time course for the development of slow inactivation. The time course for reduction of channel availability in the presence of lidocaine and carbamazepine was clearly biphasic, with a fast component that probably represents development of block of fast-inactivated channels and a slow component that was consistent with the time course for development of slow inactivation in control cells. The time course for reduction in channel availability in the presence of lacosamide showed a different pattern. The rate of development of block was 4 to 7-fold faster than the time course for development of slow inactivation, but the shape of the curve was very similar to that for slow inactivation.
Recovery from Inactivation/Block
We also examined the rate at which Nav1.3 and Nav1.7 channels recovered from inactivation and/or block by lidocaine, carbamazepine, and lacosamide (all at 1 mM; n = 5–6 for each condition). In the first set of experiments, cells were held at –120 mV, depolarized to 0 mV for 10 ms and then hyperpolarized to –120 mV for variable increasing durations before testing the available current with a step depolarization to 0 mV (Fig. 6C). The time course for recovery from the 10-ms depolarization was not different from control for currents recorded in the presence of 1 mM lacosamide, but the time course was more complex and slower in the presence of lidocaine and carbamazepine, indicating that these two compounds can rapidly alter Nav1.7 (Fig. 6A) and Nav1.3 (Fig. 6B) channel activity. In the second set of experiments, cells (n = 5–9) were held at –120 mV, depolarized to 0 mV for 5 s, then hyperpolarized to –120 mV for a variable duration before testing the available current with a step depolarization to 0 mV. The 5-s depolarization provides time for the channels to undergo slow inactivation. The time course for recovery from the 5-s depolarization was not different from control for currents recorded in the presence of 1 mM lidocaine and carbamazepine. However, the time constant for recovery from the 5-s prepulse was significantly greater in the presence of 1 mM lacosamide (p < 0.01 at 500 ms; Fig. 6, D and E).

Effects of lacosamide, lidocaine, and carbamazepine on the voltage dependence of steady-state fast and slow inactivation for Nav1.7 and Nav1.3 channels. A and B, lidocaine and CBZ caused a significant hyperpolarizing shift in the voltage dependence of steady-state fast inactivation for Nav1.7 (A) and Nav1.3 (B) channels. LCM had no effect on the voltage dependence of steady-state fast inactivation for either channel. Midpoints of Nav1.7 steady-state inactivation in A were –76.1 ± 1.9 (n = 6), –99.3 ± 1.5 (n = 7), –97.0 ± 3.3 (n = 5), and –78.7 ± 1.2 (n = 6) for control, +lidocaine, +carbamazepine, and +lacosamide, respectively. Midpoints of Nav1.3 steady-state inactivation in A were –62.1 ± 2.7 (n = 9), –82.5 ± 1.6 (n = 9), –81.0 ± 2.6 (n = 8), and –65.4 ± 1.8 (n = 11) for control, + lidocaine, + carbamazepine, and + lacosamide, respectively. C, voltage protocol for steady-state fast inactivation is shown. Data sweeps were acquired at 0.2 Hz. D and E, lidocaine and LCM caused a significant hyperpolarizing shift in the voltage dependence of slow inactivation for Nav1.7 (D) and Nav1.3 (E) channels. CBZ had no effect on the voltage dependence of slow inactivation for either channel. F, voltage protocol for steady-state slow inactivation is shown. The 100-ms pulse at –160 mV is used to allow for recovery of fast-inactivated and drug-bound fast-inactivated channels, but not slow-inactivated or drug-bound slow-inactivated channels. Data sweeps were acquired at 0.09 Hz.
Effects on TTX-R Current
We wanted to examine the effects of lacosamide, carbamazepine, and lidocaine on Nav1.8 currents and determine how the effects compared with those on Nav1.3 and Nav1.7 currents. Nav1.8 currents are very difficult to record from HEK293 cells; therefore, we recorded Nav1.8-type currents from rat L4/L5 DRG neurons 12 to 18 h after being cultured. Two distinct types of TTX-R currents (Nav1.8- and Nav1.9-type) have been identified in DRG neurons (Akopian et al., 1996; Cummins et al., 1999; Dib-Hajj et al., 1999). We focused on Nav1.8 currents because its role in pain is much better understood than that of Nav1.9 and because it is difficult to obtain stable recordings of Nav1.9 currents, even in DRG sensory neurons (Leffler et al., 2005). In our recordings, we isolated Nav1.8-type currents by using 500 nM TTX to eliminate TTX-S currents and a holding potential (–80 mV) that is efficient at eliminating the persistent TTX-R current carried by Nav1.9 (Cummins et al., 1999; Priest et al., 2005; Amaya et al., 2006). The effects of lacosamide, carbamazepine, and lidocaine were tested on resting and inactivated channels. Peak sodium current amplitude was measured before and after application of drug (300 μM) and projected IC50 values from our results using the equation: (percentage remaining current)/((1 – percentage remaining current) × [drug]). As before, resting channels were tested by holding the neurons at –80 mV and pulsing to –120 mV for 10 s (to allow all channels to move to the resting (closed state) and then eliciting currents with a 20-ms step depolarization to 0 mV. Using this protocol, we observed decreases of 7.3 ± 4.3% (n = 6), 26.4 ± 3.2% (n = 4), and 48.6 ± 7.1% (n = 5) for lacosamide, carbamazepine, and lidocaine, respectively (Fig. 7A). Based on these values, the estimated IC50 values of lacosamide, carbamazepine, and lidocaine for resting Nav1.8-type TTX-R channels are 4 mM, 840 μM, and 320 μM, respectively.
To test the ability of lacosamide, lidocaine, and carbamazepine to inhibit inactivated Nav1.8-type TTX-R channels, we pulsed the neurons to –20 mV (which produces ∼95% inactivation) for 10 s (to allow all channels to move to the inactivated state and to allow for drug binding to reach equilibrium) and then stepped the cells to –120 mV for 100 ms (to allow channels without drug bound to recover from inactivation; Cummins and Waxman, 1997) and finally elicited currents with a 20-ms step depolarization to 0 mV to estimate channel availability. Peak sodium current amplitudes were measured before and after application of lacosamide (n = 6), carbamazepine (n = 8), or lidocaine (n = 9), all at a concentration of 300 μM. Using this protocol, we observed decreases of 95.1 ± 1.3, 68.5 ± 5.1, and 89.1 ± 2.6% for lacosamide, carbamazepine, and lidocaine, respectively. Based on these values, we estimate that the IC50 for inhibition of inactivated Nav1.8-type TTX-R channels is 16 μM for lacosamide, 138 μM for carbamazepine, and 37 μM for lidocaine. Figure 7B shows the comparison of current inhibition on inactivated Nav1.7-, Nav1.3-, and Nav1.8-type channels by all three drugs at concentrations of 300 μM. In this case, each channel subtype was held at different prepulse potentials, due to differences in their voltage dependence of steady-state inactivation, to ensure ≥95% inactivation. Figure 7C shows the comparison of current inhibition of Nav1.7-, Nav1.3-, and Nav1.8-type channels held at –50 mV, which would simulate a slightly depolarized potential for a sensory neuron. Lacosamide inhibition at –50 mV was similar for Nav1.8-type, Nav1.7, and Nav1.3 currents. Lidocaine inhibition at –50 mV was also similar for all three types of currents. However, differences were still observed between the current subtypes for carbamazepine inhibition at –50 mV.

Effects of lacosamide, lidocaine, and carbamazepine on the development of slow inactivation/inhibition for Nav1.7 and Nav1.3 channels. For the control channels, a brief (20-ms) hyperpolarizing pulse to –120 mV was given after the inactivating pulse to 0 mV at increasing durations. A and B, lidocaine and CBZ caused an increase in the rate of development of inactivation/inhibition at prepulse durations ranging from 1 to 100 ms before slowing at prepulse durations greater than 100 ms for both Nav1.7 (A) and Nav1.3 (B) channels. An increase in the rate of development of inactivation/inhibition for both Nav1.7 (A) and Nav1.3 (B) channels was observed in the presence of LCM but at prepulse durations longer than those seen with lidocaine and carbamazepine. C, voltage protocol is shown. Following a variable conditioning pulse to 0 mV, a 20-ms pulse to –120 mV allows recovery from fast inactivation (but not block) before the fraction of current available with the 20-ms pulse to 0 mV. Data sweeps were acquired at 0.5 Hz for short prepulse durations and at slower rates for the longer prepulse durations.
We further investigated the effects lacosamide, lidocaine, and carbamazepine on Nav1.8-type channels by looking at their effects on the voltage dependence of steady-state fast and slow inactivation. We used 1 mM for each of the compounds to help maximize the interaction of the channels with the compound. Neurons were held at –80 mV, stepped to inactivating prepulse potentials ranging from –100 to 30 mV (in 10-mV increments) for 500 ms before stepping the cells to 0 mV for 20 ms to measure the available current. To eliminate the bias of time-dependent shifts in the voltage dependence of fast inactivation, we compared data recorded 3 min after establishing the whole-cell recording configuration from control cells (in the absence of test compound; n = 9) to data recorded 3 min after establishing the whole-cell recording configuration from cells exposed to one of the three test compounds (n = 5–7). It is surprising that none of the drugs altered the voltage dependence of steady-state fast inactivation (Fig. 8A). The estimated V1/2 of steady-state fast inactivation (obtained with Boltzmann fits) of Nav1.8-type current in the presence of lacosamide (–33.4 ± 2.0 mV; slope, 3.5 ± 0.1; n = 5), lidocaine (–39.5 ± 2.5 mV; slope, 4.5 ± 0.2, n = 7), and carbamazepine (–37.4 ± 2.5 mV; slope, 4.1 ± 0.2, n = 6) was not statistically different from that of control Nav1.8-type current (–34.7 ± 1.3 mV; slope, 4.0 ± 0.1; one-way ANOVA analysis).
To study the effects of these drugs on Nav1.8-type current slow inactivation, we stepped to inactivating prepulse potentials ranging from –100 to 30 mV (in 10-mV increments) for 10 s, hyperpolarized to –140 mV for 100 ms, then stepped to 0 mV for 20 ms to measure the available current. Effects were observed with all of the drugs on the steady-state slow inactivation curve of Nav1.8-type currents (Fig. 8B). The estimated V1/2 of steady-state slow inactivation of Nav1.8-type current in the presence of lacosamide (–54.8 ± 3.2 mV; slope, 5.2 ± 0.2; n = 6), lidocaine (–57.9 ± 2.6 mV; slope, 5.1 ± 0.2; n = 7), and carbamazepine (–53.1 ± 3.2 mV; slope, 4.4 ± 0.1; n = 6) were significantly (p < 0.001) hyperpolarized from that of control Nav1.8-type current (–37.1 ± 1.2 mV; slope, 10.4 ± 0.1; n = 9; one-way ANOVA analysis). In addition, the slope of the steady-state slow inactivation curve for Nav1.8-type current was significantly (p < 0.001; one-way ANOVA analysis) different from the slope values observed in the presence of lacosamide, lidocaine, or carbamazepine. Both lacosamide and lidocaine markedly increased the maximal extent to which slow inactivation can reduce the availability of Nav1.8, i.e., at 20-mV reduction of channel availability for control and carbamazepine reached a maximum of about 50%, whereas it amounted to 80 and 90% for lidocaine and lacosamide, respectively.
Discussion
This study was aimed at determining the inhibitory effects of a new drug, lacosamide, on voltage-gated sodium channels that are expressed in peripheral nociceptive neurons and that have been identified as playing important roles in pain. We focused our efforts on examining the effects of lacosamide on Nav1.7 and Nav1.3 channels and comparing them with clinically used traditional analgesics carbamazepine and lidocaine. We found clinically relevant concentrations of lacosamide substantially reduce Nav1.7 current amplitudes at a holding potential of –80 mV, whereas Nav1.3 currents were inhibited to a slightly lesser extent, indicating that they are less sensitive. We found that lacosamide has a significantly higher IC50 for resting Nav1.3 and Nav1.7 channels than either carbamazepine or lidocaine. Lacosamide exhibited substantially greater inhibition of inactivated Nav1.7 and Nav1.3 channels than resting Nav1.7 and Nav1.3 channels. Lacosamide is more potent at inhibiting inactivated channels than carbamazepine but slightly less potent than lidocaine. These data show lacosamide has a much greater ability to discriminate between resting and inactivated Nav1.7 and Nav1.3 channels than either lidocaine or carbamazepine.
The effects of lacosamide on Nav1.8-type currents in DRG neurons were similar to those seen with Nav1.7 and Nav1.3 because lacosamide inhibited inactivated Nav1.8-type channels to a significantly greater extent then resting channels and suggest lacosamide is the most potent of the three drugs at inhibiting inactivated Nav1.8-type channels. Although the ratio of lacosamide resting IC50 to the –50 mV IC50 was smaller for Nav1.8-type channels than for Nav1.3 or Nav1.7 channels, this ratio was still greater for lacosamide than for either lidocaine or carbamazepine (Table 2). These data, showing that lacosamide is much more effective at distinguishing between resting and inactivated Nav1.7-, Nav1.3-, and Nav1.8-type channels than lidocaine and carbamazepine, suggest that lacosamide should be selective at inhibiting the activity of neurons with depolarized membrane potentials compared with neurons with normal resting membrane potentials than either lidocaine or carbamazepine. This could be an important part of the analgesic mechanism of action of lacosamide because nociceptor hyperexcitability and increased pain sensations are likely to be associated with depolarized membrane potentials in neurons.
Ratio of IC50 for closed channels to IC50 at depolarized potentials
The differential effects of lacosamide, carbamazepine, and lidocaine on the voltage dependence of steady-state fast inactivation and steady-state slow inactivation are intriguing. These data showed that the inhibition induced by the three drugs was different depending on the length of the depolarizing pulse used to inactivate the channels. When using 500-ms prepulses, we observed a significant hyperpolarizing shift in the voltage dependence of steady-state fast inactivation for Nav1.7 and Nav1.3 treated with carbamazepine and lidocaine but not when treated with lacosamide. In contrast, although carbamazepine had no effect on the voltage dependence of slow inactivation for Nav1.7 and Nav1.3, lacosamide and lidocaine both produced a significant hyperpolarizing shift in the voltage dependence of slow inactivation of these channels. These results suggest that, although carbamazepine and lidocaine interact with fast-inactivated Nav1.7 and Nav1.3 channels, lacosamide does not. Our data are consistent with the proposal that lacosamide interacts predominantly with slow-inactivated sodium channels (Errington et al., 2008) and that this effect could be a mechanistic rather than a kinetic difference compared with lidocaine and carbamazepine. However, based on these data, we cannot rule out the alternative possibility that lacosamide may bind and unbind very slowly from fast-inactivated channels. Mutagenesis experiments that selectively alter fast or slow inactivation of the sodium channels should help definitively determine the mechanism of action for lacosamide. Interestingly, none of the three drugs had any effect on the voltage dependence of Nav1.8-type channel fast inactivation. This result is not surprising for lacosamide due to its ineffectiveness on fast inactivation of Nav1.7 and Nav1.3 channels, but it is somewhat surprising for lidocaine and carbamazepine. However, previous studies have indicated that lidocaine does not alter fast inactivation of Nav1.8 channels in the same manner that it does Nav1.7 channels (Chevrier et al., 2004) and that carbamazepine might even preferentially target slow-inactivated Nav1.8 channels (Cardenas et al., 2006). This raises the possibility that access to the drug-binding site in fast-inactivated state of Nav1.8-type channels might be substantially different from the site in Nav1.7 and Nav1.3 channels.
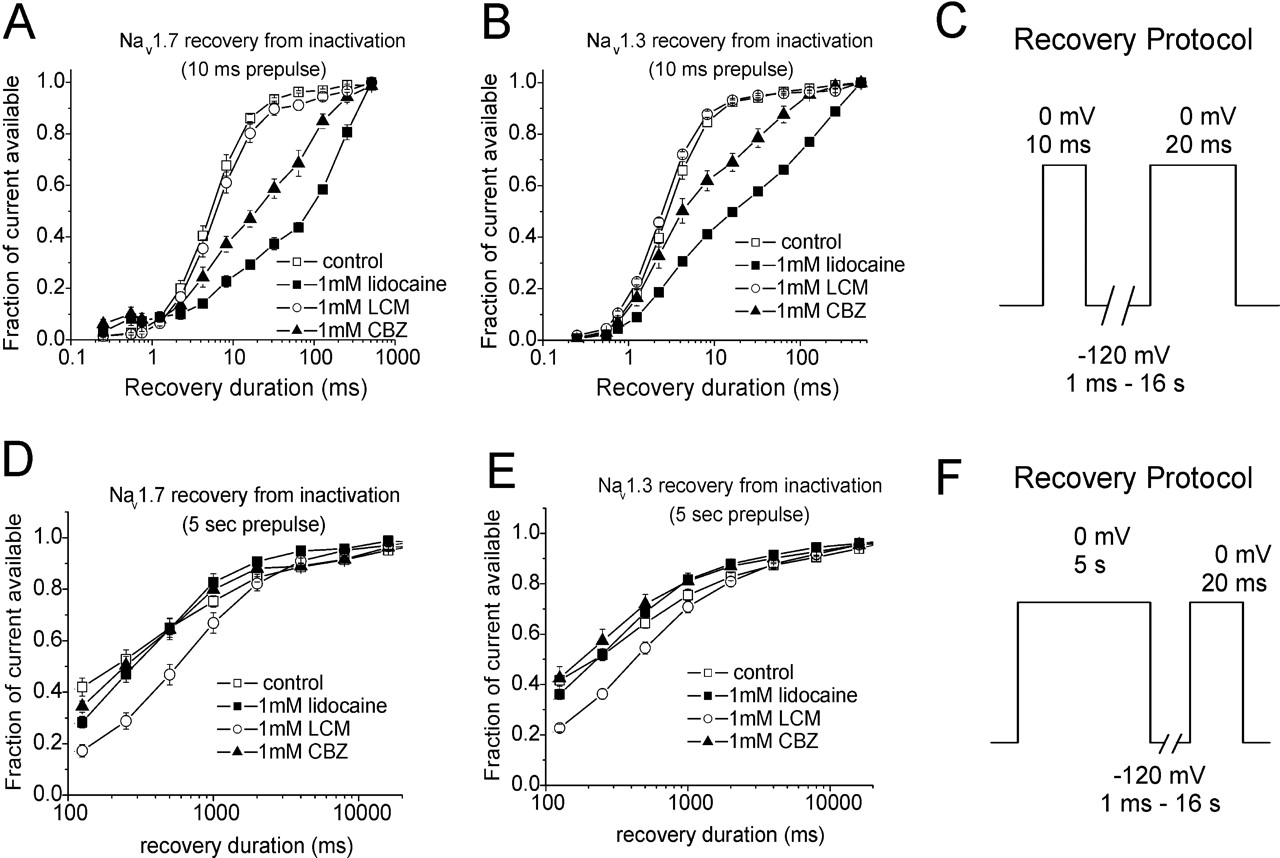
Effects of lacosamide, lidocaine, and carbamazepine on the recovery of Nav1.7 and Nav1.3 channels from inactivation. Cells were held at –120 mV, depolarized to 0 mV for 10 ms or 5s and then hyperpolarized to –120 mV for variable increasing durations before testing the available current with a step depolarization to 0 mV. A and B, lidocaine and CBZ caused a decrease in the rate of recovery from inactivation/inhibition (10 ms) for both Nav1.7 (A) and Nav1.3 (B) channels, whereas LCM had no effect. C, voltage protocol used for A and B is shown. Data sweeps were acquired at 0.5 Hz. D and E, lidocaine and CBZ did not alter recovery kinetics after induction of slow inactivation compared with control. In the presence of LCM, the fraction of available channels after 5-s depolarization was lower compared with control, lidocaine, and carbamazepine, and the duration of recovery was longer (significant at 500 ms) compared with the other conditions. F, voltage protocol used for D and E is shown. Data sweeps were acquired at 0.5 Hz for short recovery durations and at slower rates for the longer recovery durations.
An interesting finding was that carbamazepine and lidocaine show significant inhibition of Nav1.7 and Nav1.3 channels prepulsed to 0 mV for durations ranging from 3 to 100 ms (Fig. 5, A and B). In that same range of prepulse durations, lacosamide induces minimal inhibition of Nav1.7 and Nav1.3 channels. At prepulse durations ranging 100 ms to 11 s, lacosamide induces inhibition of Nav1.7 and Nav1.3, whereas inhibition by carbamazepine and lidocaine levels off. All three drugs eventually reached maximal inhibition around 9 to 11s for both channels. It is interesting that the time point where development of slow inactivation starts to occur for control channels is near the time point at which lacosamide begins to cause inhibition and also where carbamazepine and lidocaine inhibition plateaus. This suggests lacosamide interaction with Nav1.7 and Nav1.3 channels is dependent on the channels transitioning to slow-inactivated states and, in contrast, that transition to the slow-inactivated state might impede further Nav1.7 and Nav1.3 channel inhibition by carbamazepine and lidocaine. Although recovery from slow inactivation appeared to be slower in the presence of lacosamide, due to an increase in the steady-state fraction of slow-inactivated channels by lacosamide, it was not altered by lidocaine or carbamazepine, providing additional evidence that the mechanism of block by lacosamide is possibly distinct from that of lidocaine or carbamazepine.

Comparison of inhibition on resting and inactivated Nav1.7, Nav1.3, and TTX-R channels by lacosamide, lidocaine, and carbamazepine (all at 300 μM). A, LCM had the smallest inhibitory effect on resting Nav1.7, Nav1.3, and TTX-R channels compared with lidocaine and carbamazepine. Lidocaine and CBZ had the greatest inhibitory effect on resting TTX-R channels compared with Nav1.7 and Nav1.3 channels. B, effects of LCM, lidocaine, and CBZ on completely inactivated Nav1.7, Nav1.3, and TTX-R channels. For complete inactivation, the voltage of the inactivating prepulse was varied due to differences in the voltage dependence of inactivation between channels. Nav1.7 channels were prepulsed to –50 mV, Nav1.3 channels to –40 mV, and TTX-R channels to –20 mV. Lacosamide showed considerably more inhibition on TTX-R channels compared with Nav1.7 and Nav1.3 channels. Lidocaine and carbamazepine were effective at inhibiting TTX-R channels more than Nav1.3 channels but not Nav1.7 channels. C, effects of LCM, lidocaine, and CBZ on Nav1.7, Nav1.3, and TTX-R channels prepulsed to –50 mV. This prepulse voltage was chosen to theoretically represent a slightly depolarized potential for a sensory neuron. Lacosamide and lidocaine showed comparable inhibition on TTX-R channels, Nav1.7, and Nav1.3 channels. Carbamazepine displayed more inhibition on TTX-R channels compared with Nav1.3 channels. Values and statistical significance were determined using a one-way ANOVA followed with a Tukey's means comparison test (**, p < 0.01; ***, p < 0.001).

Effects of lacosamide, lidocaine, and carbamazepine (all at 1 mM) on the voltage dependence of steady-state fast and slow inactivation for TTX-R channels. A, representative family of Nav1.8-type currents recorded from a small-diameter DRG neuron. B, LCM, lidocaine, and CBZ had no effect on the voltage dependence of steady-state fast inactivation for TTX-R channels. Data were acquired at 0.2 Hz. C, LCM, lidocaine, and CBZ caused a significant hyperpolarizing shift in the voltage dependence of slow inactivation for TTX-R channels. Data were acquired at 0.09 Hz. Lacosamide caused the greatest decrease in elicited TTX-R current followed by lidocaine and carbamazepine, respectively. At 20 mV, lacosamide and lidocaine reduced available current by 90 and 80%, whereas maximal reduction by carbamazepine at this voltage was similar to control (approximately 50%).
Lacosamide has been shown in acute, inflammatory and neuropathic animal pain models (Beyreuther et al., 2006, 2007a; Hao et al., 2006), as well as in phase II clinical trials for painful diabetic neuropathy (Rauck et al., 2007), to effectively inhibit pain with few adverse side effects. Based on the data reported here, it is likely that the effects of lacosamide on sodium channel activity contribute to its analgesic activity. Tissue injury and inflammatory modulators can cause prolonged depolarization of peripheral nociceptors, and this probably contributes to inflammatory and neuropathic pain. A recent study suggests that the effectiveness of drugs at inhibiting peripheral nerve activity following nerve injury does not depend so much on the frequency at which action potentials are generated but rather on the nature of the injury and the pattern of action potential activity (Ritter et al., 2007). Given that a substantial degree of slow inactivation can be observed for TTX-R sodium channels at small, subthreshold membrane depolarizations without any spiking activity (Blair and Bean, 2003), a compound like lacosamide that targets the slow-inactivated channel might show enhanced therapeutic benefit.
Side effects related to the central nervous system can limit the usefulness of sodium channel blockers in treating pain; therefore, drugs that are better able to differentiate between normal and abnormal activity are desired. The enhanced ability of lacosamide to discriminate between resting channels and channels that have been subjected to prolonged depolarizations should contribute to a better safety profile and increased tolerability.
Overall, our data show lacosamide is effective at blocking inactivated voltage-gated Nav1.3-, Nav1.7-, and Nav1.8-type channels, voltage-gated sodium channels that have been implicated as playing important roles in pain mechanisms. Furthermore, our data show that the kinetics of lacosamide inhibition are much slower than those of two other sodium channel blockers, carbamazepine and lidocaine, at least for Nav1.3 and Nav1.7 channels. Our data are consistent with the proposal that lacosamide inhibition of voltage-gated sodium channels involves selective interactions with slow-inactivated channels (Errington et al., 2008). Ratios of resting IC50 to inactivated IC50 suggest that lacosamide should be more effective than carbamazepine and lidocaine at selectively blocking the electrical activity of neurons that are chronically depolarized while having little or no effect on neurons with normal resting potentials, and we propose that this could contribute to the therapeutic action of lacosamide.
Footnotes
-
This study was supported by Schwarz BioSciences.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.107.133413.
-
ABBREVIATIONS: Lacosamide, (2R)-2-(acetylamino)-N-benzyl-3-methoxypropanamide; DRG, dorsal root ganglion; TTX-S, tetrodotoxin-sensitive; TTX-R, tetrodotoxin-resistant; HEK, human embryonic kidney; DMEM, Dulbecco's modified Eagle's medium; DMSO, dimethyl sulfoxide; IV, current-voltage; ANOVA, analysis of variance; LCM, lacosamide; CBZ, carbamazepine.
-
↵
 The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material. -
↵1 Current affiliation: Merz Pharmaceuticals GmbH, Frankfurt am Main, Germany.
- Received October 23, 2007.
- Accepted March 28, 2008.

- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}