


Abstract
LY320135 is a selective antagonist for the brain CB1 receptor, having greater than 70-fold higher affinity for the CB1 than the peripheral CB2 receptor. The Ki values for LY320135 at the CB1 and CB2 receptors, transfected and stably expressed in cell lines, were 224 nM and >10 μM, respectively. SimilarKi values were measured in binding studies performed on cerebellum and spleen membrane preparations endogenously expressing the CB1 (203 nM) and CB2 (>10 μM) receptors, respectively. LY320135 functionally reversed anandamide-mediated adenylate cyclase inhibition in Chinese hamster ovary (CHO) cells stably expressing the CB1 receptor. Pertussis toxin treatment of CHO cells expressing the CB1 receptor attenuated the anandamide-mediated inhibition of adenylate cyclase and unmasked a stimulatory effect of anandamide on adenylate cyclase. The stimulatory component was blocked with LY320135. This compound also blocked WIN 55212–2-mediated inhibition of N-type calcium channels and activation of inwardly rectifying potassium channels in N18 and AtT-20-CB2 cells, respectively. LY320135 is a promising lead compound for the further development of novel, potent and selective cannabinoid antagonists of novel structure.
Marijuana has been used for recreational and medicinal purposes for centuries. The wide appeal of this drug lies predominantly in its ability to alter mood and behavior. The therapeutic effects of marijuana are surprisingly broad and include its ability to act as an antiemetic, anti-inflammatory, antiglaucoma, analgesic and appetite-enhancing agent. These diverse physiological effects are mediated by the active principal in marijuana, THC, for which specific receptors were identified in the brain (Devane et al., 1988). Three subtypes of cannabinoid receptors, CB1 (Matsuda et al., 1990), CB1A (Shire et al., 1995) and CB2 (Munro et al., 1993), have been identified, cloned and sequenced to date and are members of the superfamily of G-protein-coupled receptors. CB1 receptors are primarily located in the central nervous system. The CB1 receptor is functionally linked to the inhibition of adenylate cyclase (Howlett et al. 1988; Felder et al., 1992), and more recently has been shown to inhibit N- and Q-type voltage-dependent calcium channels (Mackie and Hille, 1992; Caufield and Brown, 1992;Mackie et al., 1995) and to stimulate an inwardly rectifying potassium current (Kir current) (Mackie et al., 1995). Based on the high abundance of CB1 receptors in hippocampus, cerebellum and basal ganglia (Herkenham et al., 1991), and the well known behavioral effects of cannabinoid agonists (Mechoulam, 1986), it is likely that this receptor regulates short-term memory, coordination of movement and emotions. The CB2 receptor is found predominantly in hematopoietic cells but not as yet in the brain (Munro et al., 1993; Lynn and Herkenham, 1994) and has been shown to couple to the inhibition of adenylate cyclase, but not to Q-type calcium channels or Kir current (Felderet al., 1995). The CB2 receptor may mediate some of the peripheral effects of THC, such as immunosuppression (Martin, 1986). High concentrations of THC can also activate signal transduction pathways through cannabinoid receptor-independent mechanisms (Felderet al., 1992). The discovery of brain receptors for THC led to the discovery of the endogenous cannabinoid agonist anandamide (Devane et al., 1992) and other potential cannabimimetic fatty acid amides (Felder et al., 1993; Hanus et al., 1993; Di Marzo et al., 1994). These discoveries have led to a growing interest in determining the physiological role of anandamide, other fatty acid amides and cannabinoid receptors.
Cannabinoid receptor pharmacology has been hampered by the availability of selective antagonists. Three aminoalkylindoles have been shown to display cannabinoid receptor antagonist properties. WIN 56,098 was found to be a cannabinoid antagonist in in vitroexperiments (Pacheco et al., 1991; Compton et al., 1992). More recently, two more potent antagonists, bromopravadoline (Casiano et al., 1990) and iodopravadoline (Pertwee et al., 1995), have been described. The most potent antagonist to date, SR141716A, is based on a pyrazole structure (Rinaldi-Carmona et al., 1994). SR141716A was found to be selective and potent at the CB1 receptor and blocked WIN 55212–2-mediated behavioral responses. SR141716A had a weak affinity for the CB2 receptor expressed in clonal cell lines (Felder et al., 1995). Here we describe a CB1 receptor antagonist of novel structure that is selective for the CB1 receptor.
Materials and Methods
CP 55,940 was generously provided by Dr. Larry Melvin (Pfizer Inc., Groton, CT.); WIN 55212–2 by Dr. Susan Ward (Sterling-Winthrop Research Institute, Malvern, PA); HU-210 by Dr. Raphael Mechoulam (Hebrew University, Jerusalem, Israel) and (−)Δ-9-THC by the National Institute on Drug Abuse. Anandamide was obtained from Biomol (Plymouth Meeting, PA). All other reagents were purchased from Sigma Chemical Co. (Saint Louis, MO). All assays were performed in glass test tubes which were treated by exposure to dimethyldichlorosilane vapor under vacuum overnight.
Expression of CB1 and CB2 receptors, plasma membrane preparation and radioligand binding assays.
CB1 and CB2 cDNA were stably transfected and expressed in either CHO cells or L cells as described previously (Felder et al., 1995). Plasma membranes were prepared from murine fibroblast L cells, CHO-CB2 cells, rat cerebellum and rat spleen as described previously (Felder et al., 1995). Competition binding assays were performed with 0.5 nM3H-CP 55,940 as the labeled ligand. Radioligand binding of cannabinoid agonists was measured by a previously described rapid filtration assay (Felder et al., 1993). Phenylmethylsulfonyl fluoride was included where indicated to inhibit the degradation of anandamide during radioligand binding experiments (Deutsch and Chin, 1993; Childers et al., 1994). Binding assays for data shown in table 1 were performed as described previously (Foreman et al., 1992). The Ki values for competition binding experiments and EC50 values for the inhibition of forskolin-stimulated cAMP accumulation were calculated by nonlinear regression analysis of the primary data with the computer software Graph Pad (San Diego, CA). Levels of cAMP accumulation were measured by radioimmunoassay as described previously (Felder et al., 1992).
Comparison of K values for competition of LY320135 and selected cannabinoid ligands for 0.5 nM 3H-CP 55,940 binding to the CB1 and CB2 receptors
Electrophysiological recordings.
N18 cells were cultured as described previously (Mackie et al., 1993) and were used for measurement of calcium currents. AtT-20-CB1 cells were cultured as described previously and were used to measure of potassium currents (Mackie et al., 1995). Currents were recorded by the whole-cell voltage-clamp technique (Hamill et al., 1981) with pipettes pulled from microhematocrit glass (VWR Scientific, Plainfield, NJ) and fire polished. For recording, a coverslip containing cells was transferred to a 200-μl chamber that was constantly perfused (1–2 ml/min) with the appropriate external solution. Solution reservoirs were selected by a series of solenoid valves which allowed solution changes in less than 1 min. Voltage protocols were generated, and data were digitized, recorded and analyzed with Basic-Fastlab (Indec Systems, Capitola, CA). Junction potentials were uncorrected.
Potassium currents were measured with a pipette solution containing (mM): 130,, KCl; 20, HEPES; 10, EGTA; 5, MgCl2; 3, Na2ATP; 0.6, GTP; 0.08, leupeptin, pH 7.25 with KOH. The external solution contained (mM): 40, KCl; 110, NaCl; 1, CaCl2; 25, HEPES; 10, glucose, pH 7.35 with NaOH. Fatty acid-free bovine serum albumin (3 mM) was added to decrease the adsorption of cannabinoids. The Kir current was defined as the component of the current sensitive to 1 mM Ba++ elicited during the final 150 ms of a 250-ms hyperpolarizing pulse to −100 mV from a holding potential of −45 mV. Currents were sampled at 1 kHz. The magnitude of the Kir current depended on cell size, therefore aggregate current data are presented as current densities normalized to cell capacitance.
Calcium currents (ICa) were measured with a pipette solution containing (mM): 100, CsCl; 40, HEPES; 10, EGTA; 5 MgCl2; 3, Na2ATP; 0.2, GTP; 0.08, leupeptin, pH 7.30 with CsOH. The external solution contained (mM): 160, NaCl; 10, BaCl2; 4, KCl; 1, MgCl2; 10, HEPES; 8, glucose, pH 7.35 with NaOH. Tetrodotoxin (200 nM) was added to block sodium currents and fatty acid-free bovine serum albumin was added to decrease the adsorption of cannabinoids. ICa was measured near the end of a 25-ms depolarizing pulse to 0 mV from a holding potential of −90 mV and was defined as that component of the current sensitive to 100 mM CdCl2. Currents were sampled at 4 kHz. To control for possible response variations with passage number and to avoid sources of systematic bias, experimental and control measurements were alternated whenever possible and concurrent controls were always performed. Data are expressed as the mean ± standard error of the mean.
Results
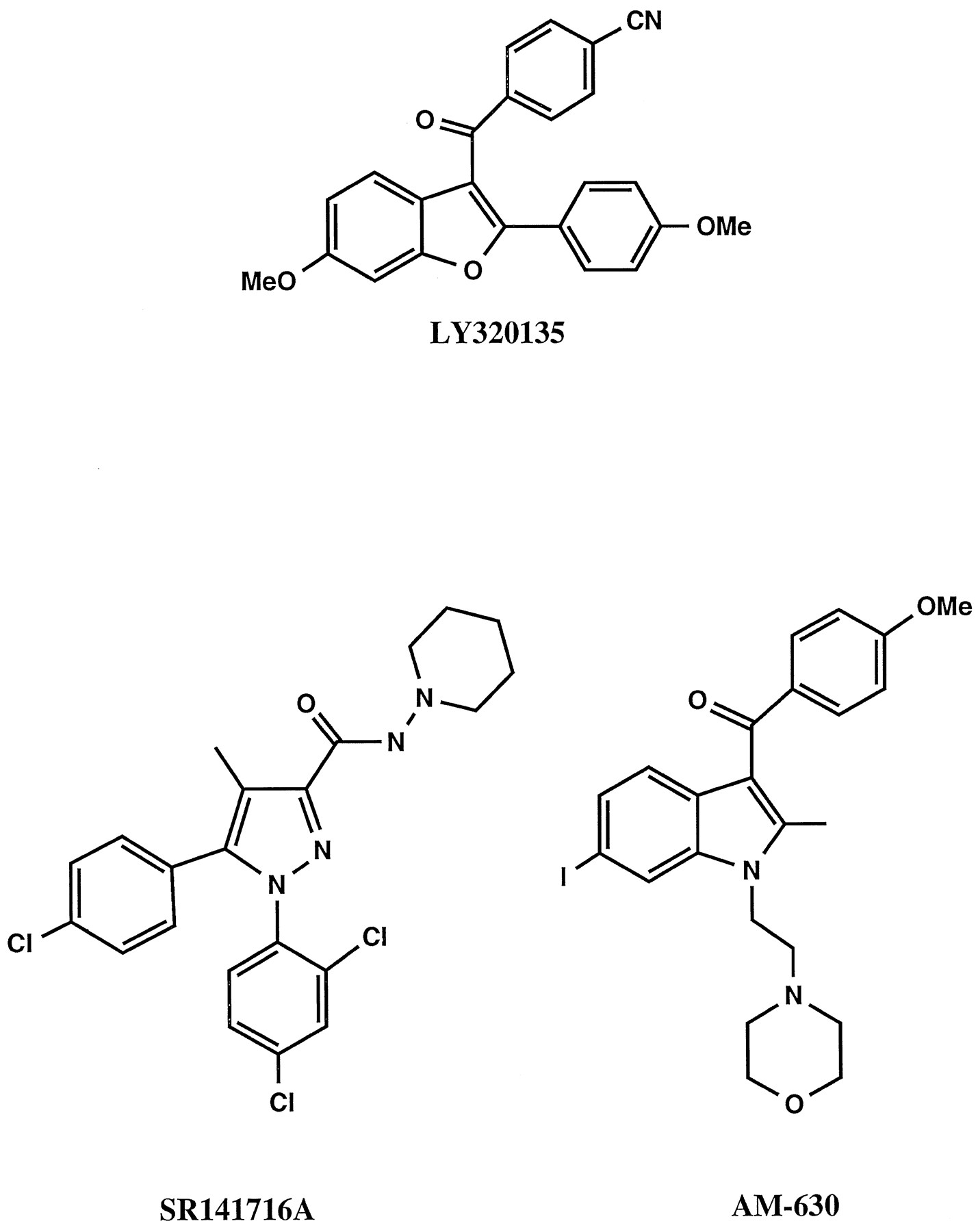
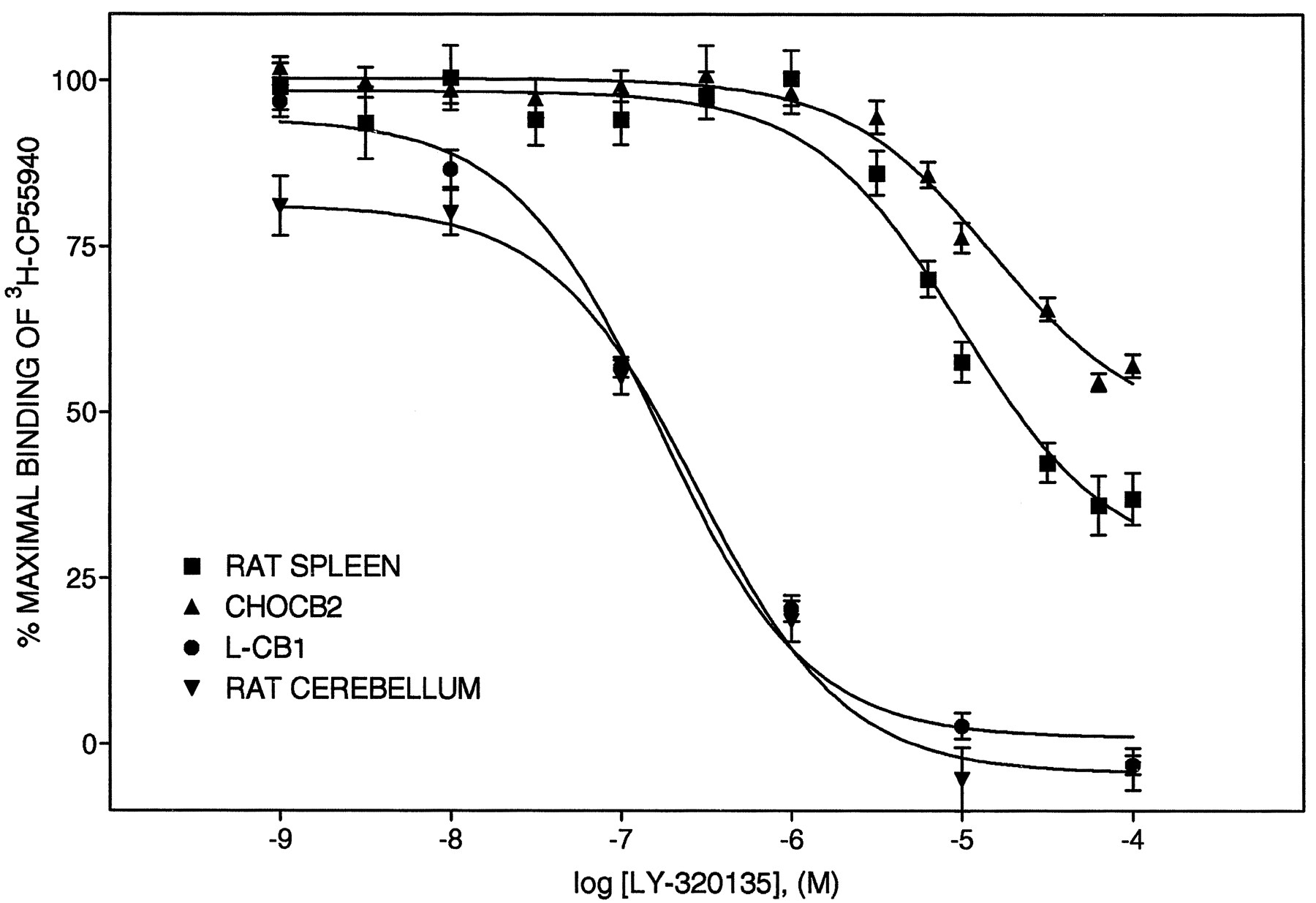
LY320135 is a substituted benzofuran which is structurally distinct from the aminoalkylindole and pyrazole type cannabinoid antagonists, AM630 and SR141716A, respectively (fig.1). LY320135 was selected from a series of related compounds as having the highest affinity for the CB1 receptor. The ability of LY320135 to compete for3H-CP55,940 binding was evaluated at the cannabinoid CB1 and CB2 receptors (fig.2). The Ki values for LY320135 are listed in table1 and are compared with theKi values for the cannabinoid antagonist, SR141716A, and selected cannabinoid agonists. LY320135 was essentially equipotent for binding to the CB1 receptor ectopically expressed in L cells (L-CB1) and to rat cerebellum (an endogenous source of CB1 receptors) (fig. 2) and completely displaced3H-CP55,940 binding at saturating concentrations. LY320135 had a greater than 70-fold higher affinity for the CB1 receptor than the CB2 receptor expressed in CHO cells (CHO-CB2) or rat spleen membranes (an endogenous source of CB2 receptors), respectively. LY320135 failed to completely displace3H-CP55,940 binding to the CB2 receptor in either spleen or CHO-CB2 membranes because of solubility limitations above 100 μM (fig. 2). Previously reported Ki values for cannabinoid receptor agonists, HU210, CP55940, THC and the endogenous cannabinoid receptor agonist anandamide, were included in table 1 to provide comparisons with known compounds by the same methodology (Felder et al., 1995). Ki ratios of CB1/CB2 receptors are listed in the right-hand column of table 1.

Structure of LY320135, SR141716A and AM-630.
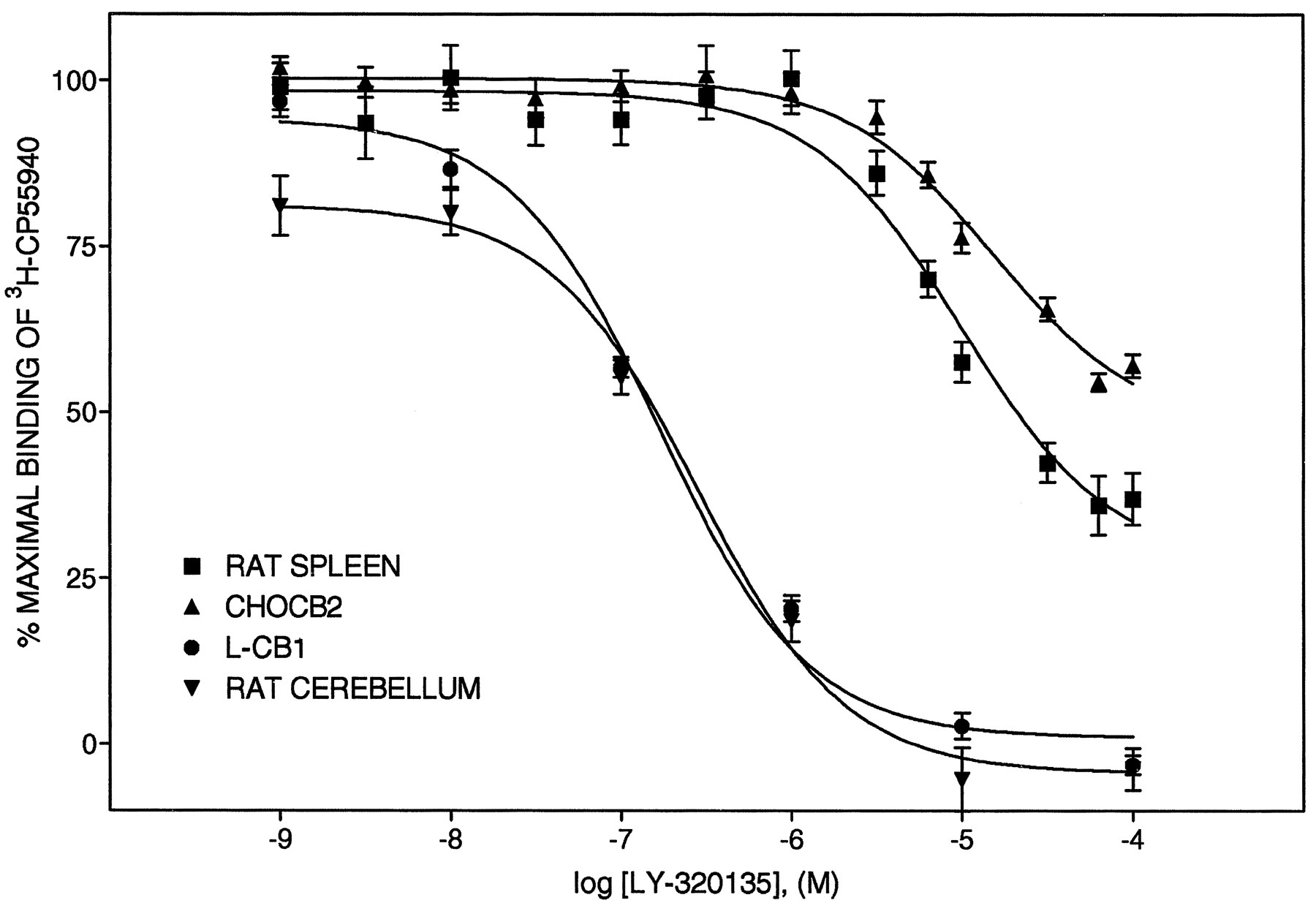
LY320135-mediated inhibition of 3H-CP 55,940 binding to membranes prepared from rat spleen, CHO-CB2 cells, rat cerebellum and L-CB1 cells, and rat spleen cell membranes. Plasma membranes were prepared from CHO and L cells stably transfected with the CB2 and CB1 receptor, respectively (CHO-CB2 and L-CB1, respectively) as well as rat spleen and cerebellum as described under “Materials and Methods.” Competition binding curves were analyzed by nonlinear regression analysis, and the derived binding constants are the mean ± S.D. of at least three experiments, each performed with triplicate determinations. Kivalues are listed in table 1. The curves shown are from representative experiments.
LY320135 was evaluated as a functional antagonist by its ability to reverse the inhibition by anandamide of forskolin-stimulated cAMP accumulation in CHO-CB1 cells (fig. 3A). Anandamide (10 μM) completely attenuated the forskolin-stimulated accumulation of cAMP (fig. 3A). LY320135 reversed the inhibitory effect of anandamide, but also caused an increase in cAMP accumulation above levels observed with forskolin alone. LY320135 (10 μM) alone had no effect on cAMP levels (data not shown), but in combination with forskolin, enhanced the accumulation of cAMP (fig. 3A). The antagonist, SR141716A, also reversed the inhibitory effects of anandamide to levels above those observed with forskolin alone (fig. 3B). SR141716A alone had no effect on cAMP accumulation (data not shown), but also displayed similar enhancement of forskolin-stimulated cAMP accumulation. CHO-CB1 cells were pretreated with pertussis toxin to determine whether Gi or Go mediated the overshoot of cAMP accumulation to levels above those achieved with forskolin alone (fig. 4). Both anandamide and WIN 55,212–2 attenuated forskolin-stimulated cAMP accumulation in CHO-CB1 cells (fig. 4). In the presence of pertussis toxin, anandamide alone had no effect on cAMP accumulation (data not shown), but augmented the forskolin-stimulated response. Similar results were observed when the aminoalkylindole agonist, WIN55212–2, was substituted for anandamide (fig. 4). Anandamide and WIN 55212–2, when applied alone, with or without pertussis toxin, did not increase cAMP accumulation above basal levels (data not shown). LY320135 blocked the anandamide-mediated augmentation of cAMP accumulation in the presence of pertussis toxin (fig. 5), which suggests that it was mediated through cannabinoid receptor stimulation (IC50 = 734 ± 122 nM). Indomethacin (10 μM) had no effect on the accumulation of cAMP in the CHO cell after anandamide and LY320135 addition (data not shown), which suggests that increases in cAMP were not mediated through prostaglandin release. Moreover, concentrations of anandamide and WIN 55212–2 used were below the those shown previously to stimulate arachidonic acid release in CHO cells (>10 μM) (Felder et al., 1995). These results suggest that the CB1 receptor couples to the stimulation of cAMP accumulation and that the stimulation is unmasked when the inhibitory response is blocked with pertussis toxin.

The effect of LY320135 (A) and SR141716A (B) on anandamide-mediated inhibition of cAMP accumulation in CHO-CB1 cells. Forskolin (500 nM)-stimulated cAMP accumulation was measured in CHO-CB1 cells, as described under “Materials and Methods,” in combination with 1 μM anandamide and increasing concentrations of either LY320135 (A) or SR141716A (B). Both LY320135 and SR141716A reversed anandamide-mediated inhibition of forskolin-stimulated cAMP accumulation in a concentration-dependent manner to levels above that observed with forskolin alone. EC50 values are provided under “Results.” Data are the mean ± S.E.M. of at least three experiments, each performed in triplicate. (FRSK, forskolin; ANM, anandamide)
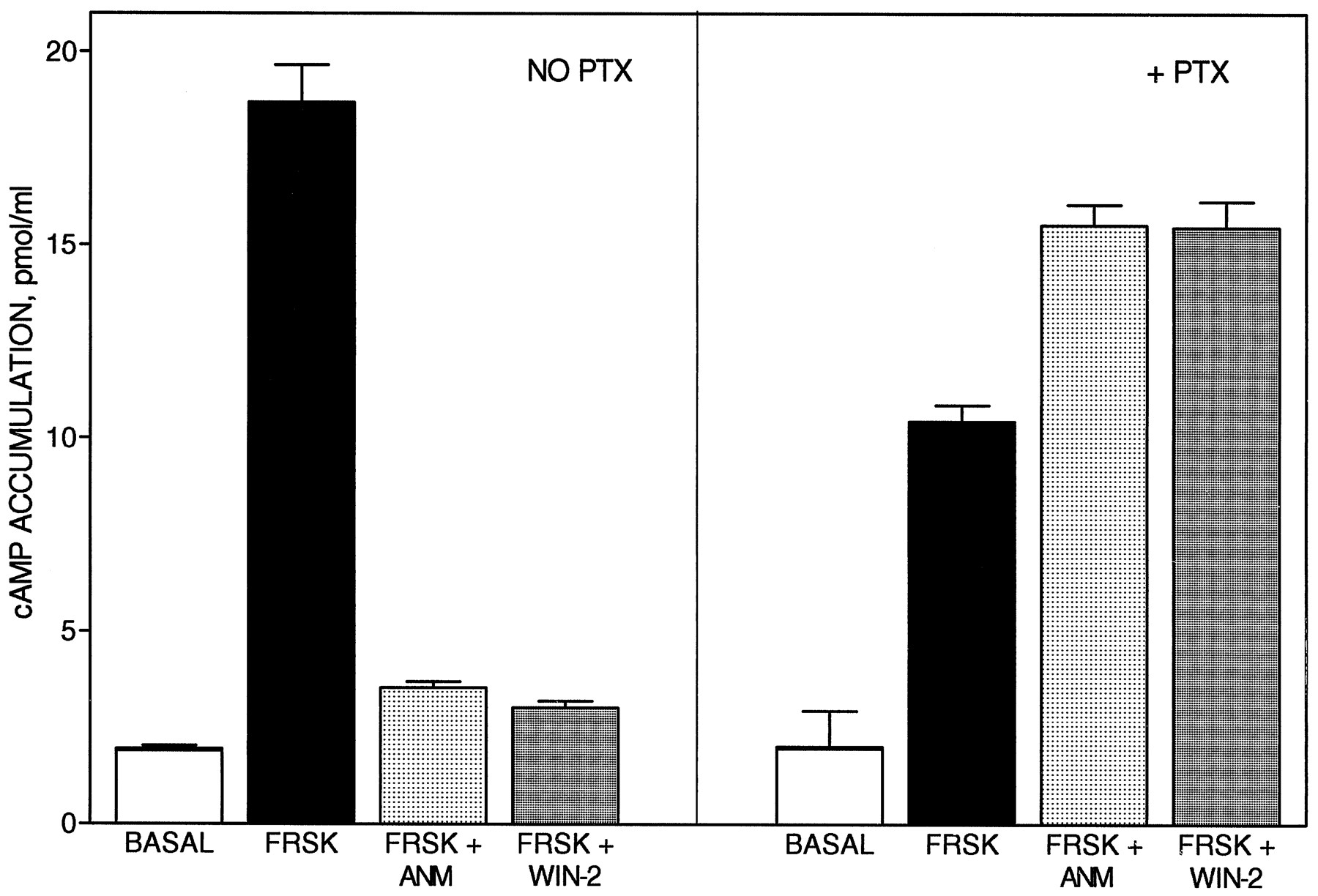
The effect of pertussis toxin on anandamide- and WIN 55212–2-mediated inhibition of forskolin-stimulated cAMP accumulation in CHO-CB1 cells. Forskolin (500 nM)-stimulated cAMP accumulation was measured in CHO-CB1 cells, in the presence and absence of anandamide or WIN 55212–2, as described under “Materials and Methods.” Pertussis toxin (10 ng/ml) was added 18 hr before the measurement of cAMP accumulation. Data are the mean ± S.E.M. of at least three experiments, each performed in triplicate. (FRSK, forskolin; ANM, 1 μM anandamide; WIN-2, 50 nM WIN 55212–2)

Effect of LY320135 on anandamide-mediated inhibition of cAMP accumulation in pertussis toxin-treated CHO-CB1 cells. Forskolin (500 nM)-stimulated cAMP accumulation was measured in CHO-CB1 cells, as described under “Materials and Methods.” Pertussis toxin (10 ng/ml) was added 18 hr before the measurement of cAMP accumulation. In the presence of pertussis toxin, LY320135 blocked the anandamide + forskolin-mediated stimulation of cAMP accumulation (IC50 = 734 ± 122 nM). Data are the mean ± S.D. of two experiments, each performed in triplicate. (FRSK, forskolin; ANM, 1 μM anandamide; LY, LY320135)
The selectivity of LY320135 for other G protein-coupled receptors was evaluated in a radioligand binding screening assay (table2). LY320135 had a 15-fold greater selectivity for the CB1 receptor than muscarinic receptors, and at least a 45-fold greater selectivity than benzodiazepine and 5-HT2 receptors. All other receptors tested in table 2 had affinities for LY320135 at least 70-fold higher than for the CB1 receptor.
Comparison of Ki values for competition of LY320135 for binding to several selected neurotransmitter receptors
The ability of LY320135 to reverse the WIN 55212–2-mediated inhibition of N-type calcium currents (ICa) and activation of Kir currents was evaluated in N18 cells and AtT-20 cells. Application of 100 nM WIN 55212–2 to N18 cells inhibited a significant fraction of the high voltage-activatedICa (fig.6A). The inhibition was quickly reversed by coapplication of 1 μM LY320135. Despite the continued application of LY320135, the muscarinic agonist, oxotremorine-M, still inhibitedIca, which suggests that LY320135, at 1 μM, doesn’t interfere with muscarinic receptors or signal transduction events distal to the cannabinoid receptor. By itself, 1 μM LY320135 had little effect on the calcium current (−2.4 ± 2.1% inhibition, n = 5). LY320135 was able to potently reverse the inhibition of ICa by 100 nM WIN 55212–2 with an IC50 of 55 ± 10 nM (fig.6B). There was a tendency for over-recovery ofICa after applications of the higher concentrations of LY320135. After the addition of 1 μM LY320135, the current returned to 116 ± 10% of its original level (n = 9, data not shown). The effects of LY320135 could not be reversed by washing for up to 5 min (data not shown).

LY320135 reverses and blocks the modulation of ion channels by cannabinoid receptor agonists. (A) LY320135 can reverse calcium current (ICa) inhibition by WIN 55212–2 in N18 cells. Application of 100 nM WIN 55212–2 inhibits a significant fraction of the ICa. Coapplication of LY320135 reverses this inhibition. In the continued presence of LY320135, inhibition by 10 μM oxotremorine-M is maintained. (B) Concentration-response curve of LY320135 reversal ofICa inhibition by 100 nM WIN 55212–2 in N-18 cells. Reversal is potent with an IC50 of 55 ± 10 nM and noncooperative with a Hill coefficient of 0.91 ± 0.16. The solid line indicates the least squares fit for the Hill equation;n = 4 to 9 for each data point. (C) LY320135 prevents activation of Kir current by WIN 55212–2 in AtT-20-CB1 cells. Prior application of 1 μM LY320135 prevents activation of Kir current by WIN 55212–2. However, 200 nM somatostatin still activates the current. (D) Summary of LY320135 effects of Kir current activation by 100 nM WIN 55212–2 in AtT-20-CB1 cells. By itself, LY320135 has little effect on Kir current activation and in control cells 100 nM WIN 55212–2 activates a substantial current. However, in cells treated with 1 μM LY320135, 100 nM WIN 55212–2 no longer activates a Kir current. For each condition, n = 4 to 6. Data are the mean ± S.E.M. (WIN, WIN 55212–2; Oxo-M; oxotremorine-M; SOM, somatostatin; LY, LY320135).
CB-1 receptors also activate an inwardly rectifying potassium current (Kir) (Mackie et al., 1995). Pretreatment of AtT-20-CB1 cells with 1 μM LY320135 blocked WIN 55212–2-mediated stimulation of Kir; however, the current was still activated by 200 nM somatostatin (fig. 6C). LY320135 by itself had little effect on Kir(0.78 ± 0.82 pA/pF, n = 4) (fig. 6D). In control cells, 100 nM WIN 55212–2 activated Kir by an average of 10.9 ± 3.2 pA/pF (n = 6). In cells exposed to 1 μM LY320135, activation of Kir by WIN 55212–2 was suppressed completely (0.08 ± 0.32 pA/pF,n = 5). However, somatostatin still activated the Kir in the presence of LY320135 (17.8 ± 2.0 pA/pF, n = 4) (data not shown).
Discussion
LY320135 was selected as a lead compound from a series of potential cannabinoid receptor antagonists because it displayed the highest affinity for the CB1 subtype of the cannabinoid receptor. This compound is a benzofuran analog structurally distinct from the previously described aminoalkylindole or pyrazole cannabinoid receptor antagonists and may provide a novel approach to cannabinoid receptor antagonist development. LY320135 displayed aKi of 141 nM for the CB1 receptor ectopically expressed in a mammalian cell line and an essentially equal affinity for CB1 receptors endogenously expressed in rat cerebellum. LY320135 had a relatively low affinity for the CB2 receptor (Ki ≥ 10 μM) and ten other unrelated receptors (table 2), and therefore offers good selectivity for the CB1 receptor. Further modifications in the structure of LY320135 are currently being evaluated for cannabinoid receptor antagonist activity. The structure of LY320135 offers considerably more accessibility for structural modification than SR141716A and may be less toxic when administered in vivo.
LY320135 was a functional antagonist for anandamide-mediated inhibition of forskolin-stimulated cAMP accumulation. During the course of these experiments, an overshoot of cAMP accumulation elicited by LY320135 was discovered which suggested that cannabinoid receptors couple to the stimulation of cAMP accumulation which is masked by the inhibitory component coupled through Gi. In the presence of pertussis toxin, LY320135 blocked the anandamide-mediated stimulation of cAMP accumulation, which demonstrates for the first time that the stimulatory response is regulated through the CB1 receptor. The LY320135-mediated reversal of the stimulatory effect had an IC50 of 734 nM which is less potent than the 141 nM Ki observed for LY320135 in competition experiments, which suggests that the functional response is complex and possibly mediated by multiple effectors. Indomethacin had no effect on the stimulatory phase of cAMP accumulation, which suggests that it was not mediated through prostaglandin production as previously demonstrated in fibroblasts (Hillard and Bloom, 1983). Additional studies have observed cannabinoid agonist-induced increases in cAMP in fibroblasts (Kelly and Butcher, 1979) and mouse brain (Dolby and Kleinsmith, 1974), but the response was not directly linked to cannabinoid receptor stimulation. Other Gi-linked receptors have been shown to stimulate cAMP accumulation after pertussis toxin pretreatment, such as alpha-2 adrenergic (Federman et al., 1992) and muscarinic m4 receptors (Joneset al., 1991). However, in CHO cells, increases in cAMP were observed only after adenylate cyclase was first stimulated with forskolin. These findings are similar to those observed in liver (Hillard et al., 1986) and cardiac cell membranes (Hillardet al., 1990) in which THC stimulated increases in cAMP, only in combination with glucagon or isoproterenol, respectively. These results are consistent with the regulation of type II or IV adenylate cyclase by βγ subunits of G proteins (Tang and Gilman, 1991). The βγ effect also requires an initial priming of the adenylate cyclase. It is not know which subtypes of adenylate cyclase are expressed in the CHO cell.
The mismatch in Ki and IC50 for LY320135 and SR141716A may be caused by the coupling of the CB1 receptor to both Gi, mediating inhibition of adenylate cyclase, and to an unknown stimulatory component resulting in increases in cAMP accumulation. The inhibitory effect appears to dominate over the stimulatory component because the stimulatory component can only be observed when the inhibitory component is reduced by either antagonist or pertussis toxin treatment. A Schild analysis would not be practical under these circumstances because of the complex nature of these second messenger responses.
LY320135 was a functional antagonist for WIN 55212–2-mediated inhibition of N-type voltage-dependent calcium currents and stimulation of Kir current. By itself, LY320135 had no effect on either current or on modulation of the currents by other agonists. Under the conditions used for evaluating N-type currents, LY320135 was potent and irreversible during the 5 min of wash perfusion. In addition, application of LY320135 resulted in an overshoot (>100% recovery in fig. 6B) in the WIN 55212–2-mediated inhibition of N-current. This result is similar to the overshoot observed in the cAMP response. The mechanism of the overshoot of calcium channel recovery may be related to activation of the cannabinoid receptor and is currently being investigated.
Development of cannabinoid receptor selective antagonists will provide the tools necessary to more fully understand the cannabinoid receptors both in the central nervous system and in the peripheral immune system. Although the CB1 and CB2 receptors have very similar pharmacological selectivity for cannabinoid agonists, it is apparent that there are significant differences in the binding pocket of these two receptors because of their differential selectivity for the currently available antagonists. Use of antagonists in studies of cannabinoid receptor biology will be useful in separating receptor-dependent from receptor-independent effects of cannabinoid agonists. A larger arsenal of cannabinoid receptor antagonists will be helpful in characterizing known as well as new cannabinoid receptor subtypes, should they be discovered.
Acknowledgments
We thank Yvonne Lai and Richard Mitchell of Panlabs (Seattle, WA) for the kind gift of AtT-20-CB2 cells used in the present study.
Footnotes
-
Send reprint requests to: Dr. Christian Felder, Eli Lilly Research Laboratories, Drop 0510, Lilly Corporate Center, Indianapolis, IN 46285.
-
↵1 The development of this study was supported in part by a SBIR Phase I grant DA09203. This study was also supported partly by the Keck Foundation, a McKnight Research Award, and National Institutes of Health grants NS01588, NS08174, and DA08934 (to K.P.M.).
- Abbreviations:
- THC
- (−)Δ9-tetrahydrocannabinol
- CHO
- Chinese hamster ovary
- HEPES
- N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid
- EGTA
- ethyleneglycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- Received November 20, 1996.
- Accepted September 23, 1997.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}