


Abstract
The β2-adrenergic receptor (β2-AR)-mediated increase in cardiac L-type Ca2+ current (ICa,L) has been documented in normal subjects. However, the role and mechanism of β2-AR activation on ICa,L in heart failure (HF) are unclear. Accordingly, we compared the effect of zinterol (ZIN), a highly selective β2-AR agonist, on ICa,L in isolated left ventricular cardiomyocytes obtained from normal control and age-matched rats with HF induced by left coronary artery ligation (4 months). ICa,L was measured by using the whole-cell voltage-clamp technique. In normal myocytes, superfusion of ZIN (10−5 M) caused a 21% increase in ICa,L (9.21 ± 0.24 versus 7.59 ± 0.20 pA/pF) (p < 0.05). In HF myocytes, the same concentration of ZIN produced a significantly greater increase (30%) in ICa,L (6.20 ± 0.24 versus 4.75 ± 0.17 pA/pF) (p < 0.01). This ZIN-induced increase in ICa,L was further augmented in both normal and HF myocytes (normal: 59 versus 21%; HF: 71 versus 30%) after the incubation of myocytes with pertussis toxin (PTX, 2 μg/ml, 36°C, 6 h). These effects were not modified by the incubation of myocytes with CGP-20712A (3 × 10−7 M), a β1-AR antagonist, but were abolished by pretreatment of myocytes with ICI-118551 (10−7 M), a β2-AR antagonist. In addition, all of the effects induced by ZIN were completely prevented in the presence of an inhibitory cAMP analog, Rp-cAMPS (100 μM, in the patch-pipette solution). In conclusion, β2-AR activation stimulates L-type Ca2+ channels and increases ICa,L in both normal and HF myocytes. In HF, β2-AR activation-induced augmentation of ICa,L was increased. These effects are likely to be mediated through a cAMP-dependent mechanism and coupled with both stimulatory G protein and PTX-sensitive G protein.
β-Adrenergic receptor (AR) stimulation modulating cardiac L-type Ca2+ current (ICa,L) plays an important role in the positive inotropic response to β-AR stimulation, and it has been shown to be impaired in heart failure (HF) myocytes (Richard et al., 1998). Besides β1-AR stimulation of L-type Ca2+ channels, which is mediated by a cAMP/PKA-signaling mechanism, the β2-AR-mediated increase in cardiac ICa,L has been documented in normal subjects (Xiao and Lakatta, 1993; Cerbai et al., 1995; Xiao et al., 1995;Skeberdis et al., 1997). It is proposed that cardiac β2-AR couples with both Gs and Gi protein (Xiao et al., 1999). The β2-AR-coupled Gi activation functionally localizes the Gs-mediated adenylyl cyclase-cAMP/PKA signaling (Zhou et al., 1997), thereby negatively regulating the Gs-mediated ICa,L and contractile response in the heart (Xiao et al., 1995). Chronic HF is associated with a marked increase in Gi protein and a selective down-regulation of β1-AR (higher β2/β1). The exaggerated β2-AR/Gisignaling may contribute to the HF-associated dysfunction of β-AR stimulation (Xiao et al., 1999). Previously, Bristow et al. (1989)reported a diminished β2-AR-mediated cardiac response by documenting a decreased adenylate cyclase stimulation. However, we and other investigators have observed an increased positive inotropic responsiveness to β2-AR stimulation in dogs with pacing-induced HF (Altschuld et al., 1995; Cheng et al., 1998) and in left ventricular (LV) myocytes from rats with infarction-induced HF (Ukai et al., 1999). This effect may be due to a β2-AR stimulation-induced alteration in the regulation of the Ca2+ channel. However, the role and mechanism of β2-AR activation on ICa,L in HF have not been previously assessed.
Accordingly, the purpose of this study was to compare the effect of β2-AR stimulation on cardiac ICa,L in LV myocytes of normal rats and rats with HF and to determine the underlying cellular mechanism. Our results indicate that the β2-AR activation-induced augmentation of ICa,L was enhanced in the HF myocytes. These effects are likely to be mediated through a cAMP-dependent mechanism and coupled with both Gsand PTX-sensitive Gi protein.
Experimental Procedures
Experimental Heart Failure Model.
The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH Publication 85-23, revised 1985).
The rat HF model was prepared by surgical ligation of the coronary artery (Dixon et al., 1990). Male Sprague-Dawley rats weighing 220 to 270 g were anesthetized with ether, intubated, and ventilated. The left coronary artery was ligated approximately 2 to 3 mm distal to its origin from the ascending aorta to produce approximately 40% infarction of the LV myocardium. The mortality was approximately 52% within 48 h. Sixteen weeks after myocardial infarction (MI), survivors (n = 21) and sham-operated rats (n = 22) were lightly anesthetized with intraperitoneal ketamine hydrochloride (50 mg/kg) and xylazine hydrochloride (10 mg/kg). Then hemodynamic measurements were performed by using a microtip pressure transducer (Millar Instruments, Houston, TX) inserted into the left ventricle through the carotid artery to verify the presence of HF in the rats with induced MI. Heart weight and lung weight were obtained after the study.
Isolation of LV Myocytes.
Myocytes were enzymatically disassociated by Langendorff perfusion as we previously described (Suzuki et al., 1998). Care was taken to excise the infarct scar in the heart of each rat with MI before the final enzymatic digestion step. With this technique, the yield of viable myocytes was greater than 80% from both control rats and rats with MI. The cells were used within 10 h.
Electrophysiological Measurement.
Membrane calcium current was recorded at 22 to 23°C with the whole-cell patch-clamp technique (Hamill et al., 1981). Axopatch 200A amplifier (Axon Instruments, Foster City, CA) was interfaced with a 12-bit A/D-D/A converter (Digidata 1200; Axon Instruments). PClamp software (PClamp 6.02; Axon Instruments) was used for data acquisition and analysis. Data were filtered by means of a 5-kHz low-pass filter and digitized at 5 kHz.
After stabilization, a drop of cell pellet containing the isolated myocytes was placed in a perfusion chamber with a volume of 0.5 ml mounted on the stage of an inverted microscope (IMT 2-F3; Olympus, Herndon, VA). Myocytes were allowed to adhere to the bottom of the chamber for 5 to 10 min and then continuously superfused at a constant rate of 2 ml/min. Only quiescent rod-shaped cells lacking membrane deformities and showing clear cross striations were studied. A borosilicate glass micropipette with an outside diameter of 1.6 mm was pulled with a two-step puller (model PP-83; Narishige, Tokyo, Japan) and heat-polished with a microfuge (MT-83; Narishige). The tip resistances were of 1.5 to 2.5 MΩ when filled with pipette (internal) solution.
Liquid junction potentials (<5 mV) were corrected before the pipette touched the cell. The formation of a GΩ seal was followed by electronic compensation of the electrode capacitance. Then the cell membrane was ruptured by means of gentle suction to establish the whole-cell configuration. Compensation of the membrane capacitance and series resistance was performed to minimize the duration of the capacitive transient. The membrane capacitance was measured, before compensation, by a 10-mV depolarizing step from a holding potential of −80 to −70 mV and integrating the area under the current transient calculated by the following formula:
Myocytes were voltage-clamped at −80 mV. Ca2+currents were elicited by stepping the membrane voltage from the holding potential to the 0 mV testing potential for 200 ms at 0.1 Hz. To avoid contamination by fast sodium channel activation and to reduce the run-down of ICa,L, a brief (60 ms in duration) prepulse was applied to −40 mV before stepping to the test potential. The average peak ICa,L run-down was about 10 to 20% during the 30 min after the initial measurement. Most (80%) of the run-down occurred within the initial 8 to 10 min. Thus, a time window of 10 to 30 min after the initial recording was chosen to measure ICa,L with respect to drug effects (Xiao and Lakatta, 1993). ICa,L was measured by the standard method as the difference between the peak inward current and the current at the end of the 200-ms pulse. For current-voltage relations, test potentials were from −35 to +60 mV at 5-mV increments and 0.1 Hz.
Solutions.
The composition of the pipette solution and that of the recording bath solution were chosen to allow isolation of ion flow through Ca2+ channels by blocking other ionic currents. Initially, the myocytes were superfused with a modified Tyrode's solution containing 137 mM NaCl, 5.4 mM KCl, 1.2 mM MgSO4, 15 mM glucose, 10 mM HEPES, and 1.5 mM CaCl2. The pH was adjusted to 7.4, with NaOH at 20 to 22°C. After formation of a GΩ seal, the perfusion buffer was changed to a patch recording bath solution, i.e., Na+- K+-free Tyrode's solution in which tetraethylammonium chloride was substituted for NaCl, 50 μM tetrodotoxin was added to eliminate sodium current, and KCl was replaced by CsCl and 3 mM 4-aminopyridine to abort the potassium current. The solution was gassed with 100% O2. The internal solution for the pipette contained 140 mM cesium aspartate, 1.0 mM MgCl2, 3 mM Na2ATP, 0.4 mM GTP, 10 mM EGTA, and 5 mM HEPES. The pH was adjusted to 7.2 (with titrated CsOH).
Materials.
Zinterol {MJ-9184-1; N-(5(2-[1,1 dimethyl-2-phenyl-ethyl] amino)-1-hydroxyethyl)-2-hydroxyphenyl]-monohydrochloride} was provided by Bristol-Myers Squibb Co. (Princeton, NJ); ICI-118,551 and CGP-20712A (CGP) was obtained from RBI/Sigma, Natick, MA; and Rp-cAMPS was from Biology Life Science Institute, La Jolla, CA. Other agents were obtained from Sigma, St. Louis, MO.
Statistical Analysis.
Data are presented as mean ± S.E.M. Statistical comparisons were performed with Student'st test or analysis of variance. A p value of <0.05 was considered significant. Prism 3.0 (GraphPad software; GraphPad, San Diego, CA) was used for the concentration-ICa,L relationship nonlinear regression analysis. As previously described by Robberecht et al. (1983) and Lands et al. (1967), the Hill equation may allow us modeling cooperatively between multiple receptor sites on each cardiomyocyte with respect to β2-AR agonist (ZIN) binding. Thus, data were fitted with the Hill equation. The best fit by the Hill equation was also compared with a fit by one-site competition equation.
Results
Verification of Experimental HF
The general hemodynamic and ICa,L features in the rats with MI are presented in Table1. LV end-diastolic pressure was increased 5-fold; left ventricular systolic pressure, LV dp/dtmax, and LV dp/dtminwere significantly decreased. The rate of LV relaxation was slowed, as indicated by a significant increase in the time constant of isovolumic LV pressure decay (τ, 196%, p < 0.05).
Characteristics of LV function and Ca2+ current of LV myocytes in myocardial-infarction-induced heart failure rats
All of the animals with MI had clear evidence of HF (anorexia, edema, and pulmonary congestion). There were no significant changes in body weight (593 ± 9 versus 598 ± 7 g, p = N.S.), whereas the heart weight (2.21 ± 0.03 versus 1.62 ± 0.02 g, p < 0.05), the calculated ratio of LV-to-body weight (2.50 ± 0.04 versus 1.85 ± 0.03 g/kg,p < 0.05), and the wet lung-to-body weight ratio (4.83 ± 0.10 versus 2.87 ± 0.07 g/kg, p < 0.05) were all significantly increased in the rats with MI. In the myocytes of rats with HF, the membrane capacitance was significantly increased (66%), indicating an existence of hypertrophy of the myocytes. After normalized by the membrane capacitance, the current density was significantly lower than that of the normal myocytes (62%,p < 0.01), indicating an absolute reduction of ICa,L.
In addition, the response of ICa,L to β-adrenergic receptor stimulation in HF myocytes was significantly attenuated. The exposure to isoproterenol (ISO, 10−7 M) increased the ICa,L by 120 ± 14% (p < 0.05, n = 10) in the normal myocytes. However, in the myocytes of rats with HF, the increase in ICa,Lwith ISO was only half (56 ± 8%, p < 0.05,n = 9). The current was blocked by nifedipine (5 × 10−6 M), a Ca2+ channel blocker, consistent with the characteristics of ICa,L. We previously showed that in rats with MI, the contractility of the myocytes was also markedly impaired (Ukai et al., 1999). These findings demonstrated the existence of an established HF in this model.
Increased Responses of ICa,L to β2-AR Stimulation in HF Myocytes
Figure 1 shows typical ICa,L responses to β2-AR stimulation in normal (Fig. 1A) and HF myocytes (Fig. 1B). Superfusion of ZIN, a highly selective β2-AR agonist (10−5 M), caused significant increases in peak ICa,L in normal myocytes (21%, 2.13 ± 0.19 versus 1.75 ± 0.15 nA, p < 0.05,n = 21). In HF myocytes, ZIN caused a greater increase in ICa,L (30%, 2.18 ± 0.14 versus 1.66 ± 0.10 nA, p < 0.01, n = 24). The ZIN-induced absolute increases in ICa,Lbetween normal (0.38 ± 0.05 nA) and HF cells (0.52 ± 0.06 nA) are statistically significant (p < 0.05). After normalized by the membrane capacitance, the ZIN-induced increases in ICa,L remained statistically different in both normal (9.21 ± 0.24 versus 7.59 ± 0.20 pA/pF) and HF cells (6.20 ± 0.24 versus 4.75 ± 0.17 pA/pF). This current was blocked by nifedipine (5 × 10−6 M). Figure1, C and D, shows the current-voltage relations for the response of ICa,L to ZIN in normal myocytes and in those of rats with HF. The stimulatory effects of ZIN on ICa,L were not accompanied by a significant change in the voltage dependence of peak ICa,Lamplitude in normal and HF myocytes.
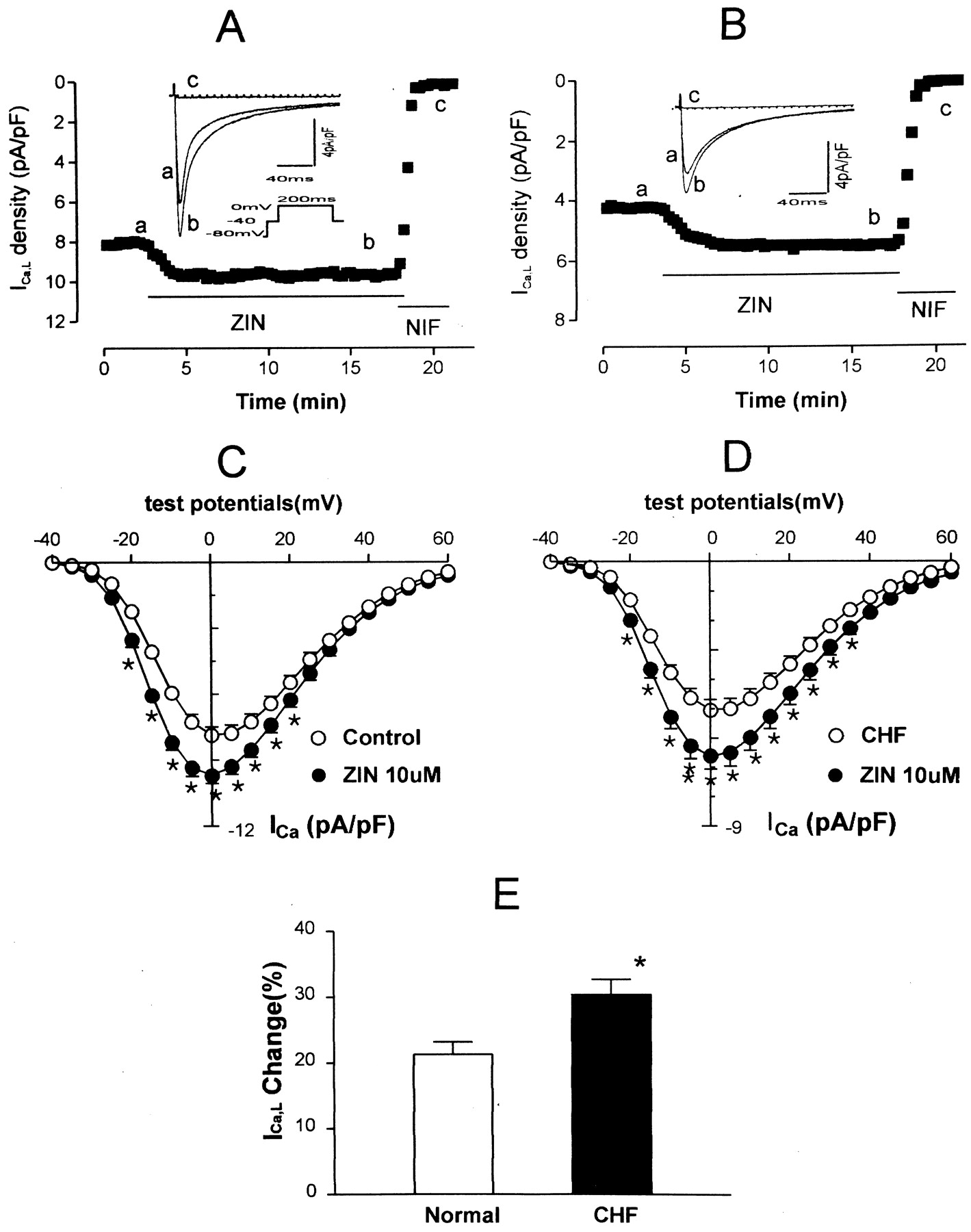
Effects of ZIN (10−5 M) on ICa,L. A, a ventricular myocyte from a normal rat was initially superfused with external solution. During the period indicated by the horizontal line, the cell was exposed to ZIN and then to nifedipine (NIF, 5 × 10−6 M). The superimposed current tracings were recorded before (a), and 10 min after (b) exposure to ZIN, and exposed to NIF (c). ICa,L were elicited by depolarization pulses from a holding potential of −80 to 0 mV for 200 ms with a brief prepulse to −40 mV (60 ms) (inset) at 0.1 Hz. B, same experiment as in A in an HF myocyte. C, current-voltage relations of ICa,L in the absence (open symbols) and presence (filled symbols) of ZIN in eight normal myocytes. ∗, indicates that the difference between pre- and postdrug is statistically significant (p < 0.05). The cells were depolarized from a holding potential of −40 mV to test potentials from −35 to +60 mV in 5-mV increments. D, current-voltage plots for seven HF cells, measured under the same experimental conditions as in C. E, average increments of ICa,L induced by ZIN (10−5 M). Values are mean ± S.E.M. forn = 21 to 24 experiments. ∗, indicates that the difference between normal and HF myocytes is statistically significant (p < 0.05). The results showed an enhanced effect of ZIN on ICa,L in HF myocytes.
Concentration-Dependent Activation of ICa,L by ZIN
Concentration-response curves of ICa,L to ZIN of myocytes from normal rats and rats with HF are compared in Fig.2. Smooth curves were obtained by fitting the data with the Hill equation. The dose-response curve was shifted upward, and the half-maximal activation concentration (EC50) of the HF myocytes was lower than that of the normal myocytes (168 versus 688 nM). The Hill coefficient of ZIN (0.83 in normal and 0.88 in HF cells) was similar as reported byRobberecht et al. (1983). Similarly, with one-site competition equation, the half-maximal activation concentration was 733 nM for normal and 174 nM for HF myocytes. There were no significant differences between the best fits achieved by both Hill and one-site competition equation.

Concentration-dependent activation of ICa,L by ZIN. Average percentage of increase of ICa,L was plotted against ZIN concentrations for normal (open symbols) and HF (filled symbols) myocytes. The cell number for each data point is indicated in parentheses. Smooth curves were obtained by fitting the data with the Hill equation. Half-maximal activation concentration was 688 nM for normal and 168 nM for HF myocytes, and the Hill coefficient was 0.83 and 0.88, respectively.
Effects of β1- and β2-AR Blockade on ZIN-Induced Increase in ICa,L
The affinity of ZIN for β2-AR is approximately 50 times over β1-AR (disassociation constant is 20 versus 1000 nM) (Minneman et al., 1979). ZIN at 10−5 M has been shown to induce maximal response in cell contraction [Ca2+]i transient and ICa,L in rat cardiomyocyte (Xiao and Lakatta, 1993; Xiao et al., 1995). This high concentration of ZIN may also stimulate β1-AR or α1-AR or work through nonadrenergic receptor system (Edwards and Whitaker-Azmitia, 1987; Parfitt and Bickford-Wimer, 1990). To determine the potential mechanism, we preincubated the myocytes with CGP (a β1-AR antagonist, 3 × 10−7 M) or ICI-118551 (ICI, a β2-AR antagonist, 10−7M) for 20 min, and ZIN was given in the presence of CGP or ICI. As shown in Fig. 3, after using CGP to block the β1-AR, the ZIN-induced increase in ICa,L persisted in both normal (7.51 ± 0.40 versus 6.20 ± 0.30 pA/pF, p < 0.05,n = 4) and HF cells (5.16 ± 0.30 versus 4.03 ± 0.20 pA/pF, p < 0.05, n = 4). In contrast, as displayed in Fig. 4, after preincubation with the β2-AR blocker, ICI, the ZIN-induced increase in ICa,L was abolished in both normal (7.22 ± 0.30 versus 7.07 ± 0.30 pA/pF,p = N.S., n = 4) and HF cells (4.52 ± 0.35 versus 4.51 ± 0.38 pA/pF, p = N.S., n = 4), indicating that ZIN increased ICa,L through β2-AR, not β1-AR, α1-AR, or other nonadrenergic system.

Effect of ZIN on ICa,L is not prevented by β1-AR blockade in normal and HF myocytes. The myocytes were preincubated with CGP (a β1-AR antagonist, 3 × 10−7 M) for 20 min. A, superimposed current tracings recorded before and 5 min after exposure to ZIN (10−5 M) in the presence of CGP in a normal myocyte. ICa,L was elicited by depolarization pulses from a holding potential of −80 to 0 mV for 200 ms with a brief prepulse to −40 mV (60 ms). B, same experiment as in A in an HF myocyte. The results showed that the effect of ZIN persisted in the presence of the β1-AR blocker (CGP 20712A). C, average effects of β1-AR blockade on ZIN-induced ICa,L responses. Values are mean ± S.E.M. for n = 4 experiments. *p < 0.05 between control and ZIN.

Effect of ZIN on ICa,L is prevented by β2-AR blockade in normal and HF myocytes. The myocytes were preincubated with ICI (a β2-AR antagonist, 10−7 M) for 20 min. A, superimposed current tracings recorded before and 5 min after exposure to ZIN (10−5 M) in the presence of ICI in a normal myocyte. ICa,L were elicited by depolarization pulses from a holding potential of −80 to 0 mV for 200 ms with a brief prepulse to −40 mV (60 ms). B, same experiment as in A in an HF myocyte. C, average effects of β2-AR blockade on ZIN-induced ICa,Lresponses. Values are mean ± S.E.M. for n = 4 experiments.
Effect of Gi Protein Blockade-Induced Increase in ICa,L
Distinct from β1-AR in signal transduction, β2-AR couples with both Gs and Gi proteins (Xiao et al., 1999). To define the role of Gi protein in β2-AR stimulation of ICa,L, we pretreated the myocytes with PTX (2 μg/ml, 36°C, 6 h). The adequacy of the complete blockage of inhibitory Gi protein in PTX-treated cells was routinely verified by the loss of the ability of acetylcholine (ACh, 10−5 M) to reverse the stimulatory effect of ISO on ICa,L.
Effect of ACh on Response to ISO.
ACh is generally thought to activate PTX-sensitive Gi protein and counteract the Gs-activating effect by β-AR stimulation. PTX-treated myocytes were compared with myocytes that had been kept at 36°C in the absence of PTX for an equal time. In the normal cells, ISO (10−7 M) increased ICa,L by 120 ± 14%, and further application of ACh (10−5 M) decreased the ISO-induced augmentation in ICa,L to 84 ± 10% (p < 0.05, n = 4). With PTX pretreatment, in the normal cells, the reduction of ISO response on ICa,L caused by ACh was prevented (an increase of 116 ± 12%, p = N.S., n = 4). This is consistent with the observations of Lauer et al. (1992). In HF cells, the increase of ICa,L by ISO was markedly attenuated (56 ± 8%), and the addition of ACh almost prevented the ISO-induced increase in ICa,L (10 ± 8%, p = N.S., n = 4). Thus, compared with normal cells, the inhibition effect of ACh on ISO response in HF cells was significantly augmented (82 versus 30%, p < 0.05, n = 4). In the PTX-treated HF cells, the reduction of ISO response caused by ACh was totally abolished (increases of 93 ± 14 versus 95 ± 10%, p = N.S., n = 4). This observation is in line with the phenomenon of PTX-restoring ISO response of contraction in cardiomyocytes from patients with HF reported by Brown and Harding (1992).
Figure 5 shows that after PTX preincubation of the myocytes to block inhibitory Gi protein, the baseline ICa,L exhibited no significant change, whereas the ZIN-induced increases in ICa,L were further augmented. In normal cells, ICa,L increased about 59% (2.28 ± 0.13 versus 1.43± 0.15 nA, p < 0.05, n = 9), and in HF cells, ICa,L increased about 71% (3.14 ± 0.27 versus 1.90± 0.16 nA, p < 0.05, n = 10). With pretreatment of PTX, the ZIN-induced absolute increase of ICa,L in HF cells (1.24 ± 0.20 nA) was significantly greater than that in normal cells (0.85 ± 0.11 nA,p < 0.05). After normalized by the membrane capacitance, the ICa,L density remained statistically different between normal (12.20 ± 0.90 versus 7.69 ± 0.32 pA/pF) and HF cells (8.90 ± 0.90 versus 5.20 ± 0.22 pA/pF).

Effect of ZIN on ICa,L is augmented by PTX in normal and HF myocytes. The myocytes were preincubated with PTX (2 μg/ml, 36°C, 6 h). A, superimposed current tracings recorded before and 5 min after exposure to ZIN (10−5 M) in a normal myocyte. ICa,L were elicited by depolarization pulses from a holding potential of −80 to 0 mV for 200 ms with a brief prepulse to −40 mV (60 ms). B, same experiment as in A in an HF myocyte. C, average effects of PTX on ZIN induced ICa,Lresponses. Values are mean ± S.E.M. for n = 9 to 10 experiments. *p < 0.05 between control and ZIN. The results showed that the effects of ZIN were enhanced by PTX pretreatment, suggesting a PTX-sensitive G protein mechanism involved in the ZIN-induced ICa,L response of ventricular myocytes.
Effects of ZIN on Response to a β1-AR Agonist.
In a subgroup of animals, we examined the effect of ZIN on ICa,L response after a β1-AR stimulation with norepinephrine (NE, 10−7 M). We found that in normal myocytes, NE caused a significant increase in ICa,L (78%, 14.05 ± 1.50 versus 8.17 ± 1.00 pA/pF, p < 0.05, n = 4). The addition of ZIN (10−5 M) caused an attenuation (43%) in NE-induced ICa,L increase (10.53 ± 1.30 versus 8.17 ± 1.00 pA/pF, p < 0.05,n = 4). In contrast, in HF myocytes, compared with normal myocytes, NE caused a much smaller increase in ICa,L (17%, 5.39 ± 1.20 versus 4.62 ± 0.90 pA/pF, p < 0.05). The application of ZIN caused an additive increase in ICa,L(approximately 8%, from 5.39 ± 1.20 to 5.75 ± 1.30 pA/pF,p < 0.05, n = 4). Thus, it appears that in normal cardiomyocytes, ZIN had an inhibitory effect when applied after the effect of β1-AR stimulation had developed; but in HF cells, ZIN exhibited a stimulatory action on ICa,L, suggesting an altered interaction between β1- and β2-AR stimulation in HF cells.
Effect of Gs-cAMP Pathway Blockade on ZIN-Induced Increase in ICa,L
Since accumulating evidence indicates that the effect of β-AR stimulation on cardiac ICa,L is mediated by a cAMP-dependent mechanism (Zhou et al., 1997), we further examined the role of cAMP-dependent PKA activation in β2-AR stimulation of ICa,L by including an inhibitory cAMP analog Rp-cAMPS (10−4 M) in pipette solution to block the stimulatory Gs protein-cAMP pathway. As shown in Fig. 6, with intracellular application of Rp-cAMPS, basal ICa,L was decreased about 20%, which is consistent with previous observations (Zhou et al., 1997). However, under this condition, ZIN-induced ICa,L increase was prevented in both normal (6.06 ± 0.32 versus 6.17 ± 0.25 pA/pF, p = N.S., n = 6) and HF (3.78 ± 0.06 versus 3.88 ± 0.10 pA/pF, p = N.S., n = 4) myocytes.

Effect of Rp-cAMPS on ZIN induced ICa,Lresponses of normal and HF myocytes. A, superimposed current tracings recorded before and 5 min after exposure to ZIN (10−5 M) in the presence of Rp-cAMPS (10−4 M) in pipette solution in a normal myocyte. ICa,L were elicited by depolarization pulses from a holding potential of −80 to 0 mV for 200 ms with a brief prepulse to −40 mV (60 ms). B, same experiment as in A in an HF myocyte. C, average effects of Rp-cAMPS on ZIN induced ICa,L responses. Values are mean ± S.E.M. forn = 4 to 6 experiments. The results showed that the effects of ZIN were abolished in the presence of Rp-cAMPS in pipette solution.
Discussion
The present study demonstrated, for the first time, that β2-AR stimulation of ICa,L by ZIN was enhanced in LV myocytes of rats with infarction-induced heart failure. These effects were mediated through a cAMP-dependent mechanism and coupled with both stimulatory Gs protein and PTX-sensitive inhibitory Gi protein.
Effects and Possible Mechanism of ZIN on ICa,L.
It has been increasingly recognized that β1- and β2-AR coexist in the heart. In large mammal hearts, β2-AR may account for approximately 40% of the total β-AR number, while in small animals, cardiac β2-ARs are less abundant. The reported ratio of β1-/β2-AR was 92–80/8–20 in adult rat hearts (Cerbai et al., 1995; Kuznetsov et al., 1995). β-AR stimulation modulating cardiac ICa,L plays an important role in the positive inotropic response to β-AR stimulation.
In the present study, consistent with previous report (Xiao and Lakatta, 1993), by using freshly isolated cardiomyocytes, we found that ZIN caused a dose-dependent increase in ICa,Lwith maximum increase (21%) at concentration of 10−5 M in normal cells. This effect was completely abolished by β2-AR antagonist, but not by the β1-AR antagonist, indicating that the alterations of ICa,L following ZIN superfusion were due to β2-AR activation. Furthermore, we found that the effect of ZIN was enhanced by pretreatment of the myocytes with PTX, indicating a negative modulation by Gi. An inhibitory cAMP analog, Rp-cAMPS, blocked the ZIN-induced increase in ICa,L. These findings suggest that the cardiac β2-adrenergic signal transduction is dual coupling to both Gsand Gi protein (Steinberg, 1999; Xiao et al., 1999) and that it may be mediated through a cAMP-dependent mechanism (Zhou et al., 1997).
These results are consistent with previous observations made in normal ventricular myocytes of several species, including adult rat (Xiao and Lakatta, 1993; Cerbai et al., 1995; Xiao et al., 1995; Zhou et al., 1997), frog (Skeberdis et al., 1997), dog (Altschuld et al., 1995), and nonfailing human atrial tissue (Kaumann et al., 1996). However, our result is inconsistent with previous reports in guinea pig cardiomyocytes (Hool and Harvey, 1997). Species difference might account for this inconsistency. Also in contrast with our observation,Nagykaldi et al. (2000) showed the absence of any increase in ICa,L in normal canine ventricular myocytes when β2-AR was stimulated alone. This discrepancy may be due to a much smaller dose of ZIN (50 nM, 200 times smaller than the dosage used by us and others) used in their study.
A novel finding in the present study is that the responsiveness of ICa,L to β2-AR stimulation was enhanced after HF. Although β2-AR-dependent signals represent only a relatively minor component of catecholamine responsiveness under normal physiological conditions, β2-ARs assume increased importance as a mechanism for inotropic support in the failing heart. Since there is a selective down-regulation of β1-ARs with resultant increase in β2/β1 ratio in HF.
In the present study, we demonstrated that in HF myocytes, the ICa,L was significantly decreased, and its response to β-AR stimulation (ISO 10-7 M) was also markedly blunted (56% versus 120% increment, p< 0.05). However, the response to ZIN (10-5 M) was significantly enhanced (30% versus 21%). The dose-response curve shifted upward, and the half-maximal activation concentration (EC50) was significantly decreased (168 versus 688 nM), indicating an increased responsiveness to β2-AR stimulation. After preincubation of the myocytes with PTX to block Gi, the increment in ICa,L by ZIN was further augmented.
These observations are supported by several recent findings (Altschuld et al., 1995). Altschuld et al. reported that the contractility of myocytes from dogs with HF were significantly more responsive to β2-AR stimulation with ZIN than were normal myocytes. In addition, we have previously demonstrated that β2-AR stimulation by ZIN caused an enhanced augmentation in ventricular contractility in conscious dogs with pacing-induced HF (Cheng et al., 1998). In cardiomyocytes from rats with infarction-induced HF (Ukai et al., 1999), we have shown that compared with normal, ZIN (10−5 M) caused an enhanced positive inotropic effect associated with an increased [Ca2+]i transient.
However, our observations are not likely to agree with the findings in the failing human heart by Bristow et al. (1989). They reported a diminished β2-AR-mediated cardiac response by documenting a decreased adenylate cyclase stimulation (32% reduction in cAMP production). It is evident that β2-AR stimulation-induced increase in cAMP is not coupled to contractility, Ca2+ dynamics, and phospholamban phosphorylation (Xiao et al., 1994). In addition, their findings might also have been complicated by the confounding effects of the tissue preparation and the use of multiple cells in their study. Furthermore, Altschuld et al. (1995) observed an increase in ICa,L without an increase in cAMP accumulation during β2-AR stimulation. Since the acute effect of ZIN on ICa,L was not examined in the Bristow et al. (1989) study, it remains unknown whether HF alters ICa,L response to β2-AR stimulation in the human heart.
Since β2-AR couples with both Gs and Gi protein, the increased Gi protein in HF may be expected to have a negative modulation on ZIN-induced changes in ICa,L. Why have others (Altschuld et al., 1995) and we observed ZIN-induced enhanced increases in cardiac contractility, [Ca2+]itransient, and ICa,L? The mechanism(s) for the ZIN-induced enhanced increase in ICa,L for HF cells versus normal cells is unclear and under investigation in our laboratory. We predict that an increase in β2-AR density on the membrane of HF cells and a differential cAMP compartmentation via altered signal transduction may contribute to our current findings.
Consistent with previous observations, in our MI rats, the cardiac hypertrophy was established, and the myocytes from these hearts were enlarged. It has been reported that the synthesis of β2-AR closely parallels cell growth (Cerbai et al., 1995); thus, previously reported preserved β2-AR density in MI rats may be associated with increased β2-AR numbers per cell. In fact, the increase of β2-AR density has been demonstrated in LV of dogs with pacing-induced HF (Kiuchi et al., 1993). Thus, the enhanced ICa,L response to ZIN in HF cells may reflect the presence of an increase in the number of β2-AR per cell, which parallels the increase in cell size. It is interesting to note that a similar observation was also obtained in neonatal myocytes. Kuznetsov et al. (1995) found that the β2-AR density per neonatal myocyte was 5-fold higher than per adult myocyte, and ZIN evoked a substantial increase in cAMP accumulation and a greater physiological effect on excitation-contraction coupling in neonatal myocytes in comparison with adult myocytes. The PTX-sensitive Gi protein was found higher in neonatal than in adult rat ventricle (Bartel et al., 1996). This similarity is consistent with the recapitulation of neonatal phenotype in HF (Feldman et al., 1991).
Our study indicates that ZIN-induced changes on ICa,L in both normal and HF cells are mediated by a cAMP-dependent mechanism. However, our subgroup study of the interaction of β1- and β2-AR stimulation and studies of others (Xiao et al., 1994; Nagykaldi et al., 2000) have demonstrated that the cAMP-dependent pathway could be significantly modified, counteracted, or paralleled by additional signaling pathways. Activation of Gi has the potential to couple β2-AR to other important signaling pathways such as the mitogen-activated protein kinase (Daaka et al., 1997).
Previous studies suggested that structural and functional compartmentation of cAMP can occur in cardiomyocytes and that the cAMP levels or the cAMP-dependent protein kinase activity in these compartments could be selectively or preferentially modulated by β2-AR stimulation (Hohl and Li, 1991; Xiao et al., 1994; Nagykaldi et al., 2000). Recently, increasing evidence further indicated that β2-AR stimulation does not produce a generalized cAMP accumulation in cardiomyocytes, but, rather, is confined in regions localized to subsarcolemmal compartments (Yanagisawa et al., 1989; Zhou et al., 1997). Thus, the enhanced ICa,L response to ZIN in HF cells observed in the present study could result from a differential cAMP elevation in distinct subcellular compartments associated with the L-type of Ca2+ channel. However, it remains to be determined how the cAMP generated by β2-AR activation is differentially compartmentalized in normal and diseased state of the heart.
The functional significance of β2-AR stimulation remains controversial (Billman et al., 1997; Cheng et al., 1998; Rockman et al., 1998; Liggett et al., 2000). We showed that the acute ICa,L response to β2-AR stimulation was enhanced in HF. This enhanced response could cause more Ca2+ influx into cardiomyocytes, enhance excitation-contraction coupling, and thus improve myocardial contractile performance in HF. Liggett et al. (2000)recently demonstrated that overexpression of β2-AR up to 100-fold in the mouse heart causes significant increased cardiac contractile force without any cardiomyopathic consequences during the 1-year study period. A gene therapy approach with overexpression of β2-ARs has also been suggested (Lefkowitz et al., 2000).
In conclusion, we found that β2-AR activation stimulates L-type Ca2+ channel and increases ICa,L in both normal and HF myocytes. In rats with infarction-induced HF, β2-AR activation-induced augmentation of ICa,L was increased. These effects are likely to be mediated through a cAMP-dependent mechanism and coupled with both stimulatory Gs protein and PTX-sensitive inhibitory Gi protein. This finding is likely to be the underlying mechanism for the improved inotropic responsiveness to β2-AR stimulation in the failing heart.
Acknowledgments
We gratefully acknowledge the help of Drs. J. Mu and R.-L. Wu on patch-clamp technique. We also acknowledge the expert editorial review of Dr. Donna Garrison, the computer programming of Dr. Ping Tan, the technical assistance of Ellen Tommasi and Mike Cross, and the secretarial assistance of Amanda Burnette.
Footnotes
-
This study was supported in part by grants from the National Institutes of Health (HL45258, HL53541, and HL12335-01A1) and the American Heart Association (9640189N). Dr. C. P. Cheng is an established investigator of the American Heart Association. An abstract of this work was presented at the 1999 American Heart Association Meeting.
- Abbreviations:
- AR
- adrenergic receptor
- ICa,L
- L-type Ca2+ current
- HF
- heart failure
- PKA
- protein kinase A
- LV
- left ventricular
- LV dp/dtmax and dp/dtmin
- maximum and minimum time derivative of LVP, respectively
- PTX
- pertussis toxin
- MI
- myocardial infarction
- ZIN
- zinterol
- CGP
- CGP-20712A
- ISO
- isoproterenol
- ICI
- ICI-118551
- Ach
- acetylcholine
- NE
- norepinephrine
- [Ca2+]i
- intracellular calcium
- Received December 26, 2000.
- Accepted March 19, 2001.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}