


Abstract
The goal of this study was to analyze the effects of mibefradil on a human cardiac K+ channel (hKv1.5) stably expressed in Chinese hamster ovary cells using the whole-cell configuration of the patch-clamp technique. Mibefradil inhibited in a concentration-dependent manner the hKv1.5 current with aKD value of 0.78 ± 0.05 μM and a Hill coefficient of 0.97 ± 0.06. Block induced by mibefradil was voltage dependent, consistent with a value of electrical distance of 0.13. The apparent association (k) and dissociation (l) rate constants measured at +50 mV were found to be 7.3 ± 0.5 × 106M−1 · s−1 and 4.3 ± 0.1 s−1, respectively. Block increased rapidly between −20 and +10 mV, coincident with channel opening and suggested an open channel block mechanism, which was confirmed by a slower deactivation time course resulting in a “crossover” phenomenon when tail currents recorded under control conditions and in the presence of mibefradil were superimposed. Shifts toward negative potentials of the maximum conductance and the activation curve were observed, confirming the voltage dependence of block. Mibefradil induced a significant use-dependent block when trains of depolarization at frequencies between 0.02 and 2 Hz were applied. In the presence of mibefradil, recovery of inactivation was faster than under control conditions, suggesting that mibefradil might compete with the inactivation gate of hKv1.5. These results indicate that mibefradil blocks hKv1.5 channels in a concentration-, voltage-, time- and use-dependent manner and the concentrations needed to observe these effects are in the therapeutic range.
Calcium channel blockers (CCBs) constitute a heterogeneous class of compounds with different potency and selectivity for voltage-dependent Ca2+ channels. According to the type of voltage-dependent Ca2+ channel they block, one can distinguish L- or T-type calcium channel antagonists. Mibefradil is a potent vasodilatator with a high selectivity for the coronary vasculature over the peripheral vasculature and the myocardium. The mechanism of action of mibefradil was characterized by the selective blockade of transient, low-voltage-activated (T-type) calcium channels over long-lasting, high-voltage-activated (L-type) calcium channels (Clozel et al., 1997). In human tissues, mibefradil has shown the highest vascular to cardiac selectivity ratio; some 3-fold higher than felodipine and some 200-fold more vascular selective than the phenylalkylamine verapamil (Sarsero et al., 1998).
Previous studies have suggested that CCBs can interact with cardiac voltage-dependent K+ channels. The humanether-à-go-go-related gene HERG product generating the rapid component of the cardiac delayed rectifier potassium current (IKr) (Sanguinetti et al., 1995), in assembly with MinK-related peptide 1 (MIRP1) (Abbott et al., 1999), is blocked in a concentration-dependent manner by verapamil and mibefradil (Chouabe et al., 1998). The slow component of the cardiac delayed rectifier potassium current (IKs) resulting from the assembly of two different proteins, KvLQT1 and IsK (Attali, 1996;Sanguinetti et al., 1996), is inhibited by bepridil and mibefradil (Chouabe et al., 1998). Verapamil and nifedipine produced a marked block of the native transient outward current (Ito) in rat ventricular myocytes (Jahnel et al., 1994). Nifedipine and verapamil actions on a human voltage-gated potassium α-subunit (hKv1.5) have been examined. Both calcium antagonists block the current expressed in HEK cells. The binding site for nifedipine would be located at the extracellular pore of the channel (Zhang et al., 1997), whereas verapamil would be a blocker of the inner pore of the channel in human myocardium (Rampe et al., 1993).
hKv1.5 is thought to underlie the 4-aminopyridine-sensitive ultra-rapidly activating delayed rectifier K+current (IKur) found in human atrial myocytes. Studies with antisense oligonucleotides provided direct evidence that hKv1.5 is essential to the expression of the human atrial IKur (Feng et al., 1997). Kv1.5 transcript (Fedida et al., 1993) and hKv1.5 protein (Mays et al., 1995) have also been detected in human ventricle despite the absence of corresponding current in human ventricle (Li et al., 1996). It is possible that the hKv1.5 α-protein contributes to K+ current in the ventricle through the formation of heteromultimeric K+ channels with other Shaker-like α-subunits (Mays et al., 1995). It has been found that IKurplays an important role in human atrial repolarization (Wang et al., 1993). Therefore, the present study was undertaken to characterize the concentration-, voltage-, state-, and use-dependent effects of mibefradil on hKv1.5 channels expressed in a stable mammalian Chinese hamster ovary (CHO) cell line.
Materials and Methods
Cell Preparation.
Effect of mibefradil has been determined using a stable mammalian cell line expressing human Kv1.5 voltage-gated potassium current (Tamkun et al., 1991). Endogenous voltage-gated potassium channels are expressed in HEK 293 cells (Yu and Kerchner, 1998) but little or no voltage-gated-like current was detected in CHO cell line (Philipson et al., 1993). Thus, we chose to express hKv1.5 in a CHO cell line. CHO cells were maintained at 37°C in minimum essential Eagle's medium alpha (Life Technologies, Basel, Switzerland) supplemented with 10% fetal bovine serum (Life Technologies) and 1% penicillin-streptomycin (Life Technologies) under a 5% CO2 atmosphere. Cells were plated on poly(d-lysine) (Sigma, Buchs, Switzerland)-treated culture dishes every 2 to 3 days after brief treatment with trypsin-EDTA (Life Technologies). For electrical recordings, cells were split, passed on acid-washed and poly(d-lysine)-coated coverslips, and used within 48 h. Nontransfected CHO cells did not display any voltage- or time-dependent current.
Electrical Recording.
CHO cells were placed in a perfused recording chamber RCP-10T (Dagan Co., Minneapolis, MN) on the stage of an inverted microscope (Eclipse; Nikon, Basel, Switzerland). Potassium outward current was measured, at 23–25°C, in the whole-cell configuration of the patch-clamp technique (Hamill et al., 1981). A quartz micromanifold (ALA Scientific Instrument Inc., New York, NY) was directed to individual cells to apply quickly various external drug-containing solutions. Pipettes were prepared from borosilicate capillary glass (Hilgenberg, Malsfeld, Germany) with a Sutter P-97 puller (Sutter Instrument Co., Novato, CA) and CPM-2 ALA microforge (ALA Scientific Instruments Inc.). Resistances of the patch pipettes were between 2 and 3 MΩ. Recordings were performed using an EPC 9 double patch-clamp amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany). Pipette potential and capacitance were nulled and a 5- to 15-GΩ seal formed with the cell membrane. Voltage-clamp protocols were applied using Pulse software (HEKA Elektronik) and whole-cell current recordings were displayed and analyzed using Pulse and Pulsefit software (HEKA Elektronik). Except when specified, the amplitude of the hKv1.5 current expressed in CHO cell line was assessed at the end of 250-ms command pulses with voltages applied every 15 s between −70 and +70 mV from a holding potential of −70 mV. Deactivation of tail current was recorded upon repolarization to −40 mV. Tail current amplitude was calculated as the difference between the peak amplitude of the tail and the sustained level of current after 200 ms of repolarization to −40 mV. Membrane potentials were corrected for a liquid junction potential between the pipette and the bath solutions. Capacitance and series resistance were optimized and ∼80% compensation was usually obtained. Average macroscopic current values were normalized for cell capacitance and expressed in picoamps per picofarad. All values are presented as mean ± S.E. Sixty-three CHO cells were clamped for this study and the average cell capacitance obtained was 19.3 ± 2.1 pF.
The standard bath solution used in the experiments contained the following: 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, 5 mM glucose; pH was adjusted to 7.3. The whole-cell pipette solution contained the following: 130 mM KCl, 1 mM MgCl2, 10 mM HEPES, 5 mM 1,2-bis(2-aminophenyoxy)ethane-N,N,N′,N′-tetracetic acid, 3 mM disodium ATP, 5 mM glucose; pH adjusted to 7.2. Disodium ATP and 1,2-bis(2-aminophenyoxy)ethane-N,N,N′,N′-tetracetic acid were obtained from Sigma. (1S,2S)-2-[{2-[3–2-benzymidazolyl)propyl]-methylamino}ethyl]-6-fluoro-1,2,3,4-tetrahydro-1- isopropyl-2-naphthyl-methoxyacetate dihydrochloride (mibefradil) was dissolved in H2O as 10 and 30 mM stock solution.
Analysis.
A first order-blocking scheme was used to describe drug-channel interaction. Apparent affinity constant,KD, and Hill coefficient,nH, were obtained from fitting of the fractional block, Y, at various drug concentrations [D]:
Voltage dependence of block was determined as follows: the current in the presence of drug was normalized to matching control to yield the fractional block Y at each control. The voltage dependence was fitted to the following equation:
Activation and inactivation curves were fitted with Boltzmann equations:
Statistical Method.
All values in the text in figures are presented as mean ± S.E. Direct comparisons between mean values in control conditions and in the presence of mibefradil for a single variable were performed by paired Student's t test. A level of P < .05 was considered to be statistically significant.
Results
Concentration-Dependent and Voltage-Dependent Block of hKv1.5 by Mibefradil.
The data in Fig. 1 show the effect of mibefradil on hKv1.5 current expressed in CHO cells under whole-cell recording conditions. Stably hKv1.5 transfected cells generated outward currents that rapidly rose to a peak and displayed no inactivation during 250-ms duration in control conditions. Outward current was followed by decaying outward tail current upon repolarization to −40 mV. No contaminating current was observed on control nontransfected cells under the experimental conditions used in this work (Fig. 1A). Representative families of whole-cell currents of a CHO cell transfected with hKv1.5 were evoked by series of 250-ms test pulses between −70 and +70 mV from a holding potential of −70 mV before and after applications of 0.1, 1, and 10 μM mibefradil. Steady-state inhibition was obtained after 2-min mibefradil application. Extracellular application of 1 μM mibefradil resulted in a statistically significant reduction of end-pulse hKv1.5 current at +50 mV (53.2 ± 6.8%; n = 6) with a marked increase in the rate of outward current relaxation in the presence of mibefradil. At 10 μM, mibefradil induced a reduction of peak and end-pulse current at +50 mV of 15.4 ± 3.8 and 94.8 ± 1.3%, respectively (n = 5). This means that steady-state currents recorded at the end of the 250-ms pulses were much more reduced, in a concentration-dependent manner, by mibefradil than the peak currents. In 10 μM mibefradil, peak current at the beginning of the trace was due to partial unblock during repolarization between depolarizing pulses. The effect of mibefradil was completely reversed upon washout of the drug (data not shown). Current-voltage relations in control conditions or in the presence of mibefradil for peak or end-pulse currents are represented in Fig. 1, B and C, respectively. All end-pulse I-V relationships were linear for depolarizations positive to −10 mV. It can be observed that end-pulse current was reduced in a concentration-dependent manner. Mibefradil (0.1 and 1 μM) reduced the amplitude of hKv1.5 at all voltages positive to 0 and −20 mV, respectively, as is evident from the change in end-pulse amplitude currents in the families of whole-cell current. At the opposite, both concentrations of mibefradil did not affect the amplitude of the peak current even at very positive voltages. In the presence of a higher concentration (10 μM) of mibefradil, the peak hKv1.5 current was also reduced but about 16-fold less than end-pulse current at +50 mV. Figure 1C shows that the two lower mibefradil concentrations induced a downward curvature of the current-voltage relationship, indicating a higher current inhibition at more positive potentials. Figure 1D shows the concentration dependence of mibefradil block of hKv1.5 in a range of concentrations between 0.1 and 30 μM. The fractional block of steady-state current measured at the end of 250-ms pulses was plotted against mibefradil concentration. A nonlinear least-squares fit of the concentration-response equation (eq. 1 underMaterials and Methods) to the individual data points yielded an apparent affinity constant (KD) of 0.78 ± 0.05 μM and a Hill coefficient of 0.97 ± 0.06. When the data were fitted with the Hill coefficient constrained to 1, a similar apparent affinity was obtained (KD= 0.79 ± 0.05 μM). These results suggested that binding of one molecule per channel was sufficient to block potassium permeation.

Inhibition by mibefradil of hKv1.5 expressed in mammalian CHO cell line. A, representative families of current traces elicited by depolarizing voltage pulses of 250 ms in 10-mV steps from a holding potential of −70 mV to + 70 mV in control conditions and in the presence of 0.1, 1, and 10 μM mibefradil. No endogenous current was recorded from nontransfected CHO cells. B and C, current amplitude measured at the peak and at the end of the depolarizing pulse plotted against the pulse potential in control and in the presence of 0.1, 1, and 10 μM mibefradil. D, dose-response relationship for mibefradil block. Mean data of end-pulse current amplitude obtained from five to seven cells using the same protocol as shown in A (+50 mV) were averaged and plotted against mibefradil concentration. A nonlinear least-squares equation best fitted the data points with a free Hill coefficient (continuous line) and with a Hill coefficient fixed to 1 (dashed line).
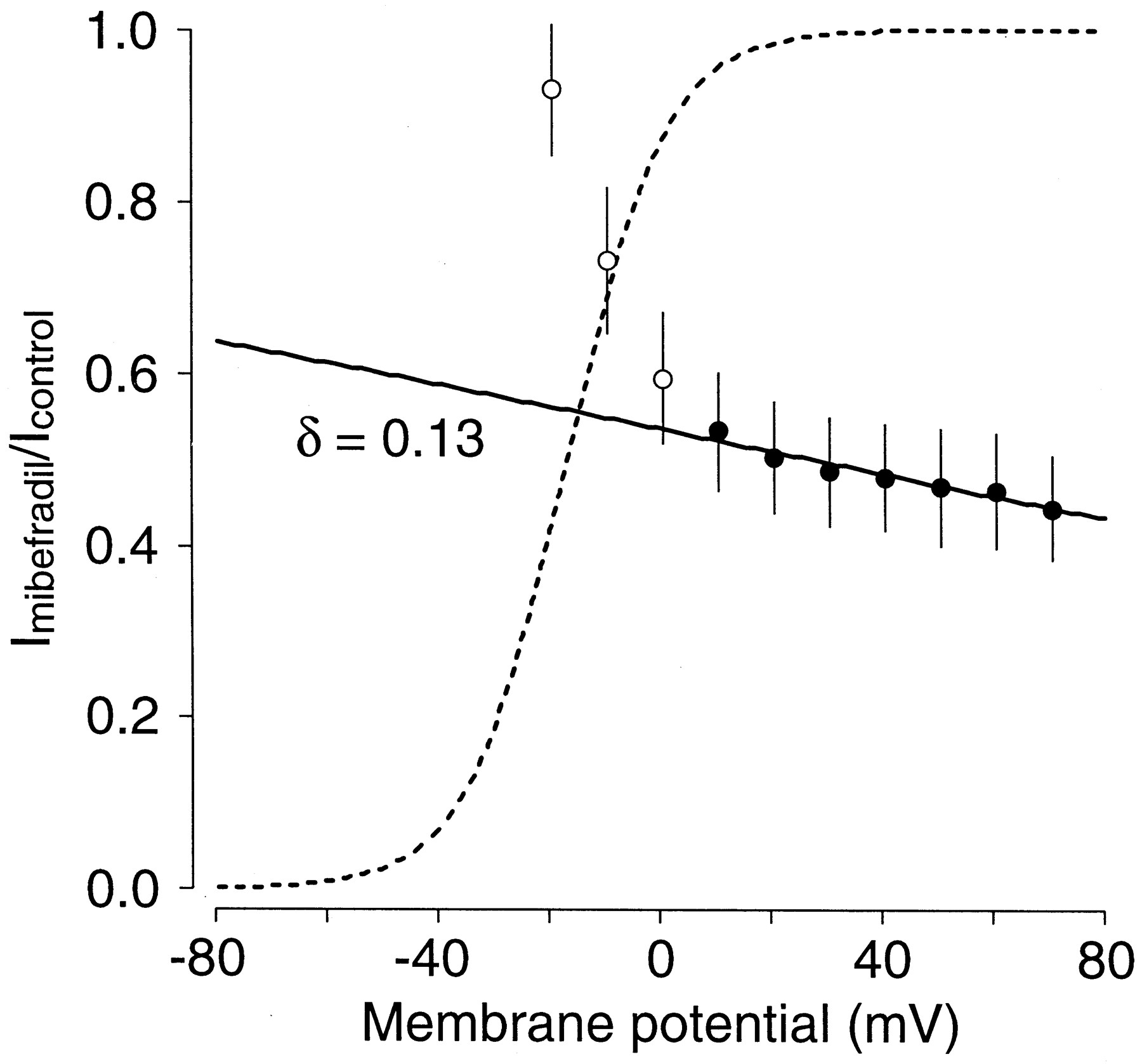
To quantify the voltage dependence of inhibition suggested by the data from Fig. 1, the relative current inhibition induced by 1 μM mibefradil was plotted as a function of voltage (Fig.2). The dotted line represents the activation curve obtained in control conditions. The current began to activate at −40 mV and the conductance of the channel was fully saturated at +10 mV. In the presence of mibefradil the blockade increased steeply between −20 and +10 mV, which corresponds to the voltage range of channel opening (Snyders et al., 1993). These results suggest that mibefradil bound preferentially to the open state of the hKv1.5 channels. Between +10 and +70 mV, block continued to increase but with a shallower voltage dependence. It is unlikely that this shallower voltage dependence at higher voltages was due to channel gating because hKv1.5 activation had reached saturation over this range of voltages. Mibefradil contains a tertiary amine with a pKa value of 8.8. Therefore, in our experiments at intracellular pH (7.2), the drug was predominantly present in the charged form. Thus, the shallow component of the voltage dependence was probably due to the influence of the transmembrane electrical field on the interaction between the charged form of the drug and the channel receptor. The simple Woodhull model (eq. 3 underMaterials and Methods), which incorporates the effect of the transmembrane electrical field could explain this effect. The parameterδ in this equation represents the fractional electrical distance, i.e., fraction of the membrane field sensed by the positive charge at the receptor site. The line in Fig. 2 represents the fit to the data points positive to +10 mV (solid symbols) and the fractional electrical distance was found to be δ = 0.13. The average value in five individual experiments was 0.13 ± 0.05.

Voltage dependence of hKv1.5 block. Relative current expressed as Imibefradil/Icontrol from data obtained in the absence and in the presence of 1 μM mibefradil. Individual data points are mean ± S.E. of five to six experiments. The dotted line shows the activation curve of the hKv1.5 channel in control conditions. Block steeply increased between −20 and +10 mV. For membrane potential positive to +10 mV, a continued but shallower voltage dependence was observed. This voltage dependence was fitted (continuous line) to eq. 3 (under Materials and Methods) and yielded a δ value of 0.13.
Time-Dependent Effects of Mibefradil on hKv1.5 Channels.
If mibefradil could access its channel receptor site only when the channel was in the open state, then inhibition of the hKv1.5 current would only develop as the channel would start to open, and block development should be visible if the blocking rate was slower than the opening rate. In the presence of mibefradil the inactivation time course displayed an additional rapid exponential component superimposed on the slow inactivation. The decay of the current was then best fitted by a biexponential function. The time constant of the rapid component was concentration dependent and it was considered to represent the time constant of development of block (τBlock). In Fig. 3, the τBlock was plotted as a function of mibefradil concentration and the experimental data were fitted to a hyperbolic function (eq. 2a under Materials and Methods). Following this approach we were able to calculate the apparent association (k) and dissociation (l) rate constants that averaged to 7.3 ± 0.5 × 106M−1 · s−1 and 4.3 ± 0.1 s−1, respectively. Following the previous assumption of a first order reaction drug/channel interaction, the ratio between l and k values would give the apparent dissociation constant, KD*(eq. 2b under Materials and Methods). This estimate was independent of the apparent KD obtained from the curve in Fig. 1D. Nevertheless, the derived value (0.58 μM) was in the same range with that calculated from the fit of the concentration-response curve. This correlation between both independent methods to estimate the affinity supports the open-channel block model used to derive the rate constants. Binding rates can also be derived from the apparent KD* and the average value of τBlock at 1 μM (0.092 ± 0.007 s). Using this procedure, the calculated k and lvalues were 6.1 × 106M−1 · s−1 and 4.8 s−1, respectively.

Kinetics of mibefradil block of hKv1.5. The mibefradil-induced fast time constant at +50 mV has been plotted against the mibefradil concentration. The best fit to the data (solid line) using the eq. 2a (under Materials and Methods) resulted in an apparent association (k) and dissociation (l) rate constant of 7.3 ± 0.5 × 106M−1 · s−1 and 4.3 ± 0.1 s−1, respectively. The calculated KD value was found to be 0.58 μM (n = 5–7 cells).
The time-dependent effects induced by mibefradil were also studied on the deactivation process of hKv1.5 channels, which represented the transition from the open to the closed state of the channel. Figure4 shows the superposition of the tail current obtained upon repolarization at −40 mV after 250-ms depolarization to +50 mV under control conditions and in the presence of 0.1 and 1 μM mibefradil. Deactivation decay was best fitted by a biexponential function. In control conditions, the channel deactivated with a fast time constant of 13.8 ± 1.1 ms (n = 6) and a slow time constant of 48.4 ± 5.7 ms (n = 6). In presence of mibefradil, the initial amplitude of the tail currents was significantly reduced. The subsequent decline of the tail current was slower than in control conditions, which resulted in a “crossover” phenomenon. This slower time course of deactivation can be attributed to the drug-induced block of the available open channels at −40 mV. Thus, in the presence of 0.1 μM mibefradil the fast component of deactivation was not significantly changed (16.6 ± 1.1 ms; n = 6) but the slow component was significantly increased to 81.9 ± 10.8 ms (n = 6;P <.05). These results indicated that drug unbinding was necessary before channels could close and supported an open channel interaction with mibefradil.

Deactivation tail current crossover caused by mibefradil. Tail currents were recorded under control conditions and in the presence of 0.1 and 1 μM mibefradil. Tail currents were obtained at −40 mV after a 250-ms depolarizing pulse to +50 mV from a holding potential of −70 mV.
Effects of Mibefradil on Activation and Inactivation Voltage Dependence.
Normalized currents generated in the presence of mibefradil showed a leftward shift of activation on the voltage axis. Activation was determined from measurements of tail currents recorded from CHO cells at −40 mV after depolarizing steps between −70 and +70 mV. Because the driving force was constant during these measurements, the activation curve reflected the fraction of channel open at each membrane potential. Tail current amplitude was normalized to peak tail current amplitude and plotted against the potential of the depolarizing step (Fig. 5A). The data were best fitted with a single Boltzmann function (eq. 4a under Materials and Methods). There was a clear concentration-dependent shift of the maximum potential to the left in the presence of mibefradil, and this effect seemed to be greater as the potential became more positive and reached 0 mV. At the foot of the curves where the channels were closed, a much smaller shift was observed. This point is illustrated by the statistically significant differences in the values for the half-activation (V0.5) and slope factor (k) of the Boltzmann functions in legend of Fig. 5. This shift could be explained by mibefradil binding preferentially to the channel open state, thus limiting a hKv1.5 conductance increase at more depolarized test potentials. After conductance of the channel was saturated, we observed a decrease of the normalized current at larger potentials. Due to more inactivation at positive potential and to the voltage dependence of mibefradil block, the magnitude of the tails was probably underestimated at higher voltages. The mid-point of the activation curve in control conditions did fit with data previously reported for hKv1.5 expressed in CHO cells (Philipson et al., 1993).

Voltage dependence of hKv1.5 block. A, activation curves were drawn by fitting normalized tail currents to the Boltzmann equation (eq. 4a, under Materials and Methods). Data are mean ± S.E. from five cells.V0.5 and slope factor (k) are −17.2 ± 0.7 mV and 8.9 ± 0.6, −24.1 ± 1.6 mV and 6.1 ± 1.3, and −29.8 ± 3.1 mV and 3.5 ± 3.8 in control conditions, and in the presence of 0.1 and 1 μM mibefradil, respectively. B, steady-state availability relations for hKv1.5 channels were obtained by plotting current amplitude during test pulse at +30 mV against the voltage of a 10-s prepulse. The data points were fitted with the Boltzmann equation (eq.4b, under Materials and Methods). Data represent mean ± S.E. from five cells. V0.5 and slope factor (k) were −26.3 ± 0.6 mV and 6.3 ± 0.5 in control conditions and −31.3 ± 0.5 mV and 6.5 ± 0.4 in the presence of 1 μM mibefradil, respectively.
Figure 5B also includes steady-state availability of the expressed current determined with a double pulse protocol. Ten-second prepulses to potentials between −80 and +30 mV in 10-mV intervals were applied every 45 s. Each prepulse was followed by a brief 5-ms hyperpolarizing step to −130 mV and a 200-ms test pulse to a constant voltage of +30 mV. Average values for peak outward current during the test pulse were normalized to the maximum peak value and plotted against prepulse potential. Due to the slow inactivation kinetic of hKv1.5 in control conditions, it was impossible to achieve a true steady-state condition for the control whole-cell current. A greater level of residual current was apparent at the end of the prepulse in control conditions (approximately 40% compared with 30% in the presence of 1 μM mibefradil). Single Boltzmann functions (eq. 4bunder Materials and Methods) were found to be the best fit for both set of data. Half-maximal inactivation occurred at the voltage of −26.3 ± 0.6 mV in control conditions. This value was in agreement with the values reported in other studies on hKv1.5 expressed in CHO cells (Philipson et al., 1993). Half-maximal inactivation occurred at the voltage of −31.3 ± 0.5 mV (n = 5; P <.05) after 1 μM mibefradil treatment.
Use-Dependent Block of hKv1.5 by Mibefradil.
We next examined whether mibefradil displayed any use-dependence effects on hKv1.5 current. In these experiments, trains of 10 depolarizing pulses of 125-ms duration from −70 mV to +50 mV were applied at three different stimulation frequencies, 0.02, 0.2, and 2 Hz, in the absence and presence of 1 μM mibefradil. Each train was separated from the others by a rest period of 2 min. Figure 6A shows original current recordings obtained after such pulse train was applied at a frequency of 2 Hz in the absence and in the presence of mibefradil. Under control conditions, the current displayed a small decline (17.7 ± 1.3%; n = 4) probably due to the fact that not enough time was allowed for full recovery of inactivation between two pulses. After 2-min exposure to 1 μM mibefradil, this pulse protocol was repeated. After the perfusion with mibefradil, the peak current amplitude elicited by the first depolarizing pulse in each train was not significantly modified, showing the absence of tonic block. In the presence of the drug, the size of the current decayed progressively until it reached a steady-state block. Measurement of the peak current amplitude showed a statistically significant use-dependent block at all frequencies of stimulation (Fig. 6B). To describe the use-dependent effect of mibefradil without complications of the decline of the current under control conditions, Fig. 6C represents the relative current (Imibefradil/Icontrol) elicited during the application of these pulse protocols as a function of the number of pulses in the train. As it is shown, the degree of frequency-dependent hKv1.5 block was increased with the driven rate.

Use-dependent block of hKv1.5 current induced by mibefradil. A, original records obtained in the absence and in the presence of 1 μM mibefradil when applying 10 depolarizing pulses (125 ms in duration) from −70 to +50 mV at a frequency of 2 Hz. B, peak outward current amplitude measured at 0.02, 0.2, and 2 Hz in control conditions and at 0.02, 0.2, and 2 Hz in the presence of 1 μM mibefradil and expressed as the pulse number in the train. C, relative current (Imibefradil/Icontrol) at 0.02, 0.2, and 2 Hz was plotted as a function of the pulse number in the train.
Recovery from Inactivation of hKV1.5.
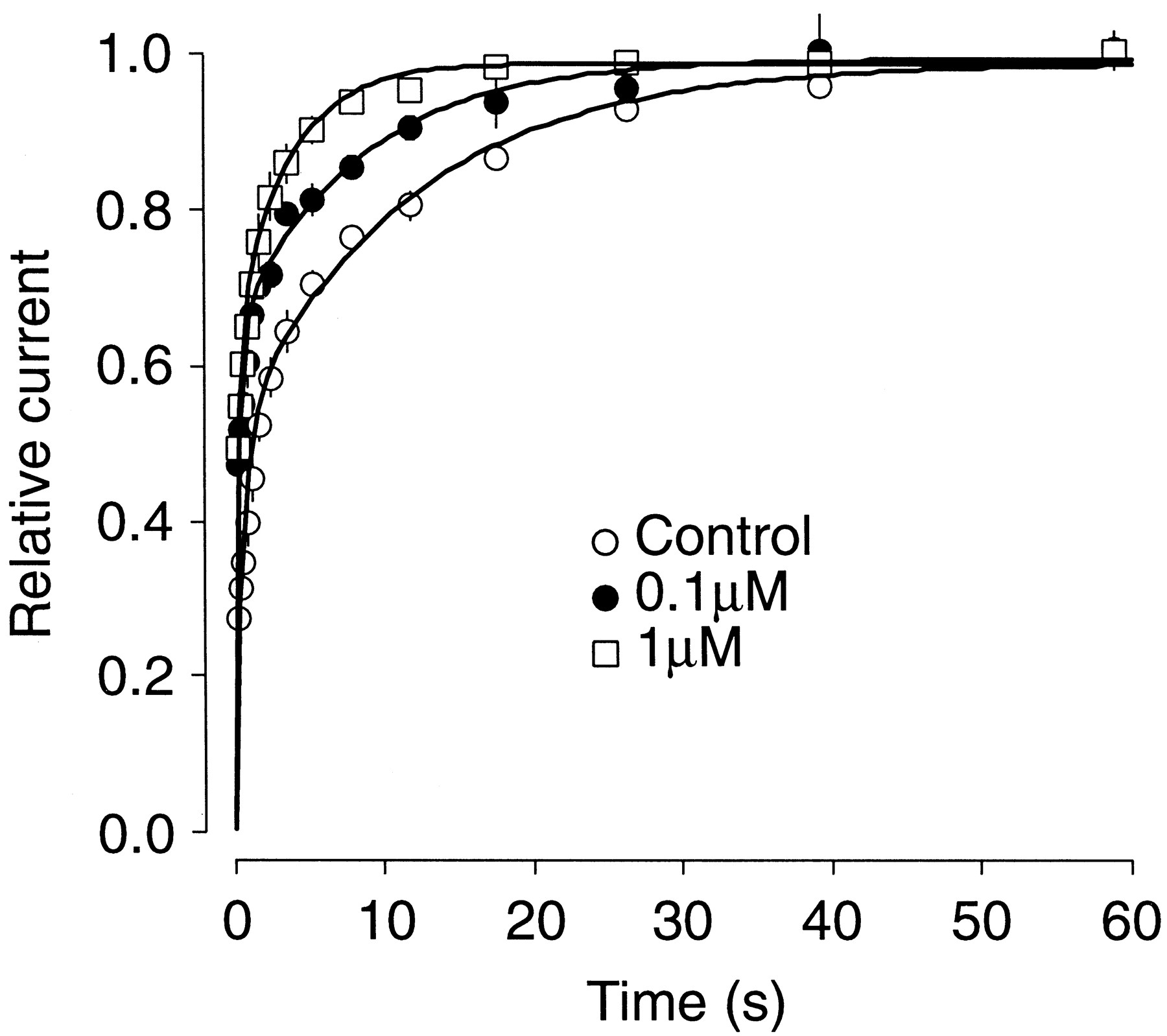
Recovery from inactivation was studied using a double-pulse protocol: whole-cell current was inactivated by a 4-s prepulse to +40 mV and the extent of recovery from inactivation determined by applying a second 0.7-s pulse to the same voltage after a variable time at −70 mV between 0.4 and 60 s. The very slow inactivation kinetic precluded measurements of recovery from true steady-state inactivation. The average percentage (n = 5) of recovery between the prepulse and each second pulse was plotted against the interpulse interval duration in Fig. 7. In control conditions, the data was best fit with a biexponential function with a slow time constant (τS) of 11.69 ± 0.11 s and a fast time constant (τF) of 0.64 ± 0.01 s (n = 5; Fig. 7). Paradoxically, in the presence of mibefradil, recovery from inactivation was faster. The recovery process was also defined by a biexponential process that exhibited a τF of 0.33 ± 0.02 and 0.30 ± 0.02 s in the presence of 0.1 and 1 μM mibefradil, respectively. τS decreased significantly to 8.22 ± 0.15 and 3.5 ± 0.26 s in the presence of 0.1 and 1 μM mibefradil, respectively (P < .05; n = 5).

Recovery from inactivation. Whole-cell current was inactivated by a 4-s prepulse to +40 mV and the extend of recovery from inactivation determined by applying a second 0.7-s pulse to the same voltage after a variable interval at −70 mV between 0.4 and 60 s. Fractional recovery from inactivation was plotted as a function of interpulse duration for hKv1.5 in control and in the presence of 0.1 and 1 μM mibefradil. The continuous lines represent double exponential functions that were the best fit to the data points (n = 5 cells).
Discussion
Mibefradil is described as a CCB with an approximately 10-fold higher sensitivity to T-type Ca2+ channels than other cloned, L-, N-, R-, P-, and Q-type channels (Bezprozvanny and Tsien, 1995). Mibefradil blockade of cloned cardiac potassium channels has also been reported (Chouabe et al., 1998). We show here that mibefradil is a potent inhibitor of hKv1.5 channels in the range of concentrations used as CCB in previous studies (Mishra and Hermsmeyer, 1994; Clozel et al., 1997).
The inhibition of hKv1.5 by mibefradil is characterized by a concentration-dependent reduction in current amplitude and an acceleration of the apparent rate of current inactivation. These effects are similar to those found with CCBs on hKv1.5 such as verapamil (Rampe et al., 1993), diltiazem (Grissmer et al., 1994), and nifedipine (Zhang et al., 1997). The characteristics of block on hKv1.5 strongly suggest that mibefradil blocks the open state of the channel. Evidence for this includes the following: 1) mibefradil accelerated the initial apparent inactivation rate of hKv1.5 with the appearance of a block time constant; 2) at the onset of the depolarizing pulse there was no inhibition of hKv1.5 at the lower dose of mibefradil, indicating that mibefradil does not bind channels in the rested state; 3) a close correlation between the voltage dependence of current activation, the steady-state availability and mibefradil-induced block; and 4) a crossover phenomenon in tail currents was observed.
Drugs that interact predominantly with open state of the channel can do so by moving into the ion-conducting pore. Mibefradil is a weak base, with a pKa of 8.8; therefore, the drug is in its charged form at pH 7.2. Access of the charged form of the drug to the receptor site likely requires that the drug moves into the electrical membrane field and the block should increase upon depolarization (Snyders et al., 1992). The voltage dependence of mibefradil block is composed of a very steep phase parallel to the voltage dependence of activation of the current and a shallower phase that reflects an additional effect of the electrical field on the charged drug. The fractional electrical distance defines the effect of the electrical field on the interaction between the drug and the receptor located in the channel. The δ value of 0.13 observed in the presence of mibefradil suggests that the drug moves about 13% into the membrane electrical field to reach the receptor.
We studied the effect of frequency on the degree of block induced by mibefradil by applying trains of depolarizations at different frequencies. Our results indicate that block of hKv1.5 increased as the frequency of stimulation was augmented. This could be explained by the mechanism of mibefradil block. Indeed, at high frequencies, more openings are available for mibefradil to interact with the channel in its conducting state. However, the peak current elicited by the first depolarization of each train of stimuli in the presence of mibefradil was similar to the peak current amplitude obtained in control conditions. This result illustrates the absence of “tonic block” with mibefradil and shows that mibefradil does not bind on the channel in the closed (resting) state.
Our data suggest that mibefradil competes with the inactivation gate of hKv1.5 channel. A key experiment that supports this proposal was the effect of mibefradil on the kinetics of the recovery from inactivation process. Under control conditions, the recovery was defined by a double exponential process that exhibited a slow and a fast time constant. In the presence of mibefradil, this recovery was also biexponential but the time constants significantly decreased, indicating a faster recovery kinetic of the hKv1.5 current. At positive potential, channel closure in the absence of drug occurs preferentially by inactivation. Hence, in control conditions, a prolonged depolarization results in a slow inactivation. Rather, in the presence of mibefradil, drug block cannot occur until the channel opens, and the decay of the current would be initially accelerated by mibefradil because the open channel can close by two pathways: inactivation and mibefradil block. If a mutually exclusive interaction exists between drug block and channel inactivation, blocked channels cannot inactivate and inactivated channels cannot be blocked. It is tempting to postulate that blocked channels are protected from inactivation. Although neither blocked channels nor inactivated channels could carry current, the blocked channels are in rapid equilibrium with the pool of open channels and thus are only “temporarily” nonconducting. Inactivated channels, on the other hand, do not return rapidly to the open channel pool and are “permanently” nonconducting. Grissmer and Cahalan (1989) showed that the slow inactivation process seen in a delayed rectifier potassium channel in lymphocytes was consistent with a model in which the channel could inactivate from the open state but not from the blocked state. Internal tetraethylammonium exerts similar effects on the inactivation process of the cloned shaker H4 potassium channel (Choi et al., 1991).
Mibefradil is accepted as a selective T-type Ca2+inhibitor (Clozel et al., 1997). Indeed, depending on the cell type, mibefradil blocks T-type Ca2+ channels 10 to 30 times more potently than L-type Ca2+ channels (Mishra and Hermsmeyer, 1994). Mibefradil is a potent vasodilator with antianginal, antihypertensive, and anti-ischemic properties. In addition, mibefradil is better tolerated than classical CCBs because it has much fewer side effects: at therapeutic doses, mibefradil decreases heart rate slightly but it neither reduces cardiac contractility nor induces reflex tachycardia (Kobrin et al., 1997). However, its interactions with other drugs, via potent cytochrome P450 inhibition, have led to the withdrawal of mibefradil from the market (Po and Zhang, 1998). T-type Ca2+ channels might be the major target of mibefradil, but given the multifunctional effects of mibefradil, it is unlikely that its action would be limited only to these channels. It has been shown that mibefradil inhibits Ca2+-activated and volume-activated Cl− channels in microvasvular cells (Nilius et al., 1997). Mibefradil also blocks in a concentration-dependent manner the cloned HERG and KvLQT1/IsK K+ channels expressed in COS cells (Chouabe et al., 1998). A study shows that mibefradil interferes with myoblast fusion, suggesting that the drug exerts this effect by inhibiting in combination L-, T-type Ca2+ channel and a delayed rectifier K+ channel, an HERG K+channel, and an inward K+ channel (Liu et al., 1999). Furthermore, data show that mibefradil can limit infarct size through a glibenclamide-sensitive mechanism (Mocanu et al., 1999).
It has been shown that, in human atrial myocytes, in which hKv1.5 is thought to underlie IKur, mibefradil shortens the plateau of action potentials but does not change the duration of the late repolarization (Bénardeau et al., 1999). This observation is consistent with a predominant block of L-type Ca2+ channel, which governs plateau duration (Coraboeuf and Nargeot, 1993). It cannot entirely exclude that block of other channels contributes to the action of mibefradil. Block of T-type Ca2+ channels could reduce the inward current early during the action potential but this channel has never been observed in human cardiomyocytes. Block of IKurmight partially counteract block of L-type Ca2+current but remains dominated by it because action potential does not lengthen in the presence of mibefradil.
The mean effective therapeutic plasma concentration of mibefradil ranged from 193 to 762 μg/l (Bernink et al., 1996; Oparil et al., 1997) and because it is about 99% bound to plasma protein (Skerjanec et al., 1996), in vitro concentrations of 0.03 to 0.2 μM probably correspond in action to clinically effective free-drug concentrations. Mibefradil (1 μM) has been found to block about 50% of T-type Ca2+ channels (Mishra and Hermsmeyer, 1994) and HERG channels (Chouabe et al., 1998), and KvLQT1/IsK channels were inhibited with a KD value of 12 μM (Chouabe et al., 1998). In our study, theKD value for mibefradil was 0.78 μM. Although the different experimental conditions could modify theseKD values, these results would suggest that mibefradil blocks Ca2+ and K+ channels only at concentrations much higher than the therapeutic plasma levels. However, it has been demonstrated that mibefradil accumulates into peripheral tissues and that the volume of distribution at steady state is larger than the plasma volume, ranging from 130 to 220 liters (Abernethy, 1997). These data suggest that mibefradil may reach cardiac concentrations that correspond to the range at which the drug can block cardiac ion channels.
It was hypothesized that many of the features of mibefradil might be related to the compound's effect on T-type Ca2+channels. Mibefradil's characteristics include a slight heart-rate-lowering effect (Veniant et al., 1993); no clinically relevant negative inotropism, presumably because normal ventricular myocytes contain mainly L-type Ca2+ channels (Bogdanov et al., 1995); coronary and peripheral vasodilatation (Orito et al., 1993); and the absence of reflex increases in neurohormones and sympathetic activity. Mibefradil was the first available compound with these characteristics and with this mechanism of action; however, the complex relation between the blockade of T-type Ca2+ channels and the resultant clinical effects have not yet been fully elucidated. From our data and previous studies, it is more likely that the beneficial effects of mibefradil involve a complex interaction of the drug with multiple ion channels.
Acknowledgments
We thank Laurence Hilfiger and Michael Weber for excellent technical assistance.
Footnotes
-
Send reprint requests to: Odile Clément-Chomienne, Preclinical Research, F. Hoffmann-La-Roche Ltd., PRBM-M Bau 70/423, CH-4070 Basel, Switzerland. E-mail:odile.chomienne{at}roche.com
- Abbreviations:
- CCB
- calcium channel blocker
- IKr
- rapid component of the cardiac delayed rectifier potassium current
- IKs
- slow component of the cardiac delayed rectifier potassium current
- Ito
- native transient outward current
- IKur
- ultra-rapidly activating cardiac delayed rectifier K+ current
- hKv1.5
- human cardiac Kv1.5 channel
- CHO
- Chinese hamster ovary
- Received April 14, 2000.
- Accepted July 5, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}