


Abstract
From the demonstration of the existence of multiple opioid receptors and the isolation of the endogenous opioid peptides in the brain, it is now clear that the activities of these receptors can be regulated at various levels. The distinct brain regional distribution of the receptor suggests a tight transcriptional regulation. Early findings of alterations in receptor binding associated with tolerance to the opioids implies that the receptor life cycle can be influenced by the presence of agonists. Until the recent reported cloning of opioid receptors, the detailed studies of the molecular mechanisms involved in their regulation could not be conducted. With the availability of the cDNA clones of the μ-, δ- and κ-opioid receptors, and the elucidation of their gene structures, it is now possible to investigate opioid receptor regulation at various levels, and to identify the specific receptors involved in the pharmacological actions of the opioids. It is now also possible to define the receptor domains responsible for the opioid ligand selectivities, agonist activation, and agonist-induced inactivation. Summarized in this report are our past efforts in defining the regulation of opioid receptor activities. Studies using heterologous expression techniques, mutational analysis of receptors to characterize transcriptional elements, and the in vivo manipulation of the receptor gene levels have made it is possible to determine the mechanisms whereby these receptors are regulated. Our studies have also identified the unique characteristics of opioid receptors as members of the superfamily of G protein-coupled receptors.
A Sincere Thanks
First of all, I feel deeply honored to receive the Otto Krayer Award and I thank ASPET, the award selection committee, and Zeneca Pharmaceuticals for the privilege of delivering this lecture. I also want to take this opportunity to thank my mentor, Professor Edward Leong Way, for getting me “hooked on morphine” for the past three decades. The enduring total support of my wife and children is an absolute essential component for our work. I also want to thank my long-term collaborator Professor Ping-Yee Law for the many contributions he has made in our laboratory. He was the chief architect for many parts of the work I will present in this talk. Of course, we must not forget that any scientific effort requires the contribution of many individuals and I am very grateful to the many outstanding and dedicated students, postdoctoral fellows, and the entire research staff with whom I was so lucky to have the privilege to work.
During the past three decades our laboratory has focused solely on the pharmacology of opioids, with specific emphasis on the neurochemical mechanism of opioid tolerance. For the presentation today, I’ll concentrate on a very limited aspect of our recent work on the regulation of opioid receptor activities.
Introduction
The independent reports on the identification of opioid receptors in 1973 by S. Snyder, E. Simon, L. Terenius, and their coworkers (Pert and Snyder, 1973; Simon et al., 1973; Terenius, 1973) marked the advent of the opioid receptor field. Using radioactive ligand binding techniques, these groups demonstrated the presence of stereoselective binding sites for opioid ligands at the synaptic plasma membrane, with ligand affinities for the binding sites paralleling their pharmacological potencies (Creese and Snyder, 1975). Brain regional localization studies using autoradiography or membranes prepared from individual brain areas indicated that opioid binding is localized to sites known to be associated with the various pharmacological properties of these drugs. Due to the parallels in receptor distribution and the sites of drug actions, it was hypothesized that the change in receptor densities and activity could be responsible for the development of tolerance during chronic exposure to morphine. However, early reports on this subject were equivocal (Klee and Streaty, 1974; Hollt et al., 1975; Rothman et al., 1986; Werling et al., 1989; Brady et al., 1989; Bhargava and Gulati, 1990). There were no data pertaining to receptor activity following chronic drug administration in these early studies.
The presence of a stereoselective binding site for plant alkaloids in neural tissues suggested that endogenous ligands must exist in animal tissues. The family of opioid peptides was established with the first isolation of enkephalin by John Hughes and his coworkers in 1975 (Hughes et al., 1975), the identification of β-endorphin by C. H. Li and his colleagues (Li and Chung, 1976), and the subsequent isolation of dynorphin by A. Goldstein and his associates (Goldstein et al., 1979). The dissimilarities among the opioid peptides for the opioid binding sites supported the existence of multiple opioid receptors, as proposed by Martin et al. (1976). Indeed, μ-, δ-, and κ-opioid receptors were ultimately characterized using selective radioactive ligands (Chang and Cuatrecasas, 1979a; Chang et al., 1979b), selective ligand alkylation protection studies (Robson and Kosterlitz, 1979; James et al., 1982), and cross-tolerance studies (Schultz et al., 1980; Porreca et al., 1982). These different opioid receptors have distinct brain regional distributions and dissimilar pharmacological properties, with the most dramatic differences being between μ- and κ-opioid receptors. For example, morphine, a μ-opioid agonist, increases vasopressin release and thereby inhibits diuresis, whereas a κ-opioid agonist, such as U50,488, decreases vasopressin release, increasing diuresis (Lelander, 1983). Thus, the differential regulation of opioid receptor activities subsequently affects the pharmacological action of a given opioid agonist.
Studies on the cellular regulation of opioid receptor activities are facilitated by the availability of clonal cell lines that express a homogeneous population of δ-opioid receptors or a mixed population of μ- and δ-opioid sites. In the early 1980s our laboratory and others demonstrated that chronic exposure of these clonal cell lines, e.g., neuroblastoma × glioma NG108-15 hybrid cells or neuroblastoma N4TG2 cells, to δ-opioid agonists revealed different types of adaptation processes (Law et al., 1982, 1983b; Chang et al., 1982). These included a decrease in the ability of agonist to regulate adenylyl cyclase activity (receptor desensitization), a decrease in the overall receptor density (receptor down-regulation), and an increase in adenylyl cyclase activity after removal of the agonist (up-regulation of the adenylyl cyclase). The down-regulation of the receptor was found to involve its internalization from the plasma membrane to the lyzosomal compartments in these clonal cells (Law et al., 1983a). Although we could subsequently demonstrate agonist-dependent down-regulation of opioid receptors in the rodent brain (Tao et al., 1987, 1988, 1990), regulation of receptor levels during the chronic administration of an opioid could not account for tolerance to the drug. Thus, the actual molecular events involved in regulating opioid receptor activities could not be elucidated, in part because of the unavailability of receptor reagents that could monitor the effects of covalent modifications of receptor protein or alterations in gene transcription.
These hurdles were overcome by the cloning of δ-opioid receptors from NG108-15 cells by Evans et al. (1992) and Kieffer et al. (1992). Subsequent cloning of μ- and κ-opioid receptors was accomplished by others based on the reported sequence of the δ-opioid receptor (Chen et al., 1993b; Fukuda et al., 1993; Li et al., 1993; Meng et al., 1993;Yasuda et al., 1993). Sequence analysis of these cloned opioid receptors demonstrated unequivocally that they belong to the superfamily of G protein-coupled receptors (GPCR) and the subfamily of rhodopsin receptors. Thus, the μ-, δ-, and κ-opioid receptors all have the putative structure of seven transmembrane domains (TMs), an extracellular N-terminus with multiple glycosylation sites, a third intracellular loop (IL-3) with multiple amphiphatic α-helixes, and an IL-4 formed by the putative palmitoylation sites at the carboxyl terminus (Evans et al., 1992; Kieffer et al., 1992; Chen et al., 1993b;Fukuda et al., 1993; Li et al., 1993; Meng et al., 1993; Yasuda et al., 1993). On the whole, these receptors are about 60% identical to each other, with the greatest homology found in the TMs (73–76%) and ILs (86–100%). The greatest divergence in amino acid sequence is found in the N-terminus (9–10%), extracellular loops (EL; 14–72%), and the C-termini (14–20%) (Chen et al., 1993a). These opioid receptors are capable of regulating the same second messengers, with activation of the cloned μ-, δ-, and κ-opioid receptors causing inhibition of adenylyl cyclase activity (Evans et al., 1992; Kieffer et al., 1992;Chen et al., 1993b; Fukuda et al., 1993; Li et al., 1993; Meng et al., 1993; Yasuda et al., 1993) and N-type (Tallent et al., 1994) and L-type (Piros et al., 1996) Ca2+ channels. Activation of these opioid receptors also increases phospholipase C activity, and causes a transient increase in the levels of intracellular Ca2+ (Johnson et al., 1994; Spencer et al., 1997), in the activation of inwardly rectifying K+ channels (Henry et al., 1995), and the mitogen-activated protein kinases Erk-1 and Erk-2 (Fukuda et al., 1996;Li and Chang, 1996).
Differences in μ- and δ-Opioid Receptor Signal Transduction
Given the similarities among the opioid receptor primary structures and their effectors, what is the reason for the existence of multiple opioid receptors with differing selectivity for various opioid ligands? To examine this issue, we decided to concentrate on the μ- and δ-opioid receptors because Met-enkephalin, an endogenous ligand, has equal affinity for these two sites, and the selective activation of these receptors results in dramatically different pharmacological responses. Because the two receptors activate the same effectors, the reason for Met-enkephalin having equal affinity for these sites is not readily apparent. When we examined the relative affinities of several ligands for μ- and δ-opioid receptors stably expressed in Chinese hamster ovary (CHO) cells, the prototypic μ-opioid ligands, such as [d-Ala2,N-MePhe4,Gly-ol5] (DAMGO) or PL017, displayed high affinity for the μ-opioid receptor, whereas the prototypic δ-opioid ligands, such as [d-Pen2,d-Pen5]-enkephalin (DPDPE) or deltorphin II, displayed high affinity for the δ-opioid receptor, although DPDPE also has a high affinity for the μ-opioid receptor as well. The ligands clearly having specificity for the expressed δ-opioid receptor are the agonist deltorphin II and the antagonist Tippψ [H-Tyr-Tic[ψ,CH2NH]Phe-Phe-OH(Tic = 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid)], whereas the ligands having specificity for the μ-opioid receptor are the agonist PL017 and the antagonist CTOP (d-Phe-Cys-Try-d-Trp-Orn-Thr-Pen-Thr-NH2) (Table 1). These findings helped define the differences within the opioid receptor structures involved in ligand recognition. In addition to differences in ligand selectivity, differences were found in the μ- and δ-opioid receptor activities. Thus, the ratios of the agonist IC50 values, or the antagonist K e values, versus theK i values for the δ-opioid receptor were all ≤1 (Table 1). This suggested that the inhibition of adenylyl cyclase activity by the δ-opioid agonist required ≤10% of the receptor occupancy, implying the presence of spare receptors. On the other hand, because the ratios of IC50, orK e values, and theK i for the μ-opioid receptor were ≥1, it appears the μ-opioid receptor inhibition of adenylyl cyclase in CHO cells requires full receptor occupancy. This difference between receptors was not due to differences in the expression levels of the δ- and μ-opioid receptor, or the amount of G proteins in the CHO cells. Rather, other components of the signal transduction cascades must be responsible for the differences in μ- and δ-opioid receptor activities.
Relative affinities and potencies of various μ- and δ-opioid receptor agonists for DOR-1 and MOR-1 stably expressed in CHO cells
Differential Regulation of μ- and δ-Opioid Receptor Signal Cascades at Level of Ligand-Receptor Interaction
One possible component in the receptor signal cascade that could be responsible for these differences is the ligand-receptor interaction site, because the differential efficiency of G protein activation could be achieved with the formation of different ligand-receptor complexes. To define the conformation of the agonist-receptor complexes, the receptor domains involved in selective ligand binding and the requirements for receptor activation must be defined. Receptor chimera studies followed by mutational analysis has revealed that the TM6 (Fukuda et al., 1995), and the EL-3 (Pepin et al., 1997), are critical for the selective, high-affinity binding of δ-opioid ligand. As for the μ-opioid receptor, there is some dispute about the domains involved in the selective recognition of DAMGO. Thus, some studies indicated that the first EL-1 (Minami et al., 1995) is involved in DAMGO recognition, whereas μ/κ chimeras studies indicated that the EL-3 was critical for DAMGO binding (Xue et al., 1995; Wang et al., 1995). When we conducted studies with the μ/δ chimeras and examined receptor affinities and the abilities of the ligands shown on Table 1to regulate adenylyl cyclase, we found that not only are different domains of the μ- and δ-opioid receptors involved in the selectivity of the ligands, but also that agonists induce different receptor conformations.
We have constructed 17 μ/δ receptor chimeras to investigate the domains involved in μ- and δ-opioid ligand selectivity and have examined the relative affinities of various agonists and antagonists, both alkaloids and peptides, for these receptor chimeras (SchemeFS1). In addition, we examined the ability of agonists to inhibit forskolin-stimulated intracellular [3H]cAMP production and determinedK e values of antagonist to reverse etorphine inhibition. Etorphine was chosen as the agonist in these studies because it displays minimal selectivity for opioid receptor subtypes. The results indicate that the progressive substitution of the δ-opioid receptor TM domains with the corresponding μ-opioid receptor TM domains resulted in the reduction of DPDPE, deltorphin II, NTB, and TIPPψ affinities (Table2). For the most part, the affinities of these ligands for the chimeras were similar to those for the δ-opioid receptor, provided that TM6 and the EL-3 was present in the chimeras. However, in the μ1δ chimera, where only the sequence of the N-terminus to the beginning of the IL-1 of the δ-opioid receptor is replaced with the complementary sequence of the μ-opioid receptor, significant reductions in the δ-opioid agonist affinities were observed (Table 2). Reverse substitution of the same sequence in the μ-opioid receptor with that of the δ-opioid receptor results in a receptor chimera, δ1μ, capable of binding the δ-opioid selective ligands with high affinity, the most dramatic of which is for TIPPψ. Thus, in this case, the δ1μ chimera exhibits nanomolar affinity for TIPPψ, whereas the wild-type μ-opioid receptor has greater than micromolar affinity for this peptide. Such data suggest the involvement of TM1 in the selectivity of δ-opioid receptor ligands. If this is the case, then substitution of this δ-opioid receptor sequence to the μ-opioid receptor should also reduce the affinity for μ-opioid receptor-selective ligands. Indeed, this appears to be case with DAMGO, PL017, oxymorphone, and CTOP affinities being reduced in the δ1μ chimera (Table 2). The only exception to this is naloxone, a ligand that does not distinguish between μ- and δ-opioid receptors. These data suggest that TM1 of the δ-opioid receptor is involved in the selective binding of δ-opioid ligands. If so, what then is the role of EL-3 and TM6 in the binding of δ-opioid ligands? Because the binding pocket of the receptor is formed by the spatial orientation of various amino acids in different TMs, the binding of receptor-selective ligands could be affected significantly by the interactions among various TMs. The relative interaction of the TM can be studied by examining the ability of the chimera δ5μ to bind δ-opioid receptor-selective ligands with high affinity. After splicing of only the EL-2 and TM5 of the δ-opioid receptor into the μ-opioid receptor, the δ5μ chimera exhibited affinities for deltorphin or TIPPψ in the 10−8 M range (greater than micromolar) instead of having very low affinity for these ligands (Table 2). Because the other receptor chimera constructs did not reveal the critical involvement of EL-2 or TM5 in δ-opioid binding, as demonstrated by the relative affinities of these ligands for the chimeras μ1–4δ, μ1–5δ, δ1–4μ, and δ1–5μ, one explanation for the high-affinity binding of the δ-opioid ligands in the δ1μ or δ5μ chimeras is the possible destabilization of μ-opioid receptor structure with the introduction of δ-opioid receptor TM domains.

Schematic representation of opioid receptor chimeras constructed from rat MOR-1 and mouse DOR-1.
Relative affinities (K i) of opioid receptor selective agonists and antagonists in CHO cells expressing DOR-1, MOR-1, or various opioid receptor chimers
Similar conclusions can be drawn from comparing the relative affinities of the μ/δ chimeras for the μ-opioid receptor-selective ligands. The influence of the TM interactions on the μ-opioid ligand binding is demonstrated by the high-affinity binding of DAMGO, PL017, naloxone, and CTOP to the δ456μ but not to the μ1–3δ receptor chimeras (Table 2). The difference between these two chimeras is that δ456μ has the TM7 sequence of the μ-opioid receptor, whereas μ1–3δ does not. However, introduction of the TM6 into the δ456μ receptor chimera reduces the affinities of these ligands for the receptor. These and other data demonstrate that the relative spatial orientation of the amino acids within the TM affect the affinities of selective ligands. As for nonselective ligands such as etorphine, the substitution of the δ-opioid receptor sequence into the μ-opioid receptor, or vice versa, does not alter their affinities.
Opioid alkaloids and opioid peptides have overlapping but distinct binding domains. This is demonstrated most dramatically by comparing the relative affinities of naloxone and CTOP for the μ/δ receptor chimeras. Thus, the presence of only the TM7 of μ-opioid receptor in the δ1–6μ chimera is sufficient for high-affinity binding of naloxone (Table 2), whereas CTOP binds with low affinity to this site (greater than micromolar). Introduction of the δ-opioid receptor sequence other than the TM1 greatly reduced the affinities of the receptor chimeras for CTOP, but not for naloxone, suggesting that these two opioid antagonists have distinct binding sites within the μ-opioid receptor. In addition, there appear to be differences among the selective agonists’ binding sites within the μ-opioid receptor. The introduction of the TM1–5 sequence of the μ-opioid receptor to the δ-opioid receptor, yielding the μ1–5δ chimera, results in DAMGO and oxymorphone, but not PL017 binding to the chimera with affinities similar to those found with the wild-type μ-opioid receptor (Table2). In the reverse receptor chimera, δ1–5μ, DAMGO and oxymorphone also display high-affinity binding, whereas PL017 exhibits minimal affinity. These data suggest that although PL017 is a μ-opioid receptor agonist, its receptor recognition site is distinct from that of the other two agonists tested.
Differences between agonist binding sites and subsequent receptor conformation is demonstrated further by one of our recent studies on receptor phosphorylation. Agonist-induced receptor phosphorylation has been proposed to be a critical step in the cellular regulation of GPCRs. The phosphorylation of the opioid receptor is reported to involve a G protein-coupled receptor kinase (GRK), but not protein kinase A (PKA; Chen and Yu, 1994; Pei et al., 1995). However, the primary sequence analysis of the opioid receptor predicts consensus sequences for the putative phosphorylation by PKA and protein kinase C (PKC). When we examined the ability of PKA to phosphorylate the μ-opioid receptor, we observed that, in the presence of morphine, PKA phosphorylates the receptor in a morphine concentration- and time-dependent manner. Moreover, this phosphorylation is naloxone reversible and is blocked by pretreating the cells with pertussis toxin (PTX). The in vitro phosphorylation is also blocked by isolating the membrane from cells pretreated with morphine in the presence of forskolin and 3-isobutyl-1-methylxanthine (IBMX), a phosphodiesterase inhibitor. Because in vitro phosphorylation is blocked by the PKA-specific inhibitor KT5720, it appears the phosphorylation of the μ-opioid receptor is, in fact, mediated by this enzyme. Surprisingly, PKA-mediated phosphorylation of the receptor in vitro is not observed in the presence of another μ-ligand, DAMGO, or other enkephalins. Rather, only μ-opioid agonists that are alkaloids, or β-endorphin, stimulate the in vitro phosphorylation of the receptor. In addition to the putative PKA sites located intracellularly, there are consensus PKA sites on the extracellular portion of the receptor or at the TM. The differences between the ability of morphine and DAMGO to induce in vitro PKA phosphorylation suggest these extracellular or TM sites are accessible to PKA in the presence of opioid alkaloids but not opioid peptides. This difference could be responsible, in part, for the differentiation of the signals induced by the μ-specific opioid alkaloid and peptide ligands.
Differential Regulation of μ- and δ-Opioid Receptor at G Protein Level
Early receptor binding and functional measurements suggested that opioid receptors belong to the super family of GPCR, and the deduced primary sequences of the cloned receptors indicate unequivocally that this is the case. One common feature of these receptors is that their activities are abolished by pretreatment with PTX, suggesting that the G proteins involved in the transduction of the receptor signals are either of the Giα group or the Goα group. Both the α-subunit and the βγ dimers of these G proteins could regulate effectors, such as adenylyl cyclase, the Ca2+ and K+ channels. Studies with other GPCRs suggest that there is specificity with the Gi/Go proteins in their coupling to the receptor and transduction of the receptor signals. Hence, it is not surprising that in earlier reports opioid receptors appear to exhibit such selectivity. Using Gi/Go α-subunit-selective antibodies and the PTX pretreatment paradigm, it has been reported that δ-opioid receptors inhibit the adenylyl cyclase in NG108-15 cells via the Gi2 (McKenzie and Milligan, 1990), whereas receptor-mediated inhibition of voltage-dependent Ca2+ channels is due to Go(Hescheler et al., 1987). Studies with the human neuroblastoma SHSY5Y cells indicated that the μ-opioid receptor inhibition of adenylyl cyclase is mediated by Go proteins (Carter and Medzihradsky, 1993).
We addressed the question of receptor-G protein interaction by directly examining the activation of Gi/Go by opioid receptor. To this end, we took advantage of the fact that activation of GPCRs leads to the exchange of GDP for GTP and the subsequent dissociation of the heterotrimeric G proteins into α-subunits and βγ dimers. This dissociation allows for the ADP-ribosylation of the α-subunits of Gi/Go by cholera toxin (Milligan and McKenzie, 1988). Furthermore, in the presence of the photolabile GTP analog, azidoanilido-[32P]GTP, receptor agonist promotes the association of this analog, making it possible to identify the G protein that interacts with the receptor. Using these two approaches, we labeled the G protein α-subunits coupled to various opioid receptors and optimized the urea/SDS-polyacrylamide gel electrophoresis system to separate Gi/Go α-subunits in a single dimension. By conducting the experiments in NG108-15, NS20Y, and N1E115 cells expressing different levels of δ-opioid receptor, or in 10 CHO clones that stably express different levels of δ-opioid receptors, we found that the ability of the δ-opioid receptor to activate multiple Gi/Go is independent of receptor density. In the cell lines tested, δ-opioid receptor activation resulted in the labeling of four different G protein α-subunits, Gαi2, Gαi3, and Gα0A, and an unknown protein (Prather et al., 1994a,b). Because the conditions used for these labeling studies favored the binding of azidoanilido-[32P]GTP to Gi/Go and not to Gz (Field et al., 1994), activation of Gz and G16, as reported by others (Garzon et al., 1997; Lee et al., 1998) using clonal cell lines stably expressing the opioid receptors, were not detected in our studies.
In addition to being independent of receptor density, the ability of other opioid receptors, μ and κ, to activate the same four G proteins is observed in CHO cells stably expressing these receptor subtypes (Chakrabarti et al., 1995b; Prather et al., 1995). Furthermore, the potencies of the μ-, δ-, and κ-agonists to activate these Gi/Goproteins is very similar in the CHO cell lines. The notable exception is the Gα subunit that we could not identify with existing antibodies.
When the type and amount of G protein being activated is examined, the multiple opioid receptors appear to exhibit some preferences (Table3). Thus, the κ-opioid receptor displays no selectivity toward the various Gi/Go, whereas both μ- and δ-opioid receptors exhibit selectivity toward Gi2 and G02. The maximal levels of Gαi2 and Gαo2labeling induced by activation of these two receptors is significantly higher than that observed with Gαi3 labeling (Table 3). Because κ-opioid receptor-induced labeling of Gαi3 is equal to that of Gαi2 and Gαo2, the preferential labeling of Gαi2 and Gαo2 induced by μ- or δ-opioid receptors cannot be attributed to differences in G protein levels in the CHO cells. These findings do not suggest, therefore, that μ- and δ-opioid receptors are associated with Gαi2and Gαo2, whereas κ-opioid receptors are coupled to all Gi/Goproteins in CHO cells. Other studies suggest that the opioid receptor exhibits changes in the association with G protein upon agonist stimulation (Law and Reisine, 1997). Hence, regulation of opioid receptor activities does not appear to involve their ability to promote the association of GTP onto the G proteins and the subsequent dissociation of heterotrimers. Accordingly, the observed differences in the G proteins involved in the opioid receptor-mediated regulation of adenylyl cyclase and Ca2+ channels must lie elsewhere.
Relative labeling intensity of Gi/Go by azidoanilido-[α-32P]GTP in presence of activated opioid receptors
In recent experiments, we observed differences in the efficiency of the receptor regulating the voltage-dependent L-type Ca2+ channels and adenylyl cyclase activity. Thus, although we demonstrated μ-opioid receptor-mediated inhibition of L-type channels in GH3 pituitary cells stably expressing the receptor, when a similar level of δ-opioid receptor is stably expressed in another GH3 clone, regulation of the L-type channels was not observed (Piros et al., 1995). In contrast, in the same two GH3 clonal cell lines both μ- and δ-opioid agonists inhibit adenylyl cyclase activity. However, when the δ-opioid receptor is overexpressed in the GH3 cell line that already expresses the μ-opioid receptor, δ-opioid receptor-mediated inhibition of the L-type Ca2+ channels is observed (J. L. Adams, L. Song, E. T. Piros, T. G. Hales, P. Y. Law, and P. L. Prather, unpublished observations). The major difference between the GH3DOR cell line and the GH3 MORDOR cell line is that there is five times more δ-opioid receptor expressed in the latter. There is also a slight difference in the G proteins that these two opioid receptors activate, with the δ-opioid receptor inducing azidoanilido-[α-32P]GTP incorporation the most in Goα1, whereas μ-opioid receptor induced GTP incorporation is greatest in Goα2(J. L. Adams et al., unpublished observations). However, with the GH3 cell lines coexpressing both the μ- and δ-opioid receptors, the δ-opioid receptor-mediated inhibition of the L-type Ca2+ channels requires a critical concentration of receptor (>0.5 pmol/mg protein) as shown by covalently inactivating the δ-opioid receptor with SUPERFIT (J. L. Adams et al., unpublished observations). At the same time, the δ-opioid receptor-mediated inhibition of the adenylyl cyclase activity remains. At this density of μ-opioid receptor, both the L-type channels and adenylyl cyclase activities are regulated by μ-opioid agonists (Piros et al., 1995). Thus, the difference in the requirement of δ-opioid receptor density to regulate these two effectors suggests different effectors are involved. The difference between the μ- and δ-opioid receptor regulation of the L-type Ca2+ channels also indicates either that different G proteins participate or that there are different efficiencies between these two opioid receptors to activate the same G protein.
There now appears to be little question of differences between the μ- and δ-opioid receptor activation of the G protein complexes. In addition to the differences in the ratios of the potencies and the affinities of various agonists (Table 1) and the activation of the L-type Ca2+ channels, there is a pronounced difference between the μ- and δ-opioid receptor-G protein complexes. In an earlier study with NG108-15 cells, we reported that after PTX pretreatment the ability of the δ-opioid agonists to induce receptor internalization and down-regulation remains unaltered, suggesting that G proteins other than the PTX substrates, such as Gi/Go, are involved in these processes (Law et al., 1985a). When we conduct similar studies with neuroblastoma N2A cells stably expressing either the μ- or δ-opioid receptor, a differential response is observed with PTX pretreatment. Similar to the results obtained with the NG108-15 cells, the PTX pretreatment does not diminish the ability of DPDPE to induce down-regulation of the δ-opioid receptor in N2A cells. On the other hand, parallel treatment of the N2A cells expressing the μ-opioid receptor with PTX results in the total elimination of DAMGO-induced receptor internalization or down-regulation (Chakrabarti et al., 1997). In both cases, the ability of the agonist to regulate adenylyl cyclase and intracellular Ca2+ level is completely eliminated.
The possibility that the agonist activates other non-PTX substrates is eliminated by mutating the conserved aspartic acid (Asp) in the TM2 to alanine. Previous studies with other GPCR and the opioid receptors (Kong et al., 1993; Surratt et al., 1994) indicates this Asp is critical for agonist activation of the receptor. When the Asp95 in the δ-opioid receptor and the Asp114 in the μ-opioid receptor are mutated to Ala and the mutants stably expressed in the N2A, the ability of the μ- or δ-opioid agonist to inhibit adenylyl cyclase activity is not observed. As with the PTX experiments, the Asp95 δ-opioid receptor mutant is down-regulated by agonist pretreatment, whereas the corresponding μ-opioid receptor mutant is not. It was also found that δ-opioid receptor remains coupled to G proteins even after PTX pretreatment or after mutation of Asp95 to Ala (Chakrabarti et al., 1997). Under the same conditions, or the equivalent mutation, the μ-opioid receptor is completely uncoupled from the G proteins and remains in the low-affinity state (Chakrabarti et al., 1997). The existence of the δ-opioid receptor in the G protein-receptor complex after PTX pretreatment or Asp95 mutation is demonstrated by the further reduction of agonist affinity after agonist or PTX treatment, respectively. Thus, these studies not only suggest the need for high-affinity states or the receptor-G protein-coupled states for agonist-induced down-regulation of the receptor, but also that the interactions between the μ-opioid receptor and δ-opioid receptor and the G proteins are different. This is surprising in view of the homology in the amino acids sequences of the IL in these two receptors. Although the greatest differences between the two receptor sequences are within the carboxyl tail, exchange of this portion of the receptor does not confer δ-opioid receptor properties to the μ-opioid receptor. That is, the μ/δ receptor chimera with only the δ-opioid receptor carboxyl tail spliced onto the μ-opioid receptor exists in the low-affinity state after PTX pretreatment. Thus, the receptor sequence that regulates the interaction between G proteins and opioid receptors remains to be elucidated.
Molecular dynamic modeling of the GPCR suggests agonist binding causes movement of the TM domains (Luo et al., 1994). Modeling, followed by mutational analysis, indicates that the molecular motion of the TM6 and TM7 is critical for transducing agonist binding signals to the IL-3 or the juxtatransmembrane portion of the carboxyl tail, the two domains known to be responsible for activation of G proteins. Our mutational and receptor chimera studies provided evidence supporting the importance of agonist-induced movements of TM in opioid receptors, as exemplified by comparing relative efficacies of agonists (Table4). Thus, the efficacies of δ-opioid agonists are >1, whereas those of the μ-opioid agonists are <1. Although the affinities of the μ-opioid agonists for the μ1–5δ chimera are less than that for the wild-type μ-opioid receptor, the potencies of DAMGO, PL017, and oxymorphone are greater with the wild type and their efficacies in this chimera are >1. The most surprising finding is that whereas PL017 had minimal affinities for the receptor chimera, it is very potent in mediating inhibition of adenylyl cyclase activities. These findings suggest that the μ-opioid receptor sequences in this chimera allow for movement of the TM with minimal agonist-induced activation energy, indicating that the efficiency of opioid receptor activation is influenced by conformational changes in the receptor brought on by agonist binding. This point is substantiated further by studies with the μ1–5δ receptor chimera. With this chimera, the affinities of the δ-opioid receptor-selective ligands are not altered significantly (Table 2), although the potencies of DPDPE and deltorphin II are greatly reduced and their efficacies are <1. Similar data were obtained with this chimera and μ-opioid receptor-selective agonists (Table 4). This suggests that activation of this chimera is inefficient compared with the wild-type δ-opioid receptor because high-affinity binding of agonists fails to fully activate this site.
Relative potencies (IC50) and efficacies of opioid receptor selective agonists to inhibit forskolin-stimulated [3H]cAMP production or K e of various opioid antagonists to reverse agonist inhibition in CHO cells expressing DOR-1, MOR-1, or various opioid receptor chimeras
The agonist-induced molecular movement of the TM, or conformational changes that lead to receptor activation, are also found with one of the receptor mutations generated by Taq polymerase. The static receptor model posits that antagonists, unlike agonists, are unable to induce conformational changes in receptors and, therefore, display no efficacy. Overexpression of cloned GPCRs exhibits basal levels of receptor activity even in the absence of agonists, as evidenced by GTP binding studies and the discovery of inverse agonists (Costa and Herz, 1989). To accommodate such observations, the ternary complex model was revised to include isomerization of the receptor R to R* (Cotecchia et al., 1993), with R* representing “activated” receptor. In this case, the agonist binding favors the formation of R*, thus enhancing association with G proteins, whereas antagonist stabilizes the R state of the receptor, reducing its interaction with G proteins. This model suggests, therefore, there is only one receptor conformation capable of activating the system. However, such a model fails to account for results obtained with opioid receptor antagonists. Thus, the structural difference between naloxone, an opioid receptor antagonist, and the corresponding agonist, oxymorphone, is at the N17 position, with oxymorphine having methyl and naloxone an allyl substitution at this position. It has long been thought that the steric hindrance resulting from the bulky group at N17 converts an opioid agonist into the corresponding antagonist. It has also been generally assumed that opioid antagonists compete with agonists for the binding pocket and that the former could not induce a conformational change in the receptor due to the steric hindrance. However, using the μ1–2δ chimera, we found that the “classical” opioid antagonists display agonistic properties (Claude et al., 1996). In this case, opioid antagonists, such as naloxone or naltrexone, do not reverse or block the effect of DPDPE. Instead, these antagonists by themselves inhibited the forskolin-stimulated adenylyl cyclase activity in CHO cells stably expressing this receptor chimera, even though their affinities for the receptor remained unchanged. Moreover, the antagonists desensitized and down-regulated the chimera receptor. Thus, in all respects, these antagonists displayed full agonistic properties in this chimera. Following complete sequencing of the receptor chimera, we determined that the Taq polymerase used to generate the receptor fragments introduces a single point mutation at TM4, resulting in the conversion of Ser196 to Leu in the μ1–2δ chimera (Claude et al., 1996). Because this serine is conserved among all three cloned opioid receptors, the mutation of this serine in TM4 to leucine should generate the desired receptor phenotype, i.e., antagonist displaying agonist properties. When Ser196, Ser177, and Ser187 in μ-, δ-, and κ-opioid receptors, respectively, are mutated to Leu, naltrexone acts like an agonist in inhibiting adenylyl cyclase activity in CHO cells stably expressing receptor mutants. In addition, this antagonist inhibits the G protein-coupled voltage-dependent inward rectifying potassium channels (GIRK) activities in Xenopus oocytes when GIRK and the receptor mutants were coexpressed in this system. Because there are no observable changes in the mutant receptor affinities for agonists and antagonists, these data suggest that the binding of antagonists to these receptor mutants causes the receptor conformational change required of an agonist. Thus, the conversion of the Ser in the TM4 to a Leu must relieve constraints that prevent G protein activation by antagonists. Because the mutation results in the removal of Ser, an amino acid that has hydrogen bonding capacity, to Leu, an amino acid that has minimal activity in this regard, it appears that the normal constraint is due to the generation of a hydrogen bond. Similar reasoning can be used with respect to the ionic bond formed between the conserved Asp in TM2 with the Asn in TM7. In this case, mutation of the Asp to Asn in TM2 completely abolishes the ability of agonist to regulate G proteins, an effect that is reversed by the mutation of Asn in TM7 to Asp. Studies are underway to determine whether there is such a partner for the conserved Ser in TM4.
The importance of the conserved Ser in TM4 for the constraint within the receptor is demonstrated further with our studies of GIRK regulation by these mutants. In these experiments, we noticed that the efficacies of the antagonists in oocytes expressing both the GIRK and the μ1–2δ chimera paralleled those of the agonists. However, when only the conserved Ser in TM4 is mutated to Leu, the antagonists exhibit only partial agonist properties in oocytes expressing both the GIRK and the mutated δ- or μ-opioid receptor (Claude et al., 1996). Furthermore, when the conserved Ser in the receptor chimeras δ456μ or δ45μ (Scheme FS1) is mutated, the antagonists once again exhibit only partial agonist properties with respect to GIRK channel activity (P. A. Claude, L. J. Erickson-Herbrandson, H. H. Loh, and P. Y. Law, unpublished observations). As seen with the conversion of Ser to Leu in other mutant receptors, the agonist affinities for the receptor, and their potencies and efficacies for regulating both adenylyl cyclase and GIRK channels, are not altered in the δ456μ or δ45μ mutant receptor chimeras. Moreover, the full agonistic properties of the antagonists in the μ12δ chimera is due to the incompatibility of the TM1 and TM7 from two different opioid receptors and not to a decrease in the receptor affinity for G proteins. In contrast, the μ1–2δ receptor chimera appears to have higher affinities for G proteins because the stable GTP analogs, GTPγS or GppNHp, are less able to decrease agonist binding in these chimeras (P. A. Claude et al., unpublished observations). Thus, the interaction between the TM1 and TM7 of the opioid receptor appears to regulate the efficacies of these ligands. Because the amino acid sequence differences between the TM7 domains of the μ12δ chimera and δ456μ chimera involves only two amino acids, it seems probable that the interaction of these amino acids in TM7 with their partners in TM1 stabilizes the receptor structure, influencing the efficacy of opioids. The identities of these amino acids are currently being deduced.
Regulation of μ- and δ-Opioid Receptor Levels by Phosphorylation and Down-Regulation
Like other GPCRs, the activities of the μ- and δ-opioid receptors are attenuated after chronic agonist treatment. Studying δ-opioid receptors in NG108-15 cells, we found that receptor desensitization and down-regulation are associated with attenuation of opioid receptor activities (Chang et al., 1982; Law et al., 1982; Law et al., 1983a b). Reports on other GPCRs, such as the β2-adrenergic receptor, indicate that agonist-induced receptor phosphorylation is the cellular mechanism responsible for receptor desensitization and down-regulation (Krupnick and Benovic, 1998). The current model suggests that β-arrestin binds to the phosphorylated receptor, competing with G protein binding for the attachment site, thereby terminating receptor function. Because the subsequent internalization of the receptor is also arrestin-dependent, it can be stimulated by receptor phosphorylation. The internalized receptor is resensitized by dephosphorylation, after which it is recycled to the membrane. Although other cellular components, such as dynamin, may be involved in the receptor internalization process (Chu et al., 1997), the critical steps for receptor desensitization and internalization are its phosphorylation attachment to β-arrestin.
Several groups, including our own, have reported agonist-induced opioid receptor phosphorylation (Arden et al., 1995; Pei et al., 1995; Yu et al., 1997; El Kouhen et al., 1999), with some suggesting that phosphorylation correlates with agonist-induced receptor desensitization. By comparing the ability of agonists to regulate GIRK in oocytes and their ability to phosphorylate receptors in CHO cells, Wang and colleagues (Yu et al., 1997) suggested that DAMGO-induced receptor desensitization is caused by phosphorylation of the μ-opioid site. Using dominant negative mutants of the GRK, and the dominant negative mutant of arrestin, Pei and coworkers (Pei et al., 1995) found that the δ-opioid agonist-mediated receptor phosphorylation is blocked by the GRK mutant and that desensitization is blocked by the arrestin mutant. The probable involvement of receptor phosphorylation in receptor desensitization is partially supported by data obtained from studying mutated receptors. Thus, Pak et al. (1997) mutated the Thr393 of the μ-opioid receptor into Ala, blunting the DAMGO-induced receptor desensitization. Devi and coworkers (Trapaidze et al., 1996;Cvejic et al., 1996) were unable to block agonist-induced internalization and down-regulation of mutant opioid receptors by mutating Thr352 . Although direct measurements of receptor phosphorylation were made in these studies, the results provide evidence supporting the role of receptor phosphorylation in the regulation of opioid receptor activities.
Others have been unable to establish a casual relationship between receptor phosphorylation and desensitization. Kovoor et al. (1997)reported a very slow rate of DAMGO-induced desensitization of μ-opioid receptor-mediated regulation of GIRK channel activity and found that the rate is not enhanced by overexpression of GRKs. Likewise, when we compared the rate of μ-opioid receptor phosphorylation and the rate of DAMGO-induced receptor desensitization in two different cell lines, we found that agonist-induced receptor phosphorylation occurred within minutes, whereas the reduction in DAMGO-mediated regulation of adenylyl cyclase activities (i.e., desensitization) took hours (El Kouhen et al., 1999). Moreover, the rate of desensitization is not enhanced by inhibition of phosphatase activity using calyculin A, or by promoting phosphorylation either by overexpression of GRKs or stimulation of endogenous protein kinases, such as PKCs. It was also found that overexpression of β-arrestin does not increase DAMGO-induced receptor phosphorylation. In other studies with human embryonic kidney cells, HEK293, which express μ- and δ-opioid receptors, overexpression of GRK-2 and β-arrestin potentiate DPDPE-induced δ-opioid receptor desensitization, but not DAMGO-induced μ-opioid receptor desensitization, even though overexpression of GRK-2 increases both the μ- and δ-opioid receptor phosphorylation. These results suggest that μ-opioid receptor phosphorylation might not be an obligatory event for desensitization of this receptor.
This conclusion is reinforced by studies involving long-term exposure to morphine. Thus, it is established that chronic administration of morphine completely desensitizes μ-opioid receptors with respect to morphine or DAMGO (Chakrabarti et al., 1995a). However, numerous reports indicate that morphine does not induce receptor phosphorylation (Arden et al., 1995; Zhang et al., 1998), even though one group did find evidence to the contrary (Yu et al., 1997). Moreover, it is generally accepted that morphine cannot induce receptor internalization (Arden et al., 1995; Zhang et al., 1998), a cellular event thought to be closely coincided with receptor phosphorylation. If in fact morphine is unable to induce receptor phosphorylation, then morphine-induced receptor desensitization must involve some other mechanisms.
The ability to induce receptor desensitization in the absence of receptor phosphorylation was demonstrated further by us and Capeyrou et al. (1997) using μ-opioid receptor mutants in which all the putative phosphorylation sites were removed. Replacement of all Ser/Thr within the carboxyl tail, or Ser/Thr within the third intracellular and carboxyl tail (Capeyrou et al., 1997), completely abolished the ability of DAMGO to induce receptor phosphorylation. This suggests that DAMGO-induced receptor phosphorylation is limited to the Ser/Thr in the carboxyl tail. This conclusion is confirmed by analysis of the cyanogen bromide cleavage of the phosphorylated receptor. Interestingly, these receptor mutants could still be desensitized by chronic exposure to agonist. Thus, these experiments suggest that receptor phosphorylation is not a prerequisite for desensitization.
For GPCRs, the receptors in the endosomes are dephosphorylated, resensitized, and recycled, suggesting that the relatively long time needed for desensitization of the μ-opioid receptor is due to recycling. Koch et al. (1998), using monensin, which traps internalized receptor within endosomes, found that the rate of DAMGO-induced desensitization of the MOR1B, a splice variant of the μ-opioid receptor, increases. Similarly, we discovered that the rate of etorphine-induced μ-opioid receptor desensitization in HEK293 cells is increased by the pretreatment with monensin, and that the rate of disappearance of cell surface receptors, as monitored by fluorescence-activated cell-sorting analysis analysis, is also increased by monensin. These data suggest that internalized μ-opioid receptors are being recycled. Because monensin, which blocks the recycling process, increases the rate of desensitization, it appears the ability of agonist to regulate adenylyl cyclase activity depends on the concentration of μ-opioid receptors on the membrane. Pak et al. (1996) proposed that μ-opioid receptor desensitization correlates with the down-regulation of the receptor. In our own experiments, down-regulation of the μ-opioid receptor did not correlate with agonist-induced desensitization. Mutation to Ala of all the Ser/Thr residues in the carboxyl tail of the μ-opioid receptor blocks etorphine-induced receptor down-regulation. However, chronic exposure of the same mutant receptor to etorphine results in a loss of agonist activity. Fluorescence-activated cell-sorting analysis of the receptor cell surface suggests that removal of the putative phosphorylation sites on the carboxyl tail of the μ-opioid receptor does not prevent its internalization. Only under conditions in which internalization of the mutant receptor is blocked by overexpression of dominant negative arrestin, is DAMGO-induced desensitization of the mutant attenuated. However, with HEK293 cells transiently expressing the wild-type μ-opioid receptor, overexpression of dominant negative arrestin does not alter DAMGO-induced receptor desensitization if internalization is inhibited. These results suggest that receptor phosphorylation and the physical removal of the receptors from the cell surface contribute to agonist-induced desensitization of the μ-opioid receptor.
The inability to block etorphine-induced μ-opioid receptor internalization by mutating all of the Ser/Thr residues in the carboxyl tail suggests that phosphorylation of the receptor is not obligatory for this event. However, the same receptor mutation does prevent etorphine-induced receptor down-regulation, indicating that the receptor sorting and redirection of the receptor traffic to the lysosomes for degradation needs a different signal than that required for receptor internalization. Thus, we systematically truncated and mutated the carboxyl tail of the μ-opioid receptor to define receptor domains that might be involved in receptor trafficking. Truncating the receptor after the putative phosphorylation sites revealed that any receptor truncation after the Ser359 does not affect the etorphine-induced down-regulation (Burd et al., 1998). However, truncation after Ser355 blocks etorphine-induced down-regulation, but not internalization, of the receptor. Because the difference between these truncations is four amino acids with the sequence of STIE, this was deleted from the wild type, and the effect on etorphine-induced receptor down-regulation examined. To our surprise, etorphine induces receptor down-regulation of this deletion mutant (Burd et al., 1998). This suggests that either more than one motif is necessary to mediate etorphine-induced receptor down-regulation, or that this region does not play a critical role in this phenomenon. For example, in the rat neurotensin receptor, Thr422 and Tyr424 are critical for agonist-induced internalization of the receptor (Chabry et al., 1995). If, however, these amino acids are modified individually, there is little or no effect on receptor internalization. Thus, it is plausible that more than one motif is involved in the etorphine-induced μ-opioid receptor down-regulation, with one motif being within the STIE, whereas the other is downstream from the Ser359. After studying numerous combinations of mutations, we found that the combination of Ser356 and Ser363 blocks the etorphine-induced down-regulation of the μ-opioid receptor (Burd et al., 1998). Because both Ser356 and Ser363 residues are putative GRK sites, it is likely these mutations block phosphorylation of the receptor. Interestingly, however, direct measurement of32P-incorporation into the receptor reveals that the mutation of Ser356 and Ser363 to Ala does not, in fact, attenuate etorphine-induced phosphorylation of the receptor. An explanation for blockade of receptor down-regulation could be that the mutations interfere with the receptor-arrestin interaction, or the formation of other receptor complexes. Our preliminary data indicate that blockade of the etorphine-induced down-regulation by Ser356 and Ser363 mutations is reversed by overexpression of arrestin or GRK-2. Therefore, the cellular processing of μ-opioid receptors requires the formation of multiple protein complexes. The identity and role of individual cellular proteins in these processes are under active investigation in our laboratory.
Regulation of Opioid Receptor Activities by Associating Proteins
Various proteins within the membrane microdomain greatly affect opioid receptor signaling. Receptor signaling through scaffold, anchoring, and adaptor proteins is a well-established phenomenon for many membrane receptors, in particular those of the tyrosine kinase family (Pawson and Scott, 1997). The recruitment of other proteins by an adaptor with multiple docking sites allows for the amplification or modulation of signals. An example of this is produced by theDrosophila InaD gene that codes for a protein with five PDZ domains (Tsunoda et al., 1997). TheInaD associates through these PDZ domains with a light-activated Ca2+ channel (TRP), phospholipase C-β, and PKC. The regulation of these effectors byInaD allows for the efficient activation of TRP by phospholipase C-β in response to stimulation of rhodopsin and Gαq, and inactivation by phosphorylation of TRP by PKC. Thus, by recruitment of cellular proteins containing particular motifs, adaptor molecules form complex signal modules that tightly regulate and amplify receptor signals.
Data are accumulating to suggest that signaling modules are created upon agonist binding to GPCR. Like the tyrosine kinase receptors that form protein scaffolds, upon agonist stimulation GPCRs are reported to dimerize (Janovich and Conn, 1996; Maggio et al., 1996; Ng et al., 1996). This dimerization appears to be crucial for receptor function because peptides that block dimerization prevent receptor activation (Hebert et al., 1996). It has also been shown that dimerization may rescue a constitutively desensitized receptor (Hebert et al., 1998). The direct recruitment of cellular proteins by a GPCR is exemplified by the association of phospholipase Cγ1 and Jun-associated kinase (JAK)2 with the angiotensin AT1 receptor (Venema et al., 1998). Recruitment of cellular proteins to the vicinity of GPCR is also accomplished with βγ dimers, as suggested by studies with GRKs (Krupnick and Benovic, 1998). Recent reports have also suggested that the βγ dimer serves as an adaptor molecule in GPCR signaling. Glutathione S-transferase fusion proteins of Rho and Rac bind βγ and thereby inhibit GTPγS binding to Rho and Rac (Harhammer et al., 1996). The amino terminus domain of GRK5 is thought to interact with calmodulin, whereas the carboxyl terminus domain interacts with βγ (Pronin et al., 1997) and the GPCRs of the rhodopsin family have been observed to interact with Rho and ADP-ribosylating factor to activate phospholipase D (Michell et al., 1998). The signal being transmitted depends upon the presence of effectors within the protein scaffold. For example, enzyme phosphorylation by PKC results in the blockade of α2-adrenergic receptor inhibition of type II, and receptor stimulation of type IV, adenylyl cyclase (Marjamaki et al., 1997). A more dramatic effect is observed with the phosphorylation of β2-adrenergic receptors that results in switching of the normal Gs receptor coupling to a Gi/Go coupling (Lefkowitz, 1998). Thus, it is possible that opioid receptor activities are regulated by the scaffolding of cellular proteins.
Opioid receptors, like other GPCRs, may dimerize, and the dimerization appears to affect receptor internalization (Cvejic and Devi, 1997). Our studies with the GH3 cells expressing both the μ- and δ-opioid receptors suggest that the formation of receptor heterodimers results in modulation of receptor activities (Adams et al., unpublished observations). Accordingly, experiments were undertaken to investigate the identities of proteins that could associate with opioid receptors. One approach is to cross-link covalently radioactive ligand onto proteins other than the receptor itself. For this purpose, we chose β-endorphin for cross-linking because it is possible that the carboxyl end of the molecule might interact with other cellular proteins. The ability to cross-link125I-β-endorphin to the opioid receptor has been reported by several laboratories, including our own. Normally, SDS-polyacrylamide gel electrophoresis analysis of the cross-linked moiety reveals two major bands, one of which is the opioid receptor and the other migrating with a molecular mass of ∼25 kDa. Previous reports suggested this 25-kDa band represents a degradation product or a component of the receptor multimers. Our recent studies indicate that the 25-kDa protein does not originate from the receptor, but rather is a component of a larger opioid receptor signaling unit. Our approach was to establish a neuro2A cell line stably expressing a μ-opioid receptor with (His)6 tagged at the N-terminus, which allows the use of Ni2+ resin for the isolation of the protein. The plasma membranes of these cells were isolated in the presence of Complete protease inhibitor cocktail (Boehringer-Mannheim, Indianapolis, IN) and were used in the cross-linking experiments with125I-β-endorphin. It was found that the ratios of the (His)6-MOR and the 25-kDa protein being cross-linked by different regents are different. The heterobifunctional cross-linker, sulfo-MBS (m-maleimidobenzoyl-N-hydroxysuccinimide ester), favored the cross-linking of 125I-β-endorphin to the receptor, suggesting the ε-amino group of lysine in125I-β-endorphin is within the vicinity of a Cys in the receptor. The differences in the ratios also suggest that the 25-kDa protein is not a degradation product of the receptor. Other evidence in support of this conclusion is that the 25-kDa protein copurifies with the receptor from the Ni2+ resin, and that the 25-kDa protein is not detected with polyclonal antibodies that recognize the carboxyl terminal of the μ-opioid receptor or the monoclonal antibodies that recognize the epitope tag spliced into the N-terminus of the receptor. Thus it appears that the 25-kDa protein, if a degradation product of the receptor, must come from receptor domains other than N- or C-terminus. Deduction of degradation products from the μ-opioid receptor sequence with known proteases does not generate a 22- to 25-kDa fragment. Moreover, incubation of isolated125I-β-endorphin cross-linked receptor with various proteases does not generate a 25-kDa fragment. The finding that cross-linking of 125I-β-endorphin with the neura2A membrane isolated from cells stably expressing δ-opioid receptor yields a 25-kDa protein suggests that it is not a specific protease degradation product of the receptor. However, without the actual protein sequence, no definite conclusion can be drawn about the source of the 25-kDa species. Nevertheless, interaction of the 25-kDa protein with the receptor can be demonstrated. Thus, electroeluted 25-kDa protein interacts with the Ni2+ resin saturated with the (His)6-MOR after renaturation, although it fails to interact with the same Ni2+resin before renaturation. Thus, it is tempting to speculate that the 25-kDa protein physically associates with the membrane and represents one of the components of the receptor signaling unit. Its identity, and its effect on opioid receptor activities, remain to be elucidated.
Regulation of Opioid Receptor at Transcriptional Level
Opioid receptor function appears to be dependent on receptor concentration at the cell surface. In addition to covalent modification, such as phosphorylation, the receptor density is controlled by opioid receptor gene transcription. Using cDNA sequences of these receptors, we and others have cloned and characterized three opioid receptor genes (Min et al., 1994; Augustine et al., 1995; Liang et al., 1995; Liu et al., 1995). Although μ-, δ-, and κ-opioid receptors are located on separate chromosomes, they have very similar genomic structures. Moreover, all opioid receptor genes have multiple introns and span large distances in the chromosomal DNA (Fig. 1).

Schematic representation of mouse genomic structures of MOR-1 and DOR-1.
Cloning and characterization of the μ-opioid receptor gene indicates it is >53 kb long with three introns (Min et al., 1994). In the case of introns 2 and 3, the exact length of these introns cannot be determined because of the lack of overlap in the isolated clones (Fig.1). Nevertheless, all the amino acid coding regions of the receptor are known, as well as the splice junction sequences. Junctions between exons are in the IL-1 (Arg95), the EL-2 (Glu213), and the cytoplasmic C-terminal region (Glu386/Leu387). The presence of introns in the position of the IL-1 and the cytoplasmic C-terminal region raises the possibility of alternative forms of MOR-1. Indeed, a splice variant of MOR-1 at the cytoplasmic C-terminal region has been reported (Kraus et al., 1995). The mouse MOR coding sequence deduced from this gene structure has 94% homology to rat MOR-1 cDNA, and is identical with the mouse cDNA sequence subsequently reported byKaufman et al. (1995).
The general genomic structure of the δ-opioid receptor gene resembles that of the μ-opioid receptor (Augustine et al., 1995), spanning 32 kb with multiple intronic structures. The splice junction is located at corresponding amino acids in the IL-1 and EL-2 in the δ-opioid receptor. The only difference between the two genes is the absence of the splice junction at the cytoplasmic C-terminus tail and the δ-opioid receptor. Extensive 3′ rapid amplification of cDNA ends (RACE) studies do not yield splice variants of the cytoplasmic C-terminus. In this case, the RACE clones differ only in the length of their polyA tracts. The amino acid coding portions of the exon sequence determined from the genomic clones of the δ-opioid receptor is 100% homologous to three of the four mouse δ-opioid receptor cDNA sequences currently in GeneBank.
The multiple exon structure of μ- and δ-opioid receptor genes is also observed with the mouse κ-opioid receptor gene, which spans at least 16 kb in the chromosome and has at least four exons (Liu et al., 1995). Exon I encodes the major portion of the 5′-untranslated region and spans a distance of 334, 340, or 716 nucleotides, depending upon the transcription initiation sites. The first intron spans a distance of 371 nucleotides. Exon II of the κ-opioid receptor gene contains 271 nucleotides, including 14 nucleotides of the 5′-untranslated sequence and splice site at Arg86. Intron II has an estimated size of 9 kb. Exon III contains 353 nucleotides and has the splice site at Val204. Intron III spans approximately 4 kb in length. Exon IV begins at Val204 and encodes the rest of the 3′ end sequence of the mouse κ-opioid receptor cDNA. The fact that the exon splice junctions of these three opioid receptor genes are at the same amino acids suggests they are evolved from a single ancestral gene.
The isolation of opioid receptor genes provides an opportunity to address the fundamental question of the importance of receptor density on pharmacological activities. We, and others, have successfully used homologous recombination to delete completely the μ-opioid receptor gene in mice (Matthes et al., 1996; Sora et al., 1997; Tian et al., 1997; Loh et al., 1998). With all knockout animals, morphine is neither lethal nor an antinociceptive agent. Depending on which exon is deleted, there are conflicting reports on the ability of the morphine metabolite, morphine-6-glucuronide (M6G), to induce an antinociceptive response. Thus, M6G is not antinociceptive in the μ-opioid receptor knockout mice generated by the deletion of exons 2 and 3 (Loh et al., 1998) but retains antinociceptive activity in mice generated by exon 1 deletion. Such observations prompted Pasternak (Leventhal et al., 1998) to postulate the existence of a μ-opioid receptor splice variant that is specific for mediating the pharmacological effects of M6G. Regardless of whether this is true, we demonstrated in the μ-opioid knockout mice that morphine regulation of gastrointestinal transit and macrophage phagocytosis and secretion of tumor necrosis factor-α is regulated by the μ-opioid receptor (Roy et al., 1998a,b). Other morphine effects on immune cell functions, such as splenic and thymic cell number and mitogen-induced proliferation, or the inhibition of IL-1 and IL-6 secretion by macrophages, are not altered by elimination of the μ-opioid receptors. Thus, alterations in μ-opioid receptor levels differentially affect the pharmacological actions of morphine.
The importance of opioid receptor density in the pharmacological actions of this drug class is supported by the established relationship between tissue (cells)-specific expression patterns of a receptor subtype and the response to opioids. Opioid gene expression is controlled by different factors in different neurons (or different brain regions), as well as in some disease states. This tissue (or neuronal)-specific expression and function of opioid receptors is ultimately dependent upon the spatial and temporal regulation of gene expression.
To investigate further how receptor levels are regulated, thecis and trans elements of the receptor genes were defined. Multiple start sites were noted using 5′ RACE and RNase protection assays or primer extension. One start site is at −268 nucleotide from the translational initiation coden ATG (designated as +1) of MOR-1 and the other is at −794 nucleotide from ATG. This latter site corresponds to the transcription start sites reported by Liang et al. (1995). In addition to these proximal and distal transcription initiation sites (TIS), we also identified several putative transcription factor binding sites by comparing the nucleotide sequence of the 1.8 kb of DNA upstream from the ATG translational start codon with those in the Transcription Factors Database (GeneBank) at 100% homology. A CCAAT box is present at positions −408 to −404, although no consensus TATA box is found between this CCAAT box and the start of transcription. This may mean that the MOR-1 promoter region does not contain a strong RNA polymerase II binding site. Consensus binding sites for Sp1 (−359 to −367), activator protein-2 (AP-2; −423 to −413, and −933 to −1040) and AP-1 (−1035 to −1029) are also observed. Additional consensus sequences found in the promoter region are a glucocorticoid/mineralcorticoid response element (−1709 to −1695), immune-cell-specific element Pu.1 (−710 to −702), and cytokine response elements nuclear factor (NF)-IL-6 (−1494 to −1486), and NF-GMβ (−652 to −646). Sequences within 1 bp of the consensus cAMP response elements are located at positions −516 to −509 and −1741 to −1744. The presence of these multiple putative transcription factor binding sites suggests that the MOR-1 gene is tightly controlled. Thus, the ability of morphine to alter levels of the proto-oncogenes c-jun and c-fos indicates a probable regulation of MOR-1 gene expression through the AP-1 binding site. The presence of putative cytokine response elements suggests a probable role for this MOR-1 gene product in immune cell functions.
In addition to the proximal TIS, transcription initiates from a distal promoter. In one group of experiments, the proximal promoter region was eliminated and the construct transfected into SHSY5Y cells. Measurement of luciferase revealed a 14-fold increase in activity as compared with promoterless vector pGL3 basic. The degree of stimulation is similar to that obtained when both, or only the proximal, promoters are present (Fig. 2). Therefore, the μ-opioid receptor gene, similar to other GPCR genes having no TATA box, initiates transcription either at a proximal or distal start site (Ko et al., 1997).

Analysis of distal and proximal promoter activities of mouse μ-opioid receptor gene in SHSY5Y cells. A, schematic representation of mouse μ-opioid receptor gene from −1083 to +20. Number refers relative nucleotide position to TIS, +1. Distal and proximal TIS, +516 and +1, are indicated by arrows. Various lengths of 5′ flanking sequences from −1083 to −362 plus 20 nucleotides from proximal TIS were inserted into promoterless luciferase (open box) vector, pGL3-basic (B). Transient transfection luciferase expression assay of the mouse μ-opioid receptor gene in SHSY5Y cells with various promoter contexts. Promoter activity of each fusion gene was normalized to β-galactosidase activity by cotransfecting internal control plasmid pCH110 and was expressed as fold activation of promoterless plasmid (pGL3-basic) activity in cells. Histograms and fold activation showed the mean ± S.E.M. of results from four or more independent duplicated transfection experiments with at least two different plasmid preparations.
In rodent brain, transcription of the μ-opioid receptor is mediated primarily by the proximal promoter and, therefore, the functional role of the distal promoter remains unclear. We have found that removal of an inhibitory regulatory region (−775 to −444 from the ATG start site), or the distal promoter regulatory sequence, restores the activity of the distal promoter (Ko et al., 1997). Detailed 3′ deletion mapping studies of the distal promoter regulatory sequence revealed a 34-bp negative cis-acting element located between −721 to −687 of the 5′ flanking region of the μ-opioid receptor gene. The S1 nuclease protection assay indicated that this 34-bpcis-acting element suppresses distal promoter activity at the transcriptional level (Choe et al., 1998). Operational characterization of this element suggests its effects are position-, promoter-, and orientation-dependent. Collectively, these data indicate this 34-bp element blocks transcription of the μ-opioid receptor gene from the distal promoter, suggesting that the distal promoter activity is under the control of this negative element.
Elements critical for controlling proximal promoter activity of the μ-opioid receptor gene have also been identified. A series of 5′ deletional constructs ranging from −4.7K to −300 bp relative to the translation initiation site (designated as +1) were constructed and transfected into SHSY5Y cells and a decrease in luciferase activity was observed until the DNA fragment was deleted to −400 bp. A dramatic decrease in luciferase activity was noted when the DNA fragment was deleted to −300 bp (Ko et al., 1997, 1998). These results suggest that the proximal promoter activity resides in −400 to −300 bp plus −299 to −249 bp, which includes the promixal transcription initiation sites. Accordingly, two cis-acting elements, the inverted GA (iGA) motif and a canonical Sp1 site located in the −300 to −400-bp region are identified. These two elements are required for the μ-opioid receptor gene proximal promoter activity (Fig.3).

5′ Deletion analysis of mouse μ-opioid receptor gene promoter in SHSY5Y cells. A, schematic representation of a series of 5′ deletions of mouse μ-opioid receptor genes from −4.5 K to −30 bp plus 20 nucleotides from proximal TIS inserted into the promoterless luciferase (open box) vector, pGL3-basic (B). Number on left of each construct refers to number of 5′ flanking nucleotide sequence. pGL3-control plasmid (C) contains SV40 promoter (gray box) and enhancer (hatched box). B, histograms of promoter activity of constructs in SHSY5Y represents mean + S.E.M. of results from four or more independent duplicated transfection experiments.
Using electrophoretic mobility shift analysis, we identified nuclear proteins that are immunologically related to Sp1 and Sp3 and specifically bind to the iGA motif. Mutation of the iGA motif results in a loss of Sp binding and a 50% decrease in promoter activity. Mutation of the canonical Sp1 binding site yields a smaller loss of activity (∼25%). Mutation of both motifs results in an approximately 70% decrease in activity. Using Drosophila SL2 cells, which do not express Sp1 or Sp1-like proteins, it was demonstrated that Sp1trans-activates the promoter containing the intact iGA and/or the canonical Sp1 motifs. Although Sp3 bound only the iGA motif, it can also trans-activate the MOR gene promoter, but to a lesser extent. Because the trans-activation of Sp1 and Sp3 is additive, the ratio of Sp1 and Sp3 molecules in cells may contribute to the regulation of transcription. Thus, these results suggest that μ-opioid receptor transcription is regulated by the cooperative interaction of Sp transcription factors.
Elements that control δ-opioid receptor gene transcription have also been analyzed. A 1.3-kb DNA fragment upstream from the ATG initiation site was sequenced and compared with the sequences in the Transcription Factors Database at 100% homology. Although there are no CCAAT or TATA boxes in the promoter region, there are numerous potential Sp1 binding sites throughout the region between 140 and 390 nucleotides upstream from the ATG initiation site. Like the μ-opioid receptor gene, multiple transcription initiation sites are observed. Also located within this region, clustered about the proximal and distal TIS, are a number of potential binding sites for AP-2. A site for NF-κB, a gene activator induced in response to various extracellular signals, is located at −163 immediately upstream of the proximal TIS. A site for nerve growth factor-induced transcription activator (NGFI-B) is located at −817 from ATG start site. Two sites for NF-IL-6, a factor that activates transcription of cytokine genes involved in immune responses and hematopoiesis, are located at −1149 and −1207 relative to ATG codon. The 80% G + C-rich sequence encompassing transcription initiation sites in the DOR promoter contains an abundance of CpG dinucleotides, suggesting this promoter may be subject to regulation by developmental and/or tissue specific methylation (Christopher and Pichon, 1994). In this context, the existence of potential AP-2 binding sites may be significant, because CpG methylation inhibits proenkephalin gene transcription through interference of AP-2 binding (Comb and Goodman, 1990), and there is an increase in the expression of AP-2 in mouse embryonic neural crest cell linkages (Michell et al., 1998). The ability of intracellular cAMP to regulate AP-2 transcription activity (Imagawa et al., 1987) and to alter δ-opioid receptor transcripts in NG108-15 cells (Gylys et al., 1997) suggests the probable involvement of AP-2 in a feedback regulation of receptor gene transcription. A combination of other transcription factors with putative binding sites in the δ-opioid receptor promoter may account for the recent reported expression of DOR in lymphocytes (Chuang et al., 1994). Moreover, NF-κB and NF-IL-6 act synergistically in response to extracellular signals to induce cytokine gene expression in lymphocytes (Kunsch et al., 1994), and Sp1 is involved in the regulation of tissue-specific gene expression in developing hematopoietic cells (Saffer et al., 1991). This combination of transcription factor activities may also account for the induction of δ-opioid receptor binding sites and mRNAs in PC12 and other neuronal cell lines (Inoue and Hatanak, 1982; Abood and Tao, 1995). Thus, the spatial and temporal regulation of δ-opioid receptor gene expression may be tightly regulated by these transcription elements.
Control of opioid receptor transcripts can be demonstrated during chronic opioid agonist treatment. In NG108-15 cells, chronic agonist exposure results in time- and concentration-dependent decreases in δ-opioid receptor binding due to internalization and degradation of receptor proteins. A parallel decrease in the steady-state level of the δ-opioid receptor mRNA during chronic opioid agonist treatment might occur because the reappearance of opioid binding is blocked by either cycloheximide or actinomycin D (Laws et al., 1985b). However, when the total mRNAs from the NG108-15 cells are analyzed by Northern blot analysis, agonist treatment (up to 24 h) does not alter the level of receptor mRNA. Analysis of the different molecular weight opioid receptor mRNA species does not yield an apparent decrease in any species. These results suggest that post-transcriptional regulation and/or translational regulation of the δ-opioid receptor may occur during chronic agonist treatment. In contrast, there was a slight, temporary increase in opioid receptor mRNA, with maximal levels attained between 4 to 8 h after initiation of chronic agonist treatment. Interestingly, when 10 μM naloxone (for 24 h) was added to the cells after 24 h of [d-Ala2,d-Leu5]-enkephalin treatment, there is an apparent decrease in the level of δ-opioid receptor mRNA. It is probable that the naloxone effect is due to an increase in cAMP, because increases in the intracellular cAMP level upon removal of agonist after chronic treatment have been widely reported. The same decrease in the opioid receptor mRNA is observed when the intracellular cAMP level in NG108-15 cells are raised with 5 μM forskolin and 0.25 mM IBMX (Fig. 4). Because the activities of putative transcription factors such as AP-2 can be greatly affected by PKA, the decrease in the opioid receptor transcription could stem from the elements in the 5′ flanking region of the gene. However, when the 1.3- or 4.7-kb leuciferase reporter gene constructs of the δ-opioid receptor are transfected into NG108-15 cells, it is not possible to demonstrate cAMP-dependent regulation of leuciferase activity. The leuciferase activity in these two constructs, containing putative PKA-sensitive elements, were not altered by forskolin and IBMX (Fig. 5). Thus, regulation of the δ-opioid receptor gene transcription by PKA must lie elsewhere.

Northern analysis of total mRNA isolated from NG108-15 treated with 10 μM forskolin and 0.25 mM IBMX for various hours. One microgram of mRNA isolated from cells thus treated was separated in a 1.2% agarose formaldyhe gel and probed with DOR-1 riboprobe in upper pannel and with random primed probe of acidic ribosyl pyrophosphorylase (ARPP) in bottom panel.

Relative leuciferase activities in NS20Y cells transfected with various DOR-1 reporter gene constructs and treated with various concentrations of forskolin and IBMX for 24 h. PDT4.3, pDT1.3, and pDT0.182 represent the −4.3 kbp, −1.3 kbp, and 0.182 kbp upstream sequences of DOR-1 gene spliced to the reporter gene leuciferase. PGL3 represents promoterless vector control and pGL3 presents leuciferase reporter gene under SV40 promoter.
The ability of NGF to increase δ-opioid receptor binding and mRNA has been studied in PC12 h cells (Inoue and Hatanak, 1982; Abood and Tao, 1995). From our 5′ upstream promoter sequence analysis, we noted a consensus sequence for an NGFI-B binding site present in the promoter region of the δ-opioid receptor gene. Thus, it is likely that the NGF effect is due to activation of this transcriptional factor, or by the NF-κB site, a transcriptional factor that is regulated by NGF. When PC12 h are treated with 50 μg/ml NGF for 3 to 7 days, increases in both the δ-opioid receptor binding and mRNA levels are found. Using the deletion analysis and transient transfection assay of the activities of all the DOR-1 reporter gene constructs, the NGF treatment does not affect leuciferase activity in these constructs (Fig.6). With both the NGFI-B and NF-κB sites in the 1.3-kb construct, 3- or 7-day NGF treatment does not increase the leuciferase activity significantly over that of the control. Because the NGF action on opioid receptor binding and mRNA levels have a delayed effect, with no apparent effect observed within the first day of treatment, it is likely the NGF effect is not a direct action of the growth factor on the δ-opioid receptor transcription, but rather results from secondary gene transcription.

Relative leuciferase activities in PC12 h cells transfected with various DOR-1 reporter gene constructs and treated with NGF for various days. DOR1.3, DOR0.89, and DOR0.26 represent length of upstream sequence of DOR-1 gene spliced to reporter leuciferase; 0.26 (mNF-kB) and 0.26 (mSP-1) represent the 0.26-bp sequence of DOR-1 gene without consensus NF-kB and SP-1 sequences. Control represents pGL3 with SV40 promoter and basic is pGL3 promoterless vector.
Conclusion
Our studies indicate that different opioid agonists yield different receptor conformations or that there are differences within the binding domains among agonists. In addition, even opioid antagonists appear to induce conformational changes within the receptor. These results force a reevaluation of the molecular models for opioid receptor activation because the static model cannot account for these findings. If opioid receptor agonists yield different receptor conformations, the molecular motion within the receptor after agonist binding must result in the same or similar changes within the intracellular domains in which activation of G proteins occurs. In addition to the probable regulation of the receptor-G protein interaction at these intracellular domains, the ability of the TM interaction to affect the efficacy of the ligands suggests other sites for the regulation of receptor activities.
Besides the regulation by the molecular interactions among receptor domains, receptor activities are influenced by cellular protein components. From our studies it appears that opioid receptor signal transduction must involve cellular components other than the receptor, G proteins, and effectors. The formation of protein scaffolds and the recruitment of other proteins that modulate receptor signals are likely events that occur after agonist-induced activation of the receptor. The fact that agonist-induced receptor desensitization requires the recruitment of GRK, arrestin, and other proteins support this hypothesis. The receptor activities are regulated subsequently by the composition of these protein scaffolds. For example, the ability to remove the receptor from the cell surface, and hence decrease agonist activity, depends upon the interaction of the receptor with arrestin. The interaction of arrestin with other cellular proteins, such as calmodulin, regulates a subtype of adenylyl cyclase and thereby greatly affects receptor activities. Hence, understanding the composition of the receptor complex is key in understanding opioid receptor signal transduction. The identities of these proteins within the signaling complexes of the opioid receptor will be the major focus of our future research.
Although we have yet to identify the elements responsible for tissue (cell)-specific expression of the opioid receptor genes, it is clear that interactions of ubiquitous transcriptional factors determine receptor gene transcription. The spatial and temporal regulation of the expression of these factors significantly affect opioid receptor levels. Also, from our studies with cAMP and NGF it is clear that regulation of receptor transcripts may not lie within the 5′-upstream element. Considering the extensive intronic sequences within these receptor genes, it is probable that regulating elements yet to be defined are located within these sequences.
In summary, our past studies reveal a complicated system for regulating the activities of opioids. Not only do factors that control receptor gene transcription and protein levels affect the pharmacology of various opioid drugs, but the interaction of the drugs within these receptors and the recruitment of cellular proteins to form the receptor signaling complexes are also important in this regard. By identifying the players involved and revealing how their activities are regulated, we may be able to elucidate the ultimate goal of our studies—the molecular mechanism of opioid tolerance and dependence.
Acknowledgments
Our appreciation is extended to the following colleagues who performed the scientific work presented in this paper: P. Y. Law, A. L. Burd, R. El Kouhen, L. Erickson, O. El Kouhen, C. Choe, H. Im, J. Ko, H. Liu, S. Minnerath, L. Wei, S. Chakrabarti, P. Claude, P. Prather, and J. Yang.
Footnotes
-
Send reprint requests to: Horace H. Loh, Department of Pharmacology, 3-249 Millard Hall, University of Minnesota Medical School, 435 Delaware St. S.E., Minneapolis, MN 55455-0347. E-mail:lohxx001{at}maroon.tc.umn.edu
-
↵1 This study was supported by National Institutes of Health Grants DA11806, DA00564, DA01583, DA07339, DA70554, and the A. and F. Stark Fund of the Minnesota Medical Foundation.
- Abbreviations:
- GPCR
- G protein-coupled receptors
- CHO
- Chinese hamster ovary
- DAMGO
- [d-Ala2,N-MePhe4,Gly-ol5]
- DPDPE
- [d-Pen2,d-Pen5]-enkephalin
- Tippψ
- H-Tyr-Tic[ψ,CH2NH]Phe-Phe-OH (Tic = 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid)
- CTOP
- d-Phe-Cys-Tyr-d-Trp-Orn-Thr-Pen-Thr-NH2
- TM6
- transmembrane domain 6
- EL
- extracellular loop
- NTB
- naltriben
- IL
- intracellular loop
- GRK
- G protein-coupled receptor kinase
- PKA
- protein kinase A
- PKC
- protein kinase C
- PTX
- pertussis toxin
- IBMX
- 3-isobutyl-1-methylxanthine
- GIRK
- G protein-coupled voltage-dependent inward rectifying potassium channels
- RACE
- rapid amplification of cDNA ends
- M6G
- morphine-6-glucuronide
- TIS
- transcription initiation sites
- AP
- activator protein
- NF
- nuclear factor
- NGFI-B
- nerve growth factor-induced transcription activator
- Received February 19, 1999.
- Accepted March 3, 1999.
- The American Society for Pharmacology and Experimental Therapeutics
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
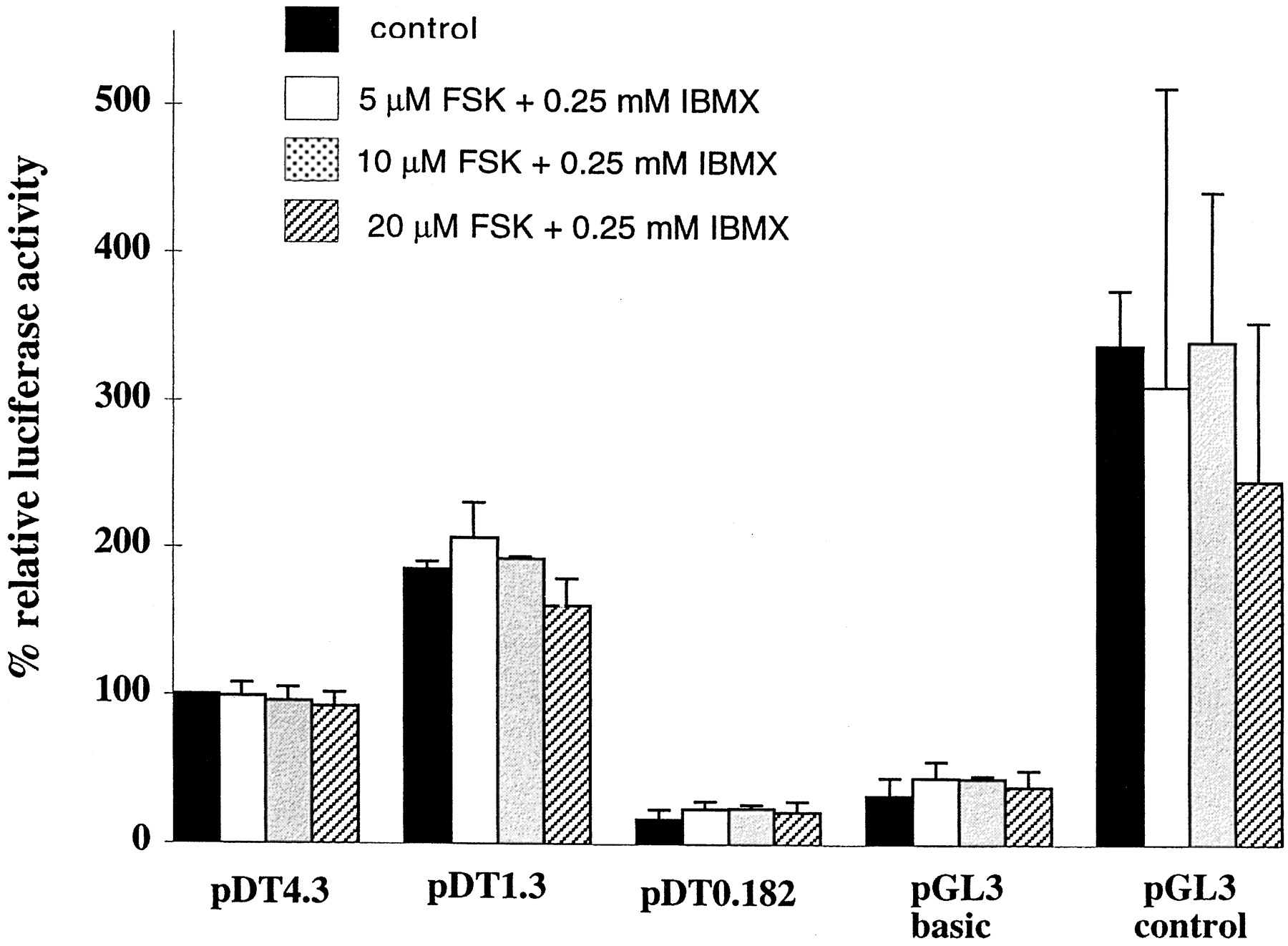{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- A Sincere Thanks
- Introduction
- Differences in μ- and δ-Opioid Receptor Signal Transduction
- Differential Regulation of μ- and δ-Opioid Receptor Signal Cascades at Level of Ligand-Receptor Interaction
- Differential Regulation of μ- and δ-Opioid Receptor at G Protein Level
- Regulation of μ- and δ-Opioid Receptor Levels by Phosphorylation and Down-Regulation
- Regulation of Opioid Receptor Activities by Associating Proteins
- Regulation of Opioid Receptor at Transcriptional Level
- Conclusion
- Acknowledgments
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters