


Abstract
It has been shown that mibefradil (Ro 40-5967) exerts a selective inhibitory effect on T-type Ca2+ currents, although at higher concentrations it can antagonize high voltage-activated Ca2+ currents. The action of mibefradil on Ca2+channels is use- and steady-state-dependent and the binding site of mibefradil on L-type Ca2+ channels is different from that of dihydropyridines. By using conventional whole-cell and perforated patch-clamp techniques, we showed that mibefradil has an inhibitory effect on both T- and L-type Ca2+currents in insulin-secreting cells. However, the effect on L-type Ca2+ currents was time-dependent and poorly reversible in perforated patch-clamp experiments. By using mass spectrometry, we demonstrated that mibefradil accumulates inside cells, and furthermore, a metabolite of mibefradil was detected. Intracellular application of this metabolite selectively blocked the L-type Ca2+ current, whereas mibefradil exerted no effect. This study demonstrates that mibefradil permeates into cells and is hydrolyzed to a metabolite that blocks L-type Ca2+ channels specifically by acting at the inner side of the channel.
It has been demonstrated that mibefradil (Ro 40-5967), a structurally novel nondihydropyridine compound, exerts a potent inhibitory effect on T-type Ca2+ currents in vascular smooth muscle cells (Mishra and Hermsmeyer, 1994; Schmitt et al., 1995), sensory neurons (Todorovic and Lingle, 1998), adrenal glomerulosa cells (Rossier et al., 1998), and spermatogenic cells (Arnoult et al., 1998). Inhibition of low voltage-activated Ca2+ currents in motor neurons occurs at a similar potency to high voltage-activated Ca2+ currents (Viana et al., 1997). A study of α1-subunits of high voltage-activated Ca2+ channels (including L-, N-, P/Q-, and R-types) expressed in Xenopus oocytes revealed steady-state- and frequency-dependent inhibitory effects of mibefradil (Bezprozvanny and Tsien, 1995). This finding suggested that the drug binds to the inactivated and open state of the channel. It has been reported that the binding site of mibefradil on Ca2+ channels is distinct from that of dihydropyridines and partially overlaps that of verapamil (Rutledge and Triggle, 1995; Ratner et al., 1996; Schuster et al., 1996). Because Ca2+ channels play important roles in insulin secretion (Keahey et al., 1989; Bhattacharjee et al., 1997) and cytokine-induced death of pancreatic β-cells (Juntti-Berggren et al., 1993; Wang et al., 1999), we were interested in characterizing the inhibitory effects of mibefradil on T- and L-type Ca2+ channels in an insulin-secreting cell line, INS-1.
Materials and Methods
Drugs.
Mibefradil ((1S,2S)-2-[2-[[3-(2-benzimidazolyl)propyl]methylamino]ethyl]-6-fluoro-1,2,3,4-tetrahydro-1-isopropyl-2-napthyl methoxyacetate dihydrochloride) was kindly provided by Dr. J.-P. Clozel (Hoffmann La Roche, Basel, Switzerland).
The free alcohol des-methoxyacetyl mibefradil (dm-mibefradil) (1S,2S)-2-[2-[[3-(2-benzimidazolyl)propyl]methylamino]ethyl]-6- fluoro-1,2,3,4-tetrahydro-1-isopropyl-2-napthyl hydroxy hydrochloride was prepared by alkaline hydrolysis: 14.2 mg mibefradil hydrochloride was dissolved in 4 ml of methanol + 1 ml of 10 N aqueous sodium hydroxide mixture (5 mM was the final concentration of mibefradil). The solution was warmed in a boiling water bath for 10 min. The reaction was followed by mass spectrometry. On completion of the hydrolysis, as determined from the mass spectra, the solution was neutralized with 5 M aqueous hydrochloric acid. The slight loss of methanol that occurred by evaporation during the reaction was corrected by adding water to keep the total volume of 5 ml.
Cell Culture.
INS-1 cells were cultured in RPMI 1640 medium containing 10% fetal bovine serum, 25 U/ml penicillin, 25 mg/ml streptomycin, and 50 μM β-mercaptoethanol in an atmosphere of 5% CO2 in air at 37°C for 2 to 5 days before recording.
Electrophysiological Recording.
The whole-cell recordings were carried out by the standard “giga-seal” patch-clamp technique (Hamill et al., 1981). The whole-cell recording pipettes were made of hemocapillaries (Warner Instrument Corp., Hamden, CT), pulled by a two-stage puller (PC-10; Narishige International, New York, NY), and heat polished with a microforge (MF-200; World Precision Instruments, Sarasota, FL) before use. Pipette resistance was in the range of 2 to 5 MΩ in our internal solution. The recordings were performed at room temperature (22–25°C). Currents were recorded using an EPC-9 patch-clamp amplifier (HEKA, Lambrecht/Pfalz, Germany) and filtered at 2.9 kHz. Data were acquired with Pulse/PulseFit software (HEKA). Voltage-dependent currents were corrected for linear leak and residual capacitance by using an on-line P/n subtraction paradigm. In whole-cell configuration, T-type Ca2+ currents were recorded at −30 mV when the holding potential was either −90 or −80 mV. For perforated patches, T-type tail currents were measured by the following protocol: cells were held at −70 mV, then stepped to +20 mV for 10 ms, and then currents were measured after a hyperpolarization at −100 mV for 200 ms. The L-type Ca2+ current was measured at +20 mV with a holding potential of −40 mV.
Solutions.
The extracellular solution used in whole-cell Ca2+ current recording contained 10 mM CaCl2, 110 mM tetraethylammonium-Cl, 10 mM CsCl, 10 mM HEPES, 40 mM sucrose, 0.5 mM 3,4-diaminopyridine, pH 7.3. The intracellular solution contained 130 mMN-methyl-d-glucamine, 20 mM EGTA (free acid), 5 mM 1,2-bis(2-aminophenoxy)ethane-N,N,N′, N′-tetraacetate (BAPTA), 10 mM HEPES, 6 mM MgCl2, 4 mM Ca(OH)2, pH adjusted to 7.4 with methanesulfonate. Mg-ATP (2 mM) was included in the pipette solution to minimize “run-down” of L-type Ca2+ currents. For perforated-patch experiments, nystatin (200 μg/ml) was used. The pipette was filled with nystatin-containing intracellular solution and gentle suction was used to achieve gigaohm resistance. The access resistance gradually decreased within 5 min after the gigaohm-seal formation, and then currents were recorded after stabilization. The extracellular solution contained 26 mM sucrose, 130 mM tetraethylammonium-Cl, 10 mM HEPES, 5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, pH 7.3. The pipette solution contained 65 mM CsOH, 65 mM CsMS, 20 mM sucrose, 10 mM HEPES, 10 mM MgCl2, 1 mM Ca(OH)2, pH 7.4.
Mass Spectrometric Analysis.
A VG 70–250 SEQ instrument (VG Analytical, Manchester, UK) was used with fast atom bombardment ionization mode to obtain mass spectra of the mibefradil and dm-mibefradil. Cultured INS-1 cells were treated with 20 μM mibefradil for various lengths of time under each experimental condition. Cells were centrifuged at 1000g. The cell pellets were collected after washing three times with PBS and were resuspended in 0.5 ml of medium for mass spectrometric analysis. For a 50-μl cell sample, 20 μl of internal standard solution (40 μM verapamil, mol. wt. 454) and 5 μl of glycerol were added, and 4 μl of this mixture was used for fast atom bombardment mass spectrometry. Several positive ion spectra were recorded in the mass rangem/z 750 to 100 at a mass resolution of 1000, and a scan speed of 2 s/decade. For mibefradil, m/z496 was the dominant ion (M + H)+ accompanied by a less intense sodiated molecular ion m/z 518. The concentrations of the mibefradil and hydrolyzed mibefradil were determined by calibrating the intensities of m/z496 and 424 with the intensity of m/z 455. For calibration, a standard solution of 50 μM drug was subjected to mass spectrometric analysis.
Separation of Cytosolic and Membrane Components.
After washing out mibefradil from the bath solution, the cells were collected and the membranes were broken down by vortexing the cells in a solution containing 5% acetic acid/CH3CN. The mixture was then centrifuged, and the supernatant was collected and defined as nonmembrane-associated components. Pellets were resuspended in 5× volume of 10 N NaOH/methanol (1:7) solution at 37°C for 5 min. The mixture was neutralized with 0.5 M HCl and centrifuged. The remaining pellet and the supernatant were collected separately.
Statistics.
All data are presented as mean ± S.E. Student's t test was used to calculate P values where given.
Results
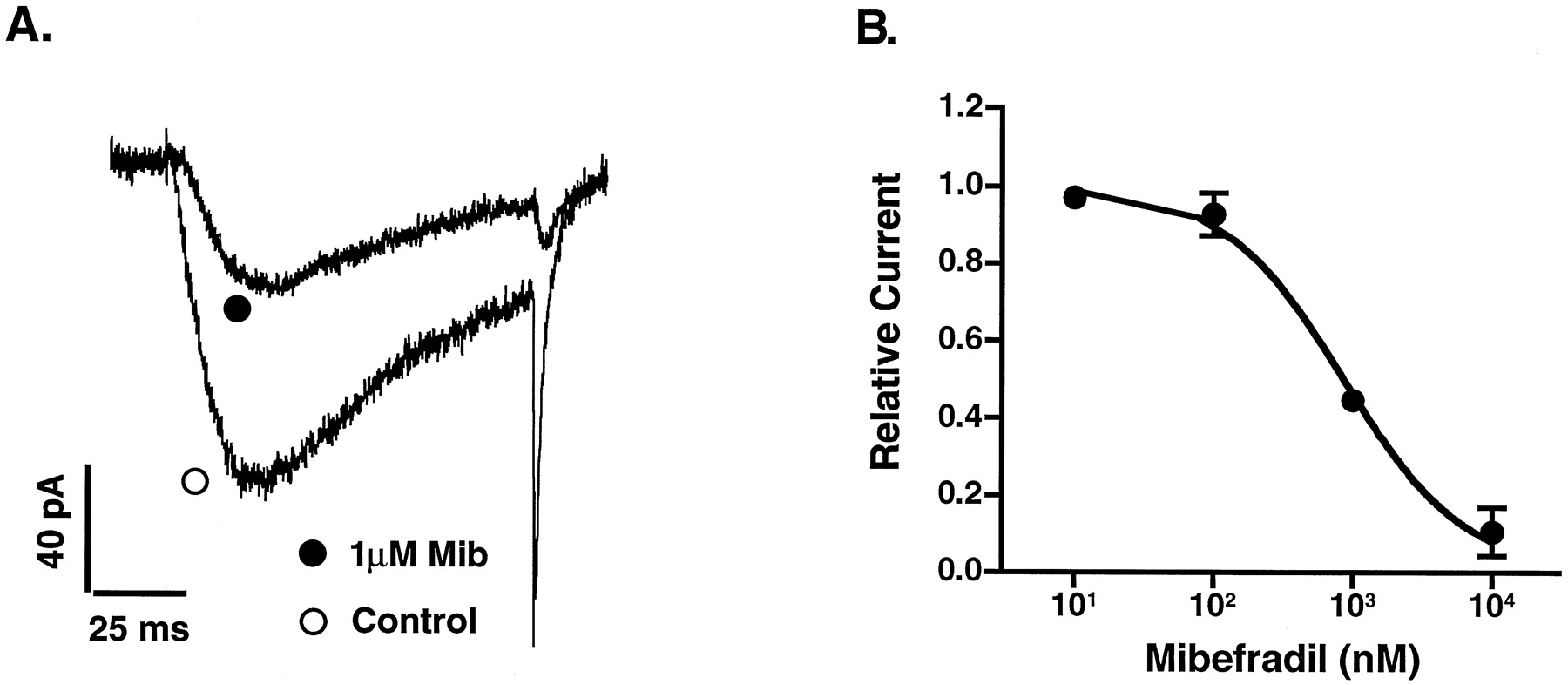
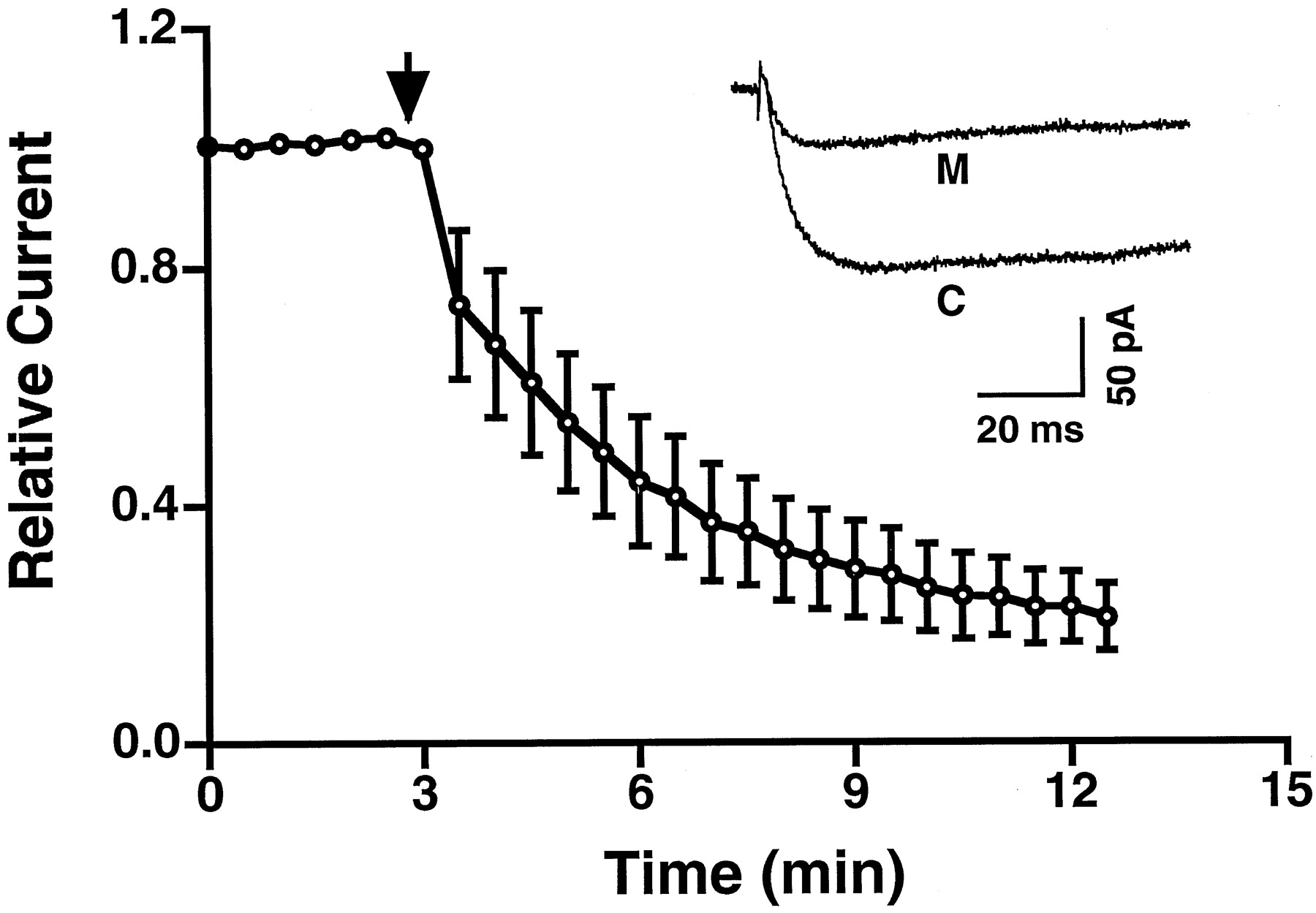
We first used whole-cell patch-clamp and a bath perfusion system to establish the dose-dependent inhibition of mibefradil on both T- and L-types of Ca2+ currents. The T-type Ca2+ current was measured at −30 mV when the membrane was held at −90 mV, and the L-type current was measured at +20 mV when the membrane was held at −40 mV. The dose-dependent inhibition of T-type Ca2+ current is shown in Fig. 1. The 50% inhibitory concentration (IC50) was 865 nM. No time-dependent effects of mibefradil on T-type Ca2+ currents were observed. In contrast, the inhibition of L-type Ca2+ currents could not be fitted with one-to-one binding curve. This was due to L-type Ca2+ current run-down in the whole-cell patch-clamp configuration. We used perforated patch-clamp to rectify run-down, however, we experienced a time-dependent inhibitory effect of mibefradil. Administration of 1 μM mibefradil progressively reduced L-type Ca2+ current up to 70% of the beginning amplitude after 10 min (Fig.2). This indicated that a more complicated pharmacological mechanism was involved in the action of mibefradil on the L-type Ca2+currents in β-cells.

A, effect of mibefradil on a T-type Ca2+current elicited at −30 mV when the membrane was held at −90 mV. Mib, mibefradil. B, inhibitory dose-response relationship of mibefradil on T-type Ca2+ currents. Currents were measured with the whole-cell patch-clamp configuration. Data from four experiments were normalized individually and then plotted as means (±S.E.). The curve was generated by fitting the data using one-to-one binding curve according to the eq. 1/(1 + [mibefradil]/Kd).

Time-dependent effect of mibefradil on L-type Ca2+ currents recorded with the perforated patch-clamp configuration (n = 5). Arrow indicates the time when 1 μM mibefradil was administered. The inset shows the inhibition of an L-type Ca2+ current by mibefradil. Traces represent L-type Ca2+currents recorded before (C) and after (M) 10-min addition of mibefradil. M, mibefradil. C, control. Error bars represent S.E.M.
We next used a drug-diffusing system to test the reversibility of the antagonism of T- and L-type Ca2+currents by mibefradil. Small volumes (approximately 2 μl) of drugs were delivered in close proximity to the recording cell with a quartz capillary positioned by a micromanipulator. After administration, drugs diffused throughout the entire recording chamber containing 2 ml of bath solution. Using this system, we found that the inhibition of mibefradil on the T-type Ca2+ current was clearly reversible. In contrast, the inhibition of the L-type Ca2+ current was poorly reversible (Fig.3).
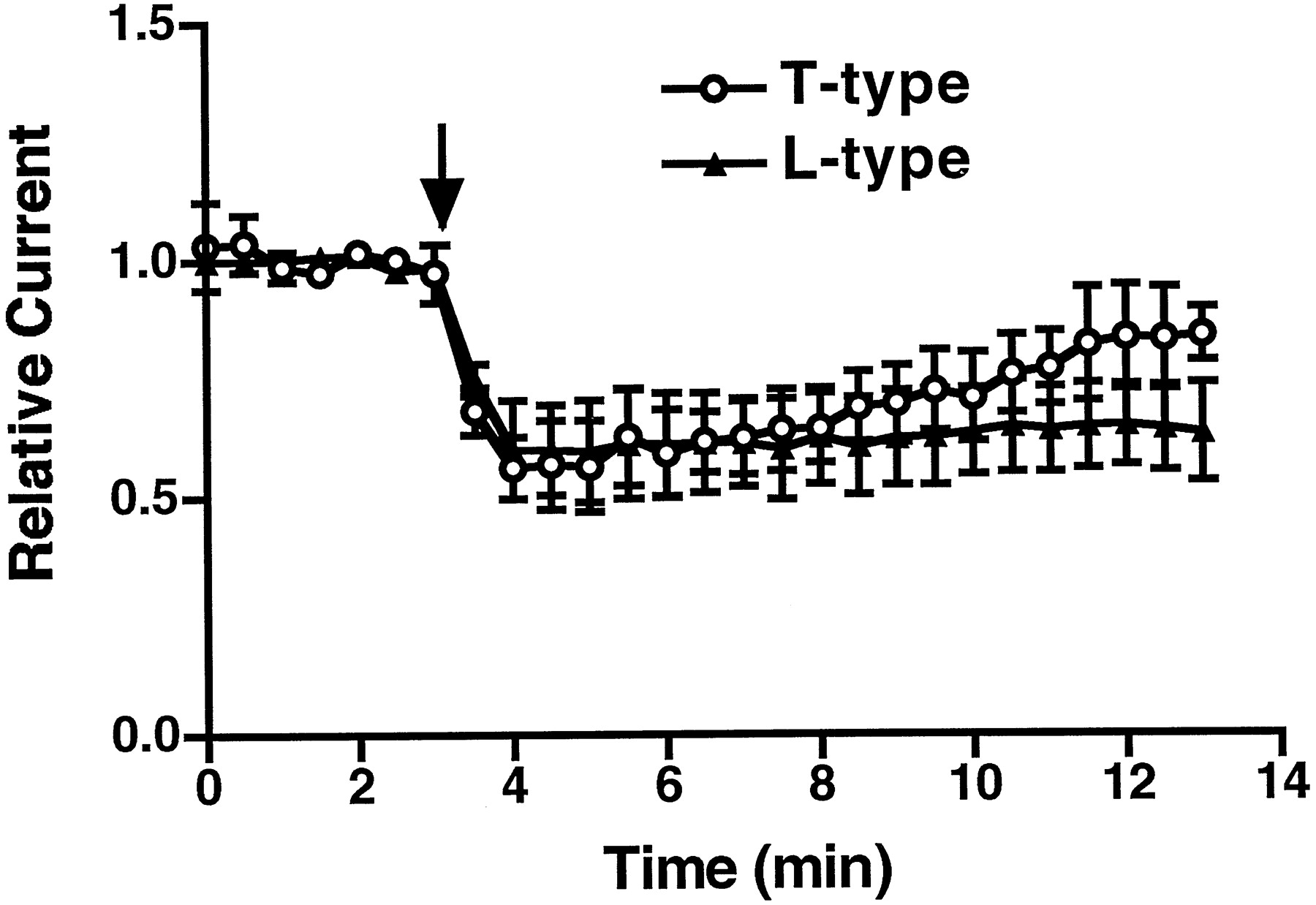
Testing of mibefradil reversibility. Open circles and solid triangles represent normalized T- and L-type currents recorded before and after mibefradil (2 μl of 10 μM) was administered, respectively. The L-type currents were recorded at +20 mV when held at −40 mV in the perforated patch-clamp configuration. The T-type Ca2+ current was measured at −30 mV when held at −80 mV in the whole-cell configuration. Arrow indicates the time when the drugs were delivered. n= 3 for each group of experiments. Error bars represent S.E.M.
The poor reversibility and time-dependent inhibition of the L-type Ca2+ current by mibefradil suggested that this drug had an accumulation effect over time. We have tested this hypothesis by applying a very low dose of mibefradil on cells and recording the L-type Ca2+currents for a longer length of time in the perforated patch-clamp configuration. As shown in Fig.4A, after 25 min of 10 nM mibefradil administration, the relative currents were reduced by 30%, whereas the currents remained unchanged for control patches. Cells preincubated with10 nM mibefradil for 2 h resulted in a reduction of current density as recorded by perforated patches (Fig. 4B). At a concentration of 10 nM, mibefradil exhibited no long-term effect on the T-type Ca2+ current.

The long-term effect of mibefradil (10 nM) on L- and T-type Ca2+ currents. Recordings were made in the perforated patch-clamp configuration. A, open circles represent the L-type Ca2+ current recorded in the cells without administration of mibefradil. Solid circles and squares represent T- and L-type Ca2+ currents recorded in the cells after administering mibefradil, respectively. T-type Ca2+ tail currents were measured with a test potential of −100 mV immediately after a depolarizing pulse to +20 mV for 10 ms. Mibefradil was delivered at time zero (n = 4 for each group of experiments). B, current densities of L-type Ca2+ channels measured in the cells that were cultured in medium with or without preincubation with 10 nM mibefradil for 2 h. Recordings were conducted in the perforated patch-clamp configuration in a bath solution that contained no drug (n = 14 for each group experiments). *P < .01 by Student's t test.
What is the mechanism by which mibefradil exerts its time-dependent inhibitory effect on L-type Ca2+currents? Because the main difference between the conventional whole-cell and the perforated-patch configuration is that the latter provides a relatively intact intracellular environment, we hypothesized that mibefradil may diffuse across the cell membrane into the cytoplasm and accumulate inside cells. To test this hypothesis, the presence of mibefradil was examined in cells preincubated with 20 μM mibefradil by using mass spectrometry. After three washes, mibefradil (peak = 496 mol. wt.) was still detected in cells (as shown in Fig.5, inset). The concentration of intracellular mibefradil after 1-min incubation was 3.18 ± 0.78 μM (n = 3). The localization of mibefradil in cells was examined by measuring the concentration of mibefradil in the pellets and supernatants after lysis of the cells. Most of the mibefradil (92%) was detected in the supernatants and none (0%) was found in the pellets after washing cells with methanol, indicating that mibefradil was retained in the cytoplasm. In addition, we detected a peak (mol. wt. = 423) that represented dm-mibefradil, a hydrolyzed metabolite of mibefradil, which is a major metabolite as documented previously (Wiltshire et al., 1992). By varying the time of preincubation, we found that dm-mibefradil accumulated inside the cells in a time-dependent manner (Fig. 5). This accumulation is consistent with the concept that dm-mibefradil has lower membrane permeability than its precursor mibefradil.
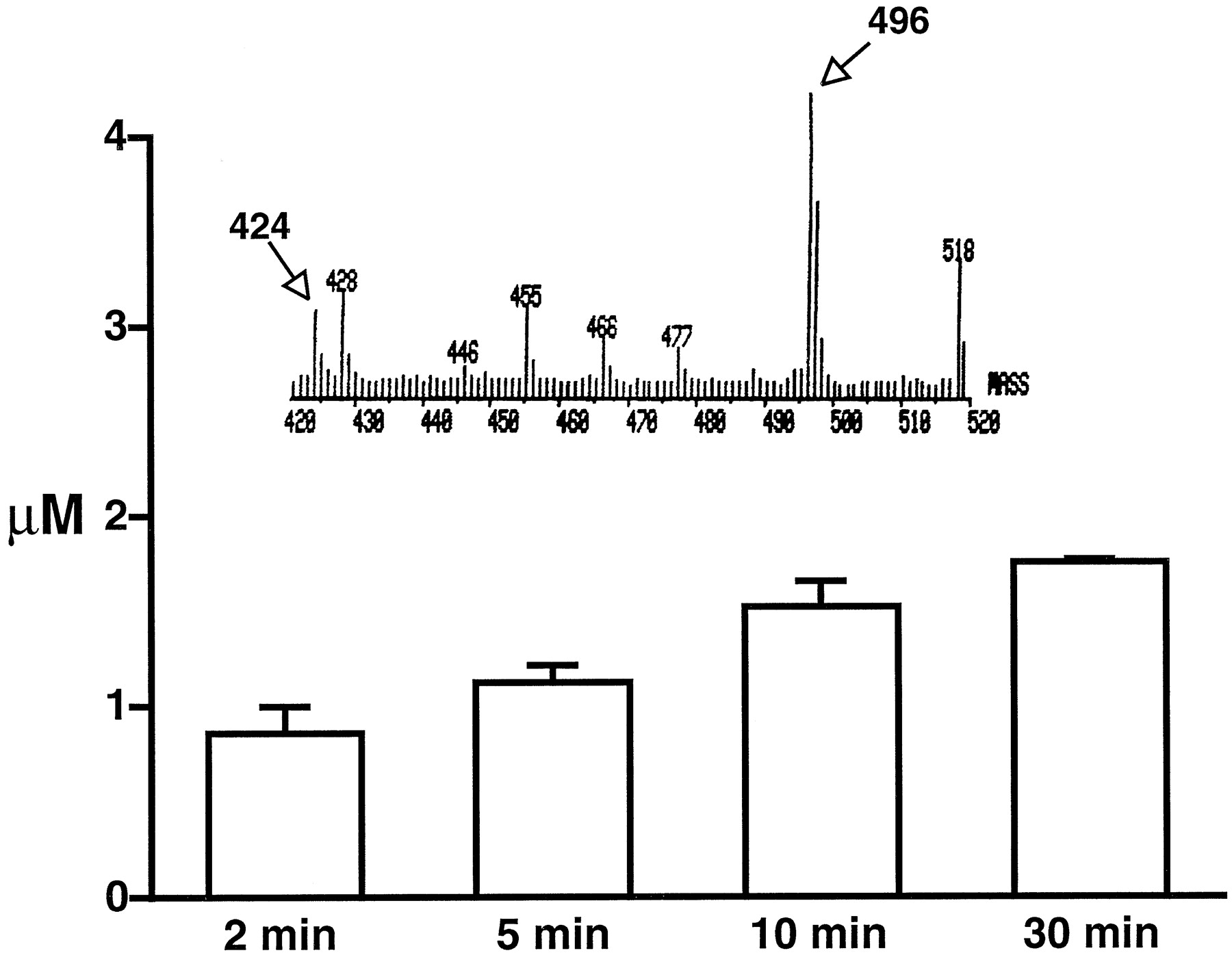
Accumulation of dm-mibefradil in cells measured with mass spectrometry. The cells were preincubated with mibefradil (20 μM) for the length of time indicated in the figure (n = 3) and washed three times before the measurements were taken. The inset shows the primary data of mass spectrometry with peaks at 496 and 424, which correspond to mibefradil and dm-mibefradil, respectively.
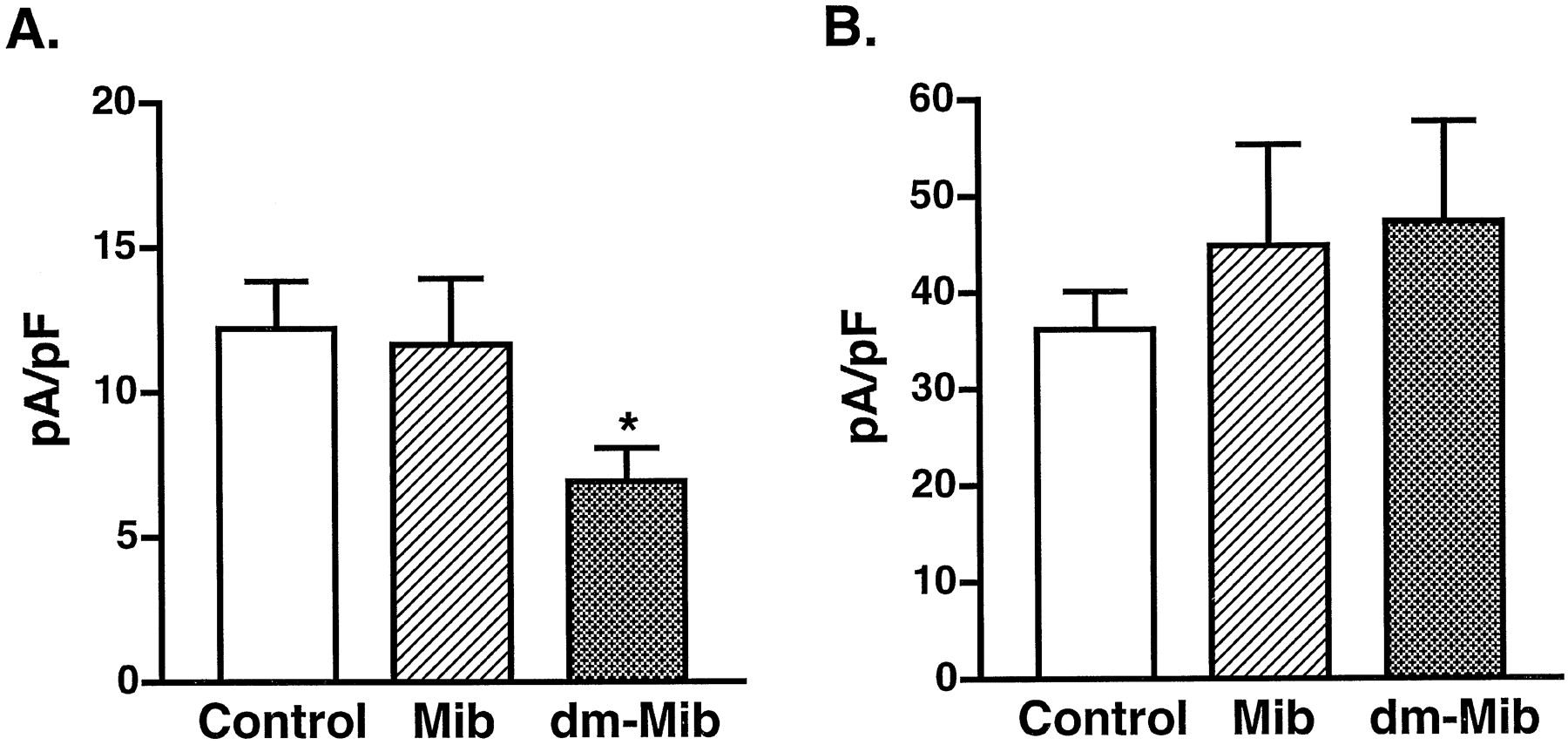
We then tested whether mibefradil or dm-mibefradil inhibits L- or T-type Ca2+ currents from inside of cells. Both L- and T-type currents were measured in the whole-cell patch-clamp configuration when 1 μM mibefradil or dm-mibefradil was included in the pipette solution. The highest amplitudes of currents were measured during the entire course of the recordings and were used to compare current densities between the control and drug experiments, allowing the variation that results from L-type Ca2+ current run-down to be normalized. As shown in Fig. 6, intracellular application of 1 μM mibefradil did not have an inhibitory effect on either L-type or T-type Ca2+currents, whereas the same concentration of dm-mibefradil specifically blocked the L-type Ca2+ current. Because the bath solution contained no drug in this series of experiments, the inhibitory effect of dm-mibefradil should be considered as acting on the inside domain of L-type Ca2+ channels.

A, the effect of mibefradil and dm-mibefradil on L-type Ca2+ currents from inside cells (n = 8); *P < .01 by Student's t test. B, the effect of mibefradil or dm-mibefradil on T-type Ca2+ current from inside cells (n = 4). All data were collected at 5 min after formation of a whole-cell patch. The pipette solution contained 1 μM drug.
Discussion
It has been suggested that mibefradil has significant therapeutic advantages in reducing blood pressure (Hefti et al., 1990; Bernink et al., 1996) and preventing blood pressure-related arterial hypertrophy (Li and Schiffrin, 1997), in lowering heart rate (Clozel et al., 1991), and in preventing and reducing hyperinsulinemia (Verma et al., 1997). Our results show that mibefradil accumulates inside β-cells and is hydrolyzed to a metabolite that blocks L-type Ca2+ currents. This mechanism indicates that in addition to its effects on T-type Ca2+ channels, mibefradil is also a potent L-type Ca2+ channel antagonist in β-cells.
In the experiment summarized in Fig. 6, we used the whole-cell patch configuration to introduce drugs inside cells via pipette. One of the major drawbacks of this technique, however, is that we have less control of the final intracellular drug concentration, especially in this case. Because L-type Ca2+channels quickly exhibited run-down, we had to record the peak current within 5 min after break-in. Within this short period of time, one can predict that the actual free concentration of mibefradil adjacent to L-type Ca2+ channels is lower than the drug concentration in the pipette. However, Fig. 6 does demonstrate that a synthesized metabolite of mibefradil has statistically significant inhibitory effects on L-type Ca2+ channels from inside of cells.
The inhibitory effect of dm-mibefradil on T-type Ca2+ currents was similar to the effect of mibefradil when it was applied in the bath solution. This suggests that the methoxyacetyl group of mibefradil does not play a key role in binding to the extracellular receptor site of T-type Ca2+ channel protein (structures of mibefradil and dm-mibefradil are shown in Fig. 7). This methoxyacetyl group, however, is necessary for blocking L-type Ca2+ channels from the inside of cells, indicating that a modification in the methoxyacetyl group of mibefradil may result in a more selective antagonist of T-type Ca2+ channels.

Chemical structures of mibefradil (1) and des-methoxyacetyl mibefradil (2).
Acknowledgments
The mass spectrometric analyses were carried out in the Mass Spectrometry and Protein Structure Laboratory of the University of South Alabama, College of Medicine. We thank Drs. Ann Abraham and F. Aladar Bencsath for the analyses, spectral interpretations, and useful discussions.
Footnotes
-
Send reprint requests to: Ming Li, Ph.D., Department of Pharmacology, University of South Alabama, College of Medicine, Mobile, AL 36688. E-mail: mli{at}jaguar1.usouthal.edu
-
1 This work was supported by National Institutes of Health Grant DK50151 and Juvenile Diabetes Foundation International Grant no. 197037.
- Abbreviations:
- dm-mibefradil
- (1S,2S)-2-[2-[[3-(2-benzimidazolyl)propyl]methylamino]ethyl]-6-fluoro-1,2,3,4-tetrahydro-1-isopropyl-2-napthyl hydroxy hydrochloride
- Received July 28, 1999.
- Accepted October 25, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}