


Abstract
Cannabinoid receptors couple to both Gs and Giproteins and can consequently stimulate or inhibit the formation of cAMP. To test whether there is specificity among cannabinoid receptor agonists in activating Gs- or Gi-coupled pathways, the potency and intrinsic activity of various cannabinoid receptor ligands in stimulating or inhibiting cAMP accumulation were quantified. The rank order of potencies of cannabinoid receptor agonists in increasing or inhibiting forskolin-stimulated cAMP accumulation, in CHO cells expressing hCB1 receptors, was identical (HU-210 > CP-55,940 > THC > WIN-55212–2 > anandamide). However, the activities of these agonists were different in the two assays with anandamide and CP-55,940 being markedly less efficacious in stimulating the accumulation of cAMP than in inhibiting its formation. Studies examining the effects of forskolin on cannabinoid receptor mediated stimulation of adenyly cyclase also revealed differences among agonists in as much as forskolin enhanced the potency of HU-210 and CP-55,940 by ∼100-fold but, by contrast, had no effect on the potency of WIN-55212–2 or anandamide. Taken together these findings demonstrate marked differences among cannabinoid receptor agonists in their activation of intracellular transduction pathways. This provides support for the emerging concept of agonist-specific trafficking of cellular responses and further suggests strategies for developing receptor agonists with increased therapeutic utility.
CB receptor agonists can produce analgesic, antiemetic and anxyolitic actions. However, because of their psychoactive properties and their other adverse effects on cognition and motor behavior, the therapeutic utility of the currently available agonists is limited (Abood and Martin, 1996; Adams and Martin, 1996; Hollister, 1986; Howlett, 1995;Pertwee, 1995). Moreover, because all of the behavioral effects of CB receptor agonists have thus far been attributed to the same (CB1) receptor subtype (Compton et al., 1993), it is unlikely that development of subtype selective agonists will yield centrally active therapeutic agents devoid of adverse effects (Matsuda, 1997; Matsuda and Bonner, 1995).
Like other G protein-coupled receptors, CB1receptors couple to multiple intracellular signal transduction pathways. CB1 receptor agonists inhibit forskolin-stimulated adenylyl cyclase by activation of a pertussis toxin-sensitive Gi/o protein (Howlett and Fleming, 1984). Activation of Gi/o proteins also modifies the function of potassium and calcium channels and,via beta-gamma subunits, stimulate MAP kinases (Bouaboula et al., 1995; Childers and Deadwyler, 1996;Deadwyler et al., 1995; Twitchell et al., 1997). More recently, CB1 receptors have also been shown to positively couple to adenylyl cyclase via pertussis toxin-insensitive Gs proteins. This dual coupling of CB receptors to G proteins with opposing effects on adenylyl cyclase has been demonstrated with both native and recombinant receptors (Felder et al., 1998; Glass and Felder, 1997; Maneuf and Brotchie, 1997) and is similar to what has been previously found for several other G protein-coupled receptors (Eason et al., 1992; Negishi et al., 1995).
Given the complexity of CB receptor-mediated signaling, it is uncertain whether all of the behavioral effects of CB receptor agonists arise via activation of the same intracellular processes. If different transduction mechanisms contribute to the expression of different behaviors, then by developing agonists that selectively target different transduction pathways, specificity in drug action may be achieved. Because such “agonist trafficking” of cellular responses (Kenakin, 1995, 1997) has been demonstrated for other G protein-coupled receptors (Eason et al., 1994; Negishiet al., 1995), we tested whether current CB1 receptor agonists demonstrate selectivity in their activation of Gs- and Gi-coupled pathways.
Methods
Cell culture.
CHO cells stably transfected with the human CB1 receptor gene were obtained from the National Institute of Mental Health. The cells were grown in 24-well plates to ∼80% confluence in F-12 medium supplemented with 10% fetal bovine serum and 500 ng/ml G-418. Each well was washed once with 1 ml of F-12 medium supplemented with 1 mM CaCl2 and 2.5 mM MgCl2. The cells were then incubated overnight in F-12 medium supplemented with 1 mM CaCl2, 2.5 mM MgCl2 and 500 μg/ml G-418. In experiments measuring Gs activity, concomitant activation of Gi proteins was prevented by including 500 ng/ml pertussis toxin in the overnight incubation.
cAMP accumulation assays.
Cells were washed and preincubated with HBSS supplemented with 10 mM HEPES and 4 mM NaHCO3 (pH 7.4) for 5 min at 37°C. Reactions were initiated by the simultaneous addition of forskolin (1 μM), agonists and antagonists to a final assay volume of 600 μl. Rolipram (50 μM), was added 5 min before the initiation of the reactions to prevent degradation of accumulated cAMP. CB1receptor ligands were dissolved (10 mM) in DMSO. Subsequent dilutions were made in HBSS with 50 mg/ml fatty acid-free bovine serum albumin. DMSO (10 mM), equivalently diluted in HBSS, served as a vehicle control and had no effect on cAMP accumulation or forskolin-stimulated cAMP accumulation. cAMP accumulation was measured after a 10-min incubation at 37°C. Reactions were terminated by aspiration of the medium and the addition of 500 μl ice-cold ethanol. The ethanol extracts were dried under N2 gas and reconstituted in acetate buffer. cAMP concentrations were quantified using FlashPlates (NEN, Boston MA).
Radioligand binding assays.
Radioligand binding studies were conducted using membranes prepared from the transfected CHO cells essentially as previously described (Felder et al., 1995). In brief, confluent cells were washed with phosphate-buffered saline, harvested and homogenized in ice cold buffer (50 mM Tris, 5 mM MgCl2, 2.5 mM EDTA, pH 7.4). The homogenate was centrifuged at 2000×g for 15 min at 4°C. The supernatant was collected and centrifuged at 43,000×g for 30 min at 4°C. The membranes were resuspended in buffer and stored at −80°C until used in binding assays. Competition binding studies were conducted by incubating membranes and competing ligands with 1.0 nM [3H]CP-55,940 in buffer containing 0.05% fatty acid-free bovine serum albumin, at 30°C for 60 min. Nonspecific binding was determined in the presence of 5 μM nonradioactive CP-55,940 (5 μM HU-210 produced an equivalent measure of nonspecific binding). In the absence of competing ligand, specific binding accounted for >75% of total binding.
Data analysis.
Data obtained in cAMP accumulation assays were expressed as the percentage of basal or forskolin-stimulated cAMP accumulation. The midpoints (EC50 values) and plateaus of the concentration-response curves were determined by iterative nonlinear regression (Prism, GraphPAD, San Diego, CA). A minimum of six concentration-response curves were generated for each condition. Each concentration-response curve was generated using at six to eight concentrations of agonist, measured as single points. For competition radioligand binding assays, IC50values were obtained from curves generated with at least eight concentrations of competing agent measured in triplicate.Ki values were then calculated using theCheng and Prusoff (1973) equation. Data were presented as pKi (the negative log of the molarKi ) or pEC50 (the negative log of the molar EC50). Analysis of Variance (ANOVA) was conducted using the statistical programs in GraphPAD Prism.
To confirm that the effects of the CB receptor agonists on cAMP accumulation were CB1 receptor-mediated, parallel assays were conducted in the presence of the cannabinoid receptor antagonist SR141716A (10–20 μM) (Rinaldi-Carmona et al., 1994). In several instances a component of the concentration response curve was found to be insensitive to the actions of SR141716A. In these cases, only the SR141716A sensitive component was taken to be CB1 receptor mediated.
Materials.
Anandamide (arachindonylethanolamide) and WIN-55212–2 (R(+)-[2,3-dihydro-5-methyl-3-[(morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazin-yl]-(1-napthalenyl)methanone mesylate) were obtained from Research Biochemicals International (Natick, MA). HU-210 ((−)-11-hydroxy-Δ8-tetrahydrocannabinol-dimethylheptyl) and SR141617A (N-(piperidino-1-yl)-5-(4-chlorphenyl)-1-(2,4-dichlorophenyl)-4-methylpyrazole-3–3-carboxamide, hydrochloride) were obtained from Tocris Cookson (Ballwin, MO). Δ9-Tetrahydrocannabinol (THC) was obtained from Sigma Chemical (St. Louis, MO). CP-55,940 ([1α,2β-(R)-5α]-(-)-5-(1,1-demethylheptyl)-2-[5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]-phenol) was synthesized in the Department of Medicinal Chemistry, Roche Bioscience (Palo Alto, CA). [3H]CP-55,940 (165 Ci/mmol) was purchased from NEN Life Sciences (Boston, MA). Forskolin, pertussis toxin and other chemical reagents were obtained from Sigma Chemical. Tissue culture medium was obtained from GIBCO BRL Life Technologies (Gaithersburg, MD).
Results
In CHO cells expressing hCB1 receptors, CB receptor agonists concentration-dependently inhibited forskolin-stimulated cAMP accumulation (fig.1A). The rank order of potency of the CB1 receptor agonists was the same as the rank order of their affinities as determined in binding studies (HU-210 > CP-55,940 > THC > WIN-55212–2 > anandamide). THC was a partial agonist in this assay, inhibiting 47% of the forskolin-stimulated cAMP accumulation, whereas WIN-55212–2 and CP-55940 were virtually full agonists (table1).

A, Inhibition of forskolin-stimulated (1 μM) cAMP accumulation by CB receptor agonists in CHO cells expressing hCB1 receptors. B, Enhancement of forskolin-stimulated (1 μM) cAMP accumulation by CB receptor agonists in CHO cells pretreated overnight with pertussis toxin. For each condition, data have been pooled from at least six different concentration-response curves. ○, HU-210; ■, CP-55,944; ▪, THC; ▵, WIN-55212–2; ♦, anandamide.
Effect of CB receptor agonists on forskolin-stimulated cAMP accumulation in CHO cells expressing the hCB1 cannabinoid receptor
Conversely, in the presence of forskolin, in cells pretreated with pertussis toxin, CB receptor agonists concentration-dependently stimulated cAMP accumulation (fig. 1B). The potencies of agonists in stimulating cAMP accumulation were 5- to 10-fold less than they were in inhibiting its formation. However, the rank order of potency of CB receptor agonists in the two assays was identical (fig.2A). Relative to WIN-55212–2, anandamide, HU-210, CP-55,940 and THC were partial agonists (table 1). Thus, while THC and WIN-55212–2 had similar activities in both assays, anandamide and CP-55,940 were less efficacious in stimulating the accumulation of cAMP as compared with inhibiting its formation (table1). Differences in relative intrinsic activities of agonists in the two assays were shown by the absence of a statistically significant correlation in intrinsic activity values (fig. 2B).
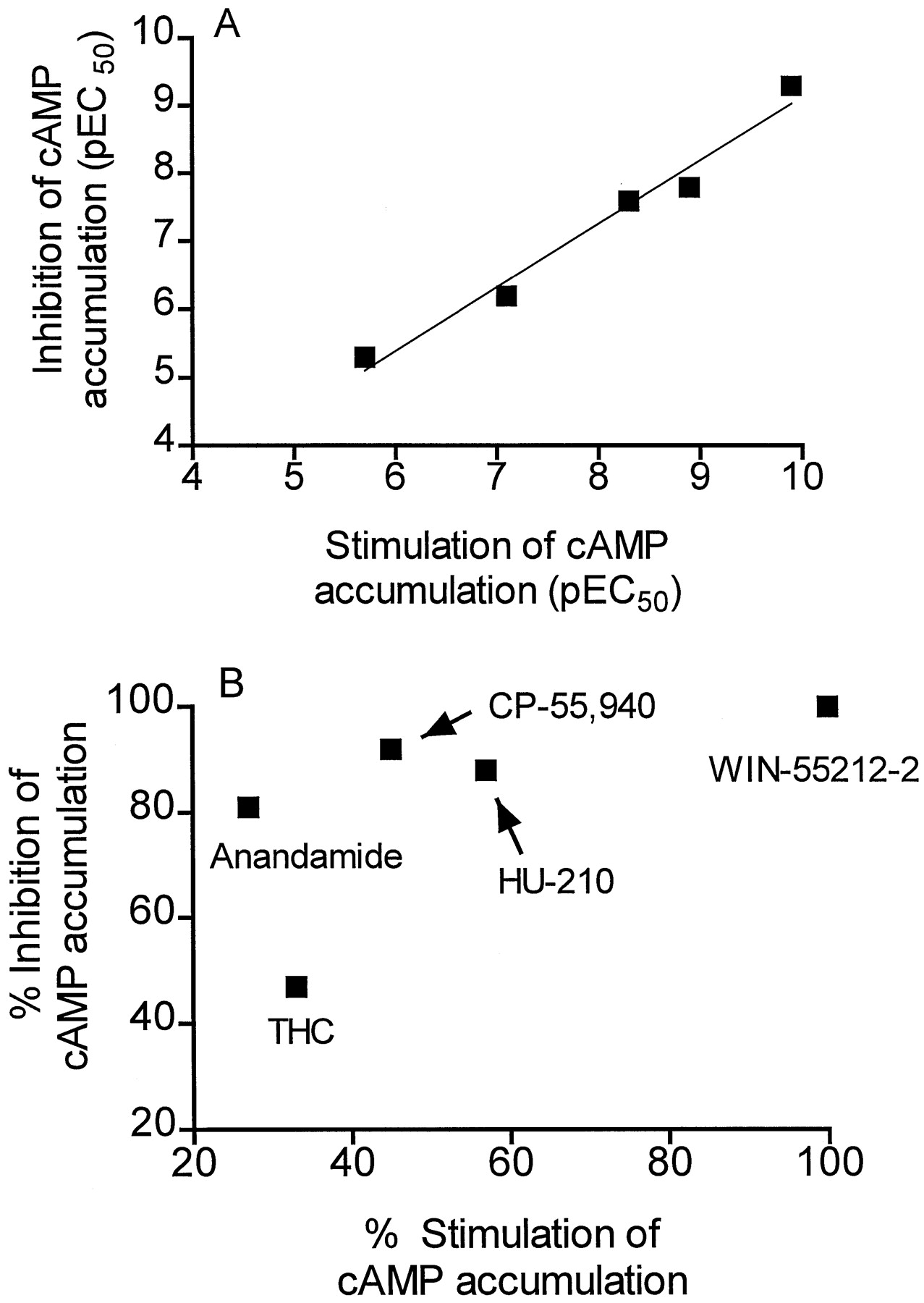
A, Correlation in the potencies of CB1receptor agonists in stimulating or inhibiting forskolin-stimulated (1 μM) cAMP accumulation (R2 = .97, P < .002). B, Absence of correlation (P = .2) in the intrinsic activities of CB1 receptor agonists in stimulating or inhibiting cAMP accumulation. Values are normalized to the maximum response produced by WIN-55212–2 and (in the case of anandamide) reflect only the SR-141716A-sensitive component of the response.
A stimulatory effect of CB1 receptor agonists on cAMP accumulation was also detected in the absence of forskolin. The potencies of most CB1 receptor agonists, including HU-210 and CP55,940, were 50- to 100-fold lower in the absence of forskolin than in its presence (table2). However, by contrast, the potency of WIN-55212–2 was not modified by forskolin (fig.3).
Effect of CB1 receptor agonists on basal cAMP accumulation in CHO cells expressing the hCB1 cannabinoid receptor

Concentration-response curves for HU-210 (A) and WIN-55212–2 (B) in CHO cells expressing the hCB1 receptor in the presence (circles) or absence (squares) of 1 μM forskolin. For each condition, data have been pooled from at least six different concentration-response curves. Note that to facilitate comparison of these curves, the maximum response within each curve was normalized to 100%.
The effects of the cannabinoid receptor antagonist SR141716A on cAMP accumulation were examined. SR141716A, at concentrations up to 20 μM, had no stimulatory or inhibitory effect on cAMP accumulation, either in the presence or absence of forskolin (data not shown). However, SR141716A (10–20 μM) blocked both the inhibitory and stimulatory effects of HU-210, CP-55,940 and WIN-55212–2 on forskolin-stimulated cAMP accumulation (fig.4). SR141716A also blocked the effects of HU-210, CP-55,940 and WIN-55212–2 on basal cAMP accumulation (measured in the absence of forskolin, data not shown). The potency of SR-141716A was consistent with a specific effect at the CB1receptor (pKB values for SR141716A blockade of HU-210, CP-55,940 and WIN-55212–2 inhibition of forskolin stimulated cAMP accumulation were 8.1 ± 0.1, 8.1 ± 0.02 and 8.3 ± 0.2, respectively). However, in contrast to the complete block of the effects of HU-210, CP-55,940 and WIN-55212–2, the stimulatory effect of anandamide on cAMP accumulation was only partially blocked by SR141716A, with ∼80% of the response being insensitive to SR141716A (fig. 4). A similar SR141716A-insensitive stimulatory effect of anandamide on cAMP accumulation was also detected in untransfected CHO cells (data not shown) and thus was attributed to a non-CB1 receptor-mediated mechanism. No SR141716A-sensitive stimulatory or inhibitory effects of cannabinoids on cAMP accumulation were detected in untransfected CHO cells.

A–E. Effects of CB1 receptor agonists on forskolin-stimulated (1 μM) cAMP accumulation in the absence (closed symbols) or presence (open symbols) of SR141716A. Data depicted by circles were generated in cells pretreated overnight with pertussis toxin. Data have been pooled from at least six different concentration-response curves. F, The SR-141716A-sensitive component of anandamide evoked stimulation of cAMP accumulation (♦) was obtained by subtraction of the SR-141716A-insensitive component.
Discussion
In CHO cells expressing the hCB1 receptor, CB receptor agonists concentration-dependently inhibited forskolin-stimulated cAMP accumulation by an SR141716A-sensitive mechanism. This inhibitory effect was not detected in cells pretreated with pertussis toxin, nor was it detected in untransfected cells. Conversely, when cells expressing the hCB1receptor were pretreated with pertussis toxin, an SR141716A-sensitive, stimulatory effect of CB receptor agonists on cAMP accumulation was revealed. Because the stimulatory effects of CB receptor agonists were (for all agonists except anandamide) fully reversed by SR141716A and were not detected in untransfected cells, they were not the consequence of a nonspecific action of the agonists. Moreover, because these stimulatory effects were detected in cells pretreated with pertussis toxin they were not mediated via activation the Gi/o pathway, as has been proposed for similar phenomena involving adrenergic or opioid receptors (Avidor-Reisset al., 1997; Federman et al., 1992). Thus these finding confirm the ability of CB1 receptors to functionally couple, in the same cell system, to both Gs and Gi protein-linked transduction pathways (Felder et al., 1998; Glass and Felder, 1997; Howlett, 1985; Maneuf and Brotchie, 1997).
The rank orders of potencies of agonists in stimulating or inhibiting forskolin-stimulated cAMP accumulation were identical. However, there were marked differences among cannabinoid receptor agonists in their intrinsic activities in the two assays. Thus, CP-55,940 demonstrated only 45% of the activity of WIN-55212–2 in the Gs-linked assay but 92% of WIN-55212–2’s activity in the Gi-linked assay. Similarly, anandamide demonstrated only 27% the activity of WIN-55212–2 in the Gs assay but 81% the activity of WIN-55212–2 in the Gi assay. Because these assays were conducted with cells from the same passage, differences in receptor density cannot account for the differences in intrinsic activity. Thus, these findings indicate that there is specificity among CB1 receptor agonists in their relative abilities to activate Gs- and Gi-coupled transduction pathways.
The mechanism underlying the different relative intrinsic activities of CB1 receptor agonists is not clear. One possibility could have been that the agonists had different affinities for Gs- and Gi-coupled CB1 receptors. However, if this were the case, it would have been expected that differences in potency as well as activity would have been observed (Kenakin 1997). Moreover, also contrary to the data, it might also have been expected that agonists with the greatest potency and activity in the Gi-coupled pathway would have had the lowest potency or activity in the Gs-coupled pathway. Thus, the direct linear correlation in potencies of CB receptor agonists for the Gs- and Gi-coupled responses suggests that more complex mechanisms are responsible for the differences in relative intrinsic activities.
An additional level of complexity in the actions of CB receptor agonists was revealed by studies comparing CB receptor-mediated stimulation adenylyl cyclase in the absence or presence of forskolin. Forskolin, acting directly on the cyclase, can synergistically enhance the action of the Gsalpha subunit in activating adenylyl cyclase (Sutkowski et al., 1994). Consistent with this synergistic interaction, HU-210 and CP-55,940 were 50- to 100-fold more potent in stimulating the formation of cAMP in the presence of forskolin than in its absence. However by striking contrast, forskolin had no effect on the potency of WIN-55212–2 or anandamide (and enhanced the potency of THC only 10-fold). Because the stimulatory effects of HU-210, CP55,940 and WIN-55212–2 on both basal and forskolin stimulated cAMP accumulation were fully blocked by SR141716A and because these compounds had no effect on cAMP accumulation in untransfected cells, the differences among agonists cannot easily be ascribed to nonspecific actions on the cyclase. One explanation may be that WIN-55212–2 predominately activated isoforms of the cyclase, which do not show large synergistic interactions between the Gs protein and forskolin (e.g., type I adenylyl cyclase), whereas HU-210 and CP-55,940 may have predominately activated isoforms of the cyclase that show a large synergistic interaction (e.g., type II adenylyl cyclase) (Pieroni et al., 1993; Sunaharaet al., 1996; Sutkowski et al., 1994). However, because the specific isoforms of adenylyl cyclase that are expressed in these cells are unknown, this idea remains entirely speculative. Nevertheless, it is intriguing to note that WIN-55212–2 binds to the CB1 receptor in a manner different from CP-55,940 and HU-210 (Song and Bonner, 1996), and this is at least consistent with the possibility that WIN-55212–2 stabilized different activated conformations of the CB1 receptor than did CP-55,940 or HU-210 and thus activated different sets of intracellular processes.
In summary, these findings confirm that recombinant hCB1 receptors in CHO cells couple both positively and negatively to adenylyl cyclase, extend previous studies by demonstrating differences among agonists in their relative intrinsic activities in Gs and Gicoupled pathways and have revealed intriguing differences among CB receptor agonists in their receptor-mediated activation of adenylyl cyclase(s). Whether these differences among agonists in their profile of intracellular signal transduction are biologically relevant remains to be determined. The comparisons of intrinsic activity in the Gs- and Gi-coupled pathways were made in the presence of forskolin. Intrinsic activities may be different in more physiological settings and may also be subject to numerous additional modulating influences. Nevertheless, these findings, together with the demonstration of dual coupling of native CB1 receptors (Glass and Felder, 1997) and the finding of pharmacological differences among the adenylyl cyclases activated by endogenous CB1 receptors (Pachecoet al., 1994), strengthen the possibility that specificity in intracellular trafficking by different CB1receptor agonists may confer different behavioral effects. This in turn provides a rationale for developing CB1 receptor agonists with increased selectivity for specific intracellular transduction pathways as potential therapeutic agents with diminished adverse effects.
Footnotes
-
Send reprint requests to: Douglas W. Bonhaus, Ph.D., Department of Molecular Pharmacology, Roche Bioscience, Neurobiology Unit, 3401 Hillview Avenue, Building R2–101, Palo Alto, CA 94304. E-mail: Doug.Bonhaus{at}roche.com
- Abbreviations:
- cAMP
- cyclic AMP
- CB
- cannabinoid
- CP-55
- 940, [1α,2β-(R)-5α]-(−)-5 (1,1-dimethylheptyl-2-[5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]-phenol
- HBSS
- Hanks’ balanced salt solution
- HEPES
- 4-(2-hydroxyethyl)-1-piperaineethanesulfonic acid
- HU-210
- (−)-11-hydroxy-Δ8-tetrahydrocannabinol-dimethylheptyl
- SR141617A
- N-(piperidino-1-yl)-5-(4-chlorphenyl)-1-(2,4-dichlorophenyl)-4-methylpyrazole-3–3-carboxamide hydrochloride
- THC
- Δ9-tetrahydrocannabinol
- WIN-55212–2
- R(+)-[2,3-dihydro-5-methyl-3-[(morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazin-yl]-(1-napthalenyl)methanone mesylate
- Received March 30, 1998.
- Accepted July 13, 1998.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}