


Abstract
Elevated cAMP in NRK-52E and L6 cells causes a marked reduction in the phosphorylation of numerous phosphoproteins, as detected initially with phosphoserine-specific antibodies. Here, we show that elevation of cAMP in NRK cells by forskolin/3-isobutyl-1-methylxanthine (IBMX) treatment decreased phosphorylation of substrates for different protein kinases, pointing to a common protein phosphatase as a target for cAMP-dependent regulation. Forskolin/IBMX treatment completely dephosphorylated a selective protein phosphatase 2A (PP2A) substrate, elongation factor-2 (EF-2), at its Ca2+ calmodulin-dependent kinase site, and decreased phosphorylation of substrates for cyclin-dependent kinases, including retinoblastoma (Rb) protein. As reported before, forskolin/IBMX also decreased phosphorylation of a protein kinase C substrate, the Na,K-ATPase. The cAMP-stimulated dephosphorylation was blocked by the protein phosphatases 1 (PP1) and PP2A inhibitor okadaic acid at concentrations selective for PP2A but was not blocked by tautomycin at concentrations selective for PP1. The data implicate PP2A as a cAMP-activated phosphatase. Contrary to expectation, we found evidence that cAMP-dependent activation of PP2A did not depend on protein kinase A (PKA). Pretreatment of cells with the PKA inhibitor H89 abolished PKA activity measured in cell extracts and significantly decreased cAMP-activated phosphorylation of a known PKA substrate, ARPP-19, in cells, but failed to block the cAMP-stimulated dephosphorylation of EF-2, Rb, and other proteins. This novel pathway of PP2A activation, acting on the time scale of minutes, represents yet another example of a cAMP-mediated, PKA-independent signaling mechanism. Because PP2A is active toward a variety of endogenous substrates, cAMP-stimulated dephosphorylation may have complicated the interpretation of many prior studies.
The common practice of investigating signal transduction pathways with protein-specific probes produces outstandingly rigorous molecular information. It should be recognized, however, that when the focus is assumed to be exclusively on identified elements, unanticipated interactions between pathways may go undetected. Our recent work on regulation of the Na,K-ATPase began with the hypothesis that there was allosteric cross-talk between identified serine phosphorylation sites for PKC and PKA (Feschenko et al., 2000). With a site-specific, phosphorylation-sensitive antibody, the predicted cAMP-dependent reduction in phosphorylation by PKC was observed. With antibodies that reacted with a variety of phosphoproteins, however, we found that elevation of cAMP decreased phosphorylation of many proteins in parallel to that of the Na,K-ATPase (Feschenko et al., 2000). The data suggested the existence of a cAMP-stimulated mechanism that limits or terminates cellular responses by either preventing or reversing phosphorylation.
The cAMP-mediated reduction in phosphorylation could be caused by inhibition of multiple kinases or stimulation of a phosphatase. The major serine/threonine-specific phosphatases have been classified into four main types (PP1, PP2A, PP2B, and PP2C) according to molecular structure, hydrolysis of selected substrates in vitro, requirement for cations, and sensitivity to activators and inhibitors (Oliver and Shenolikar, 1998). Natural toxins such as calyculin A, tautomycin, and okadaic acid inhibit PP1 and PP2A, which are closely related in structure (Oliver and Shenolikar, 1998). PP1 and PP2A are regulated by forming quaternary complexes with targeting subunits or regulatory subunits that localize the enzymes to particular subcellular compartments, confer substrate specificity, and in some cases affect activity (Lester and Scott, 1997). Although many such regulatory subunits have been detected, the physiological roles of only a few are known (Hunter, 1995; Oliver and Shenolikar, 1998; Bibb et al., 1999), and much remains to be discovered about protein phosphatase regulation. The results obtained in the present study suggest that cAMP activates PP2A by a novel mechanism. Because PP2A is active toward substrates for a variety of protein kinases, this cAMP-dependent mechanism may influence many signal transduction pathways.
Interestingly, Usui and collaborators (1998) have shown in vitro that phosphorylation of a B subunit of erythrocyte PP2A by PKA leads to enzyme activation. However, the mechanism reported here apparently does not depend on PKA. Various hormones and neurotransmitters and the drug forskolin are used widely to elevate cAMP and are generally assumed to act only through PKA. However, the effects of cAMP do not always require PKA. In addition to the regulatory RI and RII subunits of PKA, cyclic nucleotide-binding domains are found in several other proteins, including cyclic nucleotide-gated ion channels (Shabb and Corbin, 1992), cyclic nucleotide-regulated guanine exchange factors (cAMP-GEFs) such as Epac1 and 2 (Kawasaki et al., 1998; de Rooij et al., 1998), protein kinase G, and proteins with GAF domains (occurring in cGMP-regulated phosphodiesterases, adenylyl cyclases, and theEscherichia coli protein Fh1A) (Ho et al., 2000). The general reduction of phosphorylation observed in this study seems to extend the impact of actions of cAMP that are independent of the activity of PKA.
Materials and Methods
Chemicals.
Calyculin A, forskolin, 3-isobutyl-1-methylxanthine (IBMX), and phorbol-12-myristate-13-acetate (PMA) were from Sigma/RBI (Natick, MA); H89, okadaic acid, and KN-93 were from Alexis Biochemicals (San Diego, CA); roscovitine, butyrolactone I, tautomycin, and PD98059 were from Calbiochem (La Jolla, CA); and kemptide and inhibitor 2 were from Sigma-Aldrich (St. Louis, MO). ATP concentration determination was performed with a kit from Sigma-Aldrich.
Cell Treatments.
NRK-52E cells were maintained in DMEM (supplemented with 10% fetal bovine serum, 2 mMl-glutamine, penicillin, and streptomycin) in a 5% CO2 atmosphere. Monolayers of cells in 35-mm dishes were preincubated for 1.5 to 2 h at 37°C in 5% CO2 in DMEM without serum. Drugs or their vehicle (DMSO) were added, and the cells were incubated at 37°C. The medium was discarded, dishes were chilled on ice, and 0.2 ml of 1% SDS in 10 mM Tris-HCl buffer (pH 7.4) at 100°C was added to each dish. Cells were scraped, and the extracts were clarified by centrifugation for 30 min at 40,000 rpm. Supernatants were diluted 1:1 with Laemmli sample buffer, and aliquots (30 μl per well) were loaded on SDS gels. For32Pi metabolic labeling of intact cells, cells in 35-mm dishes were washed in phosphate-free DMEM without serum and preincubated in the same medium for 1 h at 37°C in 5% CO2. Fresh medium containing 70 μCi/ml [32P]orthophosphoric acid was then added. After 1 h of incubation, cells were washed with nonradioactive phosphate-free DMEM and used as above. All experiments shown are representative of multiple similar experiments.
Gel Electrophoresis, Electrophoretic Transfer, and Immunostaining.
Electrophoresis and Western blotting (with luminol-based detection) were performed as described previously (Feschenko and Sweadner, 1995), using 10% (for most experiments) and 16% (for ARPP-19) polyacrylamide gels. Three phosphospecific antibodies were used, 470 (Fisone et al., 1994), anti-phospho-elongation factor-2 (anti-phospho-EF-2) (Marin et al., 1997), and anti-phospho-ARPP-19 (Dulubova et al., 2001). Rb protein phosphorylation was detected by antibody stain (catalog number 554136; BD PharMingen, San Diego, CA) of bands shifted in mobility. Densitometry was with a 300A scanning densitometer (Molecular Dynamics, Sunnyvale, CA), and care was taken that the film signals were not saturated. All blots were stained with amido black after immunostaining to verify the even loading of samples.
Preparation of Cell Extracts for Enzyme Activity Measurements.
Cells were incubated with drugs in 35-mm dishes as described above. After two brief washes with ice-cold PBS, cells were lysed in 200 μl of ice-cold extraction buffer/dish containing 50 mM Tris-HCl pH 7.5, 0.1 mM EDTA, 0.1 mM EGTA, 10% (v/v) glycerol, 0.1% 2-mercaptoethanol, 1 mM benzamidine, 1 mM phenylmethylsulfonyl fluoride, 25 μg/ml leupeptin, and 25 μg/ml aprotinin. The dishes were incubated on ice for 10 to 15 min and frozen at −80°C. After thawing, cells were scraped and disrupted by repeated aspiration through a 21-gauge needle. Extracts were clarified by centrifugation at 1500 rpm for 5 min, and then supernatants were separated into aliquots and frozen at −80°C.
PKA Activity.
For measurement of PKA activity that could be detected in extracts of control or H89-treated cells, 10-μg samples of extract protein were supplemented with 2 mM MgCl2 and 100 nM calyculin A. Kemptide (2 μg) with or without 0.5 mM cAMP was added, and reactions were started by addition of [γ-32P]ATP (2000–3000 cpm/pmol) to a final concentration of 100 μM. The reactions proceeded for 10 to 15 min at 30°C and were stopped by the addition of an equal volume of 175 mM orthophosphoric acid. Aliquots of the samples were spotted onto P-81 ion exchange paper (Whatman, Clifton, NJ) and washed three to five times with 75 mM orthophosphoric acid, and the remaining radioactivity was counted with a scintillation counter. Samples without kemptide were used as blanks, and these values were deducted from the values obtained in the presence of kemptide.
Protein Phosphatase Activity.
Phosphatase activity in cell extracts was usually measured with 32P-labeled glycogen phosphorylase a (protein phosphatase assay system 13188-016; Invitrogen, Carlsbad, CA). Extract from untreated cells was diluted to 100 μg/ml in the assay buffer provided by the manufacturer. Twenty microliters of diluted extract was used per sample. Inhibitor 2 (1 μM), 4 nM okadaic acid, or 1 mM cAMP was added in 20 μl of assay buffer. Inhibitor 2 and okadaic acid were preincubated with samples for 30 min on ice, and cAMP was preincubated for 15 min at 30°C. All samples were further preincubated for 2 min at 30°C, and the reactions were started by addition of 40 μg of32P-labeled phosphorylase a in 20 μl. After 10 to 15 min at 30°C the reaction was stopped by adding 180 μl of 20% trichloroacetic acid. The tubes were vortexed and chilled on ice for 30 min and then centrifuged at 12,000 rpm for 3 min at 4°C. Two hundred microliters of each supernatant was counted with a scintillation counter. Treatment groups were normalized by protein concentration. Data are expressed as percentage of phosphate released by untreated cell lysates and are the average of three to four experiments. When measuring tautomycin-sensitive PP1 activity, phosvitin was used as a substrate according to published method (Geladopoulos et al., 1996).
Results
Activation of a cAMP-Dependent Pathway Decreased Phosphorylation of Many Proteins.
Our initial observation of cAMP-stimulated reduction in phosphorylation used phosphoserine-sensitive antibodies to detect phosphorylation (Feschenko et al., 2000). Here, we show that the same phenomenon was readily detected in NRK-52E cells prelabeled with32Pi. Figure1 compares the phosphoprotein patterns detected by 32P-autoradiography or immunoblot using antibody 470 (Fisone et al., 1994). Treatment of these cells with a combination of forskolin and IBMX elevated cAMP from about 20 pmol/mg to >10 nmol/mg (Feschenko et al., 2000), and this produced a general decrease in substrate phosphorylation, although32Pi incorporation into some proteins was not affected (Fig. 1A). Inhibition of protein phosphatases PP1 and PP2A with calyculin A resulted in a net increase in phosphorylation of many proteins. However, treatment with forskolin/IBMX before calyculin A reduced32Pi incorporation to control levels. Under basal conditions, only a few proteins were detected with the phosphoserine antibody (Fig. 1B). Calyculin A increased phosphorylation in terms of both the number of proteins and their intensity, but incubation with forskolin/IBMX before calyculin A reduced phosphorylation by 60 to 80% (by densitometry, not shown) relative to calyculin A treatment alone. IBMX in combination with 1,9-dideoxyforskolin, an analog that does not stimulate adenylyl cyclase, had no effect (not shown). Similar results were observed with a different subset of proteins stained by a commercial anti-phosphoserine antibody [Fig. 7 of Feschenko et al. (2000)]. The data indicate that elevated cAMP reduced phosphorylation in intact cells on a time scale of 15 to 30 min and that many phosphoproteins were affected.

Phosphatase inhibitor calyculin A increased phosphorylation of many proteins, and activation of a cAMP-dependent pathway counteracted it. Cells were preincubated for 15 min with 50 μM forskolin/500 μM IBMX or vehicle at 37°C, and then where indicated, 100 nM calyculin A was added and cells were further incubated for 20 min. Cells were lysed and subjected to SDS-gel electrophoresis followed by transfer to nitrocellulose. Detection of phosphorylated proteins was either by autoradiography of32P (A) or with anti-phosphoserine antibody 470 stain of the same blot (B). Samples are in duplicates. Molecular weight markers are indicated.
Because it is possible to deplete ATP in cells with extremely high production of cAMP, intracellular ATP concentrations were determined with a luciferase/luciferin assay. Values of 16.3 ± 1.6 and 11.8 ± 1.1 nmol of ATP/mg of protein were obtained in controls and samples treated with forskolin for 15 min, respectively. This final concentration in forskolin-treated cells (∼70% of control) should be sufficient for protein kinases to be fully active.
Antibody 470 was particularly good at detecting proteins affected by the cAMP-mediated reduction in phosphorylation. Although this polyclonal antibody was raised against a peptide containing a phosphorylated serine within a PKA consensus site, its specificity is obviously not limited to PKA substrates. To ascertain what kinase(s) phosphorylate these proteins, we used a battery of inhibitors with different selectivity and added calyculin A to prevent dephosphorylation. Phosphorylation of most of the bands above 70 kDa was inhibited 40 to 50% by butyrolactone 1 and roscovitine (Fig.2, A and B). Butyrolactone 1 and roscovitine are selective for several cyclin-dependent kinases, predominantly cdk1 and 2. Inhibitors of MAP-kinase cascade (PD98059), PI3-kinase (wortmannin), PKA (H89), and Ca2+/calmodulin-dependent kinases (KN93) did not block calyculin A-induced phosphorylation (Fig. 2B). Down-regulation of PKC by prolonged incubation with PMA (phorbol-12-myristate-13-acetate) was also without effect (not shown). The conclusion is that cdk substrates are among the proteins targeted by cAMP-stimulated dephosphorylation, along with the PKC site on the Na,K-ATPase. Two other identified targets, EF-2 and Rb, will be shown below.
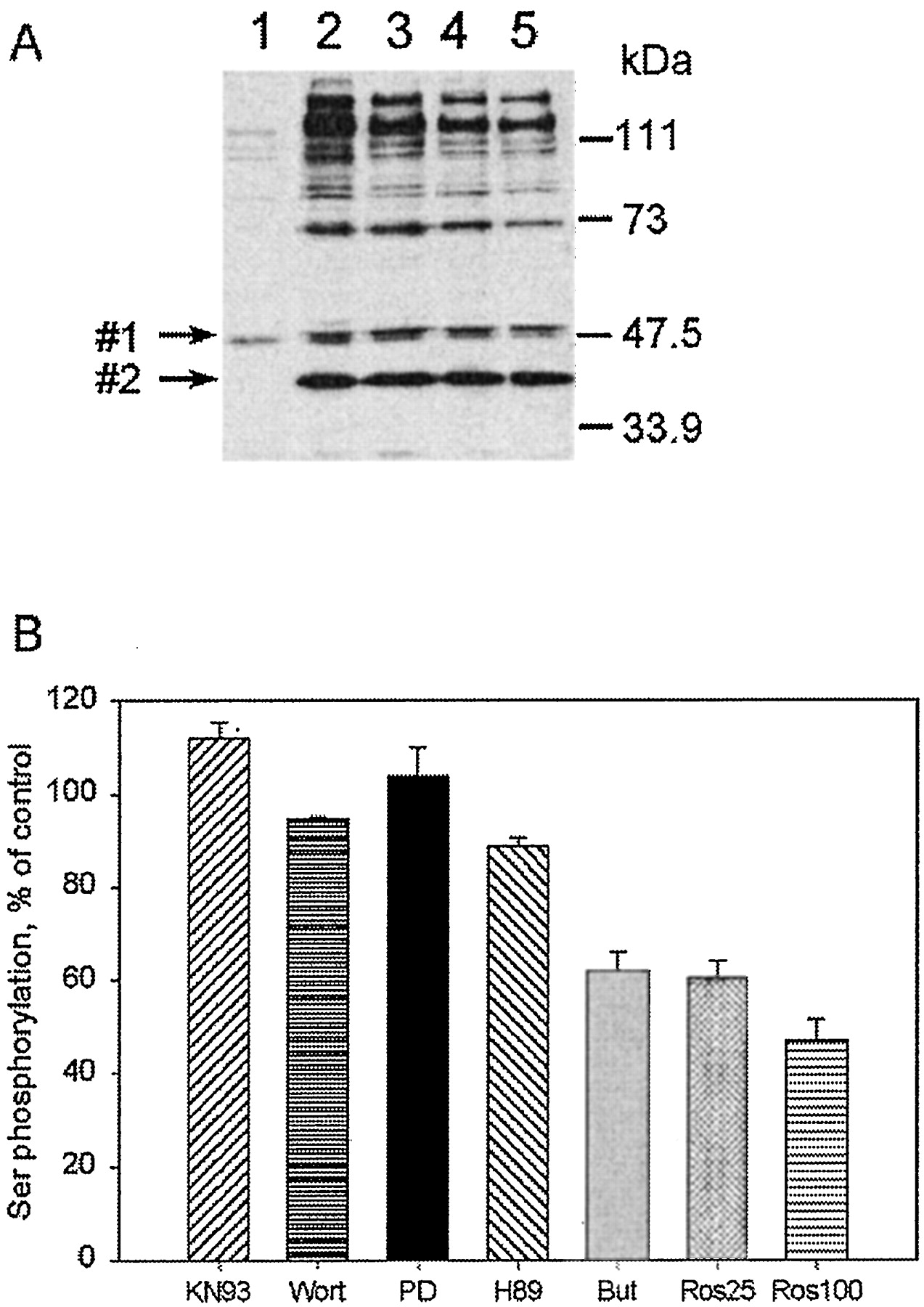
Many proteins recognized by antibody 470 are cdk substrates. A, immunodetection of serine-phosphorylated proteins before (1) and after (2–5) inhibition of phosphatase by calyculin A. Cells were preincubated for 1 h at 37°C with vehicle (lanes 1 and 2), cdk inhibitors 100 μM butyrolactone 1 (lane 3), or roscovitine at 25 μM (lane 4) or 100 μM (lane 5). Then 50 nM calyculin A or vehicle was added, and samples were incubated for an additional 20 min. Phosphoproteins were visualized by staining blots with anti-phosphoserine antibody 470. Arrows point to the proteins, marked 1 and 2, the phosphorylation of which was unaffected by cdk inhibitors. Molecular weight markers are indicated. B, densitometric evaluation (n = 3–5) of phosphorylation seen after preincubation with inhibitors of different kinases before calyculin A treatment. The values are expressed as a percentage of the phosphorylation level with calyculin A alone. Cells were preincubated for 1 h with kinase inhibitors at the following concentrations: KN93, 10 μM; wortmannin (Wort), 100 μM; PD98059 (PD), 30 μM; H89, 30 μM; butyrolactone 1 (But), 100 μM; roscovitine (Ros), 25 and 100 μM. Bands no. 1 and no. 2 were omitted from scanning.
Two proteins were notably different in their phosphorylation. Phosphorylation of the protein marked no. 1 (Mr 48 kDa) (Fig. 2A) was not affected by cdk inhibitors but was decreased by the PKA-selective inhibitor H89 (see Fig. 6A), suggesting that it is a PKA substrate. None of the inhibitors blocked the phosphorylation of protein no. 2 (Mr 40 kDa).

Phosphatase stimulation by cAMP was independent of PKA activity. A, effect of PKA inhibitor H89 on the phosphorylation of different proteins, including one (ARPP-19) that is a known PKA substrate. Cells were pretreated with or without 30 μM H89 for 1 h; then 50 μM forskolin/500 μM IBMX was added where indicated, and cells were incubated for 15 min; finally 50 nM calyculin A was added where indicated, and incubation was continued for 20 min. Four antibodies were used for detection: anti-Rb, anti-phospho-EF-2, 470 (anti-phospho-serine), and anti-phospho-ARPP-19. The arrow marks protein no. 1 (upper band); its phosphorylation was blocked by pretreatment with H89. B, densitometry (n = 3) of ARPP-19 phosphorylation, detected with specific antibody. The values are expressed as percentage of phosphorylation level in untreated cells.
PP2A Is Responsible for Dephosphorylation.
Although both stimulation of phosphatase activity and inhibition of kinase activity may contribute to cAMP-stimulated reduction in phosphorylation, two kinds of experiments suggested that the increase in protein phosphatase activity was required. When phosphorylation was first enhanced with calyculin A treatment and the drug was then washed out, with time in culture we observed a slow dephosphorylation that was accelerated by addition of forskolin/IBMX (data not shown). This is consistent with stimulation of phosphatase activity as the primary effect, but the down-regulation of multiple kinases by phosphatase-mediated kinase dephosphorylation could also contribute (Millward et al., 1999). More significantly, when calyculin A was added to cells before the addition of forskolin/IBMX (the opposite of the order of addition in Fig. 1), no cAMP-stimulated dephosphorylation of phosphoproteins was observed (data not shown). This indicates the obligatory participation of PP1 or PP2A and seems to rule out a hypothetical pathway such as direct protein kinase A-mediated inhibition of the activity of various protein kinases.
To investigate which protein phosphatase was responsible, we compared the effects of tautomycin and okadaic acid under conditions where they are selective for PP1 and PP2A, respectively (Fig.3). Okadaic acid and calyculin A are known to have higher affinity for PP2A than for PP1, and tautomycin the opposite (Favre et al., 1997; Nishi et al., 1999). Okadaic acid can be applied to cells at 1 μM and inhibit PP2A without any detectable inhibitory effect on PP1 (Millward et al., 1999). This produced a pattern of phosphoproteins identical with that obtained with calyculin A: Okadaic acid enhanced phosphorylation, but this was blocked by prior treatment with forskolin/IBMX (Fig. 3A). Tautomycin treatment resulted in very little increase in phosphorylation (Fig. 3B), indicating that the majority of the phosphoproteins detected with this antibody were substrates for PP2A. In control experiments, a 60% reduction in total phosphatase activity was measured in extracts of tautomycin-treated cells, indicating that inhibition of PP1 was successful (data not shown).

Sensitivity to selective phosphatase inhibitors. A, NRK-52E cells were preincubated either with forskolin/IBMX or vehicle for 15 min. Cells were then incubated with a PP2A-selective concentration of okadaic acid, 1 μM, for 1 h or 50 nM calyculin A for 20 min where indicated. B, NRK-52E cells were incubated either with tautomycin (5 or 10 μM) for 5 h or with 50 nM calyculin A for 30 min. Tautomycin has been shown to penetrate intact cells much more slowly than calyculin A and okadaic acid (Favre et al., 1997).
cAMP Stimulated Dephosphorylation of Elongation Factor-2.
To further test the hypothesis that PP2A is the target of cAMP, we examined the behavior of a known PP2A substrate, EF-2 (Nairn and Palfrey, 1987; Nilsson and Nygard, 1995), which is phosphorylated by a Ca2+/calmodulin-dependent kinase (EF-2 kinase). We used an anti-phospho-EF-2 antibody to detect changes in its phosphorylation levels in cells (Fig.4A). High levels of phosphorylated EF-2 were detected under basal conditions. Calyculin A did not increase this phosphorylation, suggesting that most EF-2 exists in its fully phosphorylated state in our basal conditions (deprived of serum for 1.5–2 h). Elevation of intracellular cAMP by forskolin/IBMX caused complete dephosphorylation of EF-2 in 10 to 15 min, and this effect of cAMP was blocked by preincubation with calyculin A. Figure 4B demonstrates that in NRK-52E cells phospho-EF-2 is, in fact, dephosphorylated by PP2A but not by PP1. The specific PP1 inhibitor, inhibitor-2 (I2), did not block dephosphorylation of EF-2 in NRK-52E cell lysates, whereas a low concentration of okadaic acid that selectively inhibits PP2A completely abolished it. Both PP1 and PP2A phosphatases were active in cell extracts in these conditions, and inhibitor-2 acted as an effective inhibitor of PP1 (Table1).

Dephosphorylation of elongation factor 2 (EF-2) and retinoblastoma protein (Rb) was accelerated by cAMP. A, NRK-52E cells were incubated with or without forskolin/IBMX for 10 or 20 min; with calyculin A for 10 or 20 min; or first preincubated with calyculin A for 10 min and then with forskolin/IBMX for 10 min. A high level of phosphorylated EF-2 was detected in untreated cells using an antibody against phospho-EF-2. B, NRK-52E cell lysates were preincubated in the presence of 4 nM okadaic acid (ok.a.), 1 μM inhibitor 2 (I2), or without phosphatase inhibitors for 30 min on ice, and incubated at 30°C for the times indicated. Okadaic acid prevented EF-2 dephosphorylation. C, cells were treated as described in Fig. 1. Different phospho-forms of the Rb protein were detected by gel mobility with a specific monoclonal antibody. ppRb and pRb indicate the hyper- and the hypophosphorylated forms of Rb, respectively. Duplicate samples are shown.
Protein phosphatase activity in NRK cell extracts
Rb Phosphorylation Is Reduced by cAMP.
Elevation of cAMP levels by forskolin has been shown to induce rapid dephosphorylation of retinoblastoma protein (Rb) in lymphoid cells, and the effect was mimicked by two different cAMP analogs and prevented by pretreatment with okadaic acid (Christoffersen et al., 1994). This closely resembles our observations, and so we assessed Rb phosphorylation in NRK-52E cells. Both hyperphosphorylated and hypophosphorylated forms of Rb were present in control conditions, as detected by their mobilities on SDS polyacrylamide gels (Fig. 4C). Elevation of cAMP caused a clear shift toward the less-phosphorylated form. These data are consistent with direct dephosphorylation of Rb by PP2A. PP2A may also have acted upstream to inhibit Rb phosphorylation, however, because there is evidence that the major Rb kinases cdk2, cdk4, and cdk6 can be regulated by PP2A (Millward et al., 1999; Yan and Mumby, 1999). In NRK-52E cells, however, as observed for EF-2, calyculin A did not further increase basal Rb phosphorylation, which suggests that a dephosphorylation event that regulates kinase activity is not the major determinant of phosphorylation level here.
cAMP Did Not Activate PP2A Directly.
Both PP1 and PP2A phosphatases were active in extracts from untreated cells (Table 1). Inhibitor 2 (1 μM) and a low concentration of 4 nM okadaic acid were used to selectively inhibit PP1 and PP2A, respectively. Prior treatment with forskolin/IBMX did not affect the phosphatase activity measured in vitro (data not shown). The results suggest that cell lysis may deregulate PP2A activity that is restrained by targeting mechanisms in vivo (Lester and Scott, 1997). Alternatively, phosphorylasea may not be a suitable substrate to detect cAMP-stimulated PP2A activity. In agreement with previous reports (Begum and Ragolia, 1996; Mukhopadhyay et al., 1998), addition of 1 mM cAMP to cell lysates also did not directly affect protein phosphatase activity, at least with phosphorylase a as substrate. The results suggest that cAMP activates PP2A through a process that can be disrupted by homogenization or dilution, consistent with emerging concepts of the complexity of phosphatase cell biology.
Is PKA Involved in Mediating the cAMP Effect on PP2A Activity?
The role of PKA, the usual cAMP target, in the forskolin/IBMX-induced decrease in phosphorylation was investigated using a cell-permeable PKA active site inhibitor, H89 (Chijiwa et al., 1990). To verify the ability of H89 to block PKA activity in these cells, they were incubated with or without 30 μM H89 and forskolin/IBMX and washed, and PKA activity was measured in cell extracts with a specific PKA substrate, kemptide, in the presence or absence of added cAMP (Fig. 5). Forskolin/IBMX pretreatment stimulated activity 12-fold, as did addition of cAMP to the lysate, but prior H89 treatment of the intact cells fully blocked PKA activation either by forskolin/IBMX preincubation or by cAMP addition, indicating complete and persistent inhibition.

Inhibition of PKA activity in intact NRK-52E cells by specific inhibitors. Intact cells were pretreated with 30 μM H89 or vehicle and then incubated with or without 50 μM forskolin/500 μM IBMX (F + I). PKA activity was measured in cell lysates with kemptide and [32P]ATP in the presence or absence of 0.5 mM cAMP. Densitometry is shown for n = 3–4, expressed as percentage of PKA activity in untreated cell lysates in the presence of cAMP (20–31 nmol/mg/h).
H89 was consequently used to study PKA involvement in mediating the cAMP effect on PP2A activity. The phosphorylation of Rb, EF-2, and antibody 470-stained proteins in intact cells is shown in Fig.6A. ARPP-19 protein, a known PKA substrate (Dulubova et al., 2001), was used as an internal control for the inhibition of cAMP-PKA-dependent phosphorylation. Its phosphorylation was enhanced 2- to 3-fold by forskolin/IBMX, and H89 almost abolished the increase without affecting basal phosphorylation (Fig. 6, A and B). This further documents that normal PKA activity is present in the cells and that H89 is an effective inhibitor for it in these experimental conditions. Another example of PKA-dependent phosphorylation is seen with the protein marked no. 1. Forskolin/IBMX in combination with calyculin A stimulated phosphorylation of protein no. 1, and preincubation with H89 abolished it. In contrast, in the same experiments, H89 failed to block the cAMP-dependent activation of PP2A-dependent dephosphorylation of Rb protein, EF-2, or the phosphoproteins detected by antibody 470. The results indicate that the cAMP effect is independent of the activity of the catalytic subunit of PKA, and incidentally of any other kinase that H89 may also inhibit (Davies et al., 2000).
An incidental observation was that calyculin A restored phosphorylation of EF-2 even if cells were first preincubated with forskolin (Fig. 6A). The effects of calyculin A on Rb were intermediate, whereas the phosphorylation of proteins recognized by antibody 470 was submaximal if forskolin/IBMX was added before calyculin A. A difference between these phosphoproteins is that most EF-2 was found in the phosphorylated form under basal conditions (Fig. 4A), the Rb phosphorylation was mixed (Fig. 4C), and the phosphorylation of the proteins recognized by antibody 470 was very low (Figs. 1, 2, and 6). We can hypothesize that less PP2A inhibition was needed to shift the balance toward phosphorylated EF-2 than toward phosphorylated cdk substrates. Different local concentrations of PP2A or different complexes of PP2A with other proteins may also influence PP2A's ability to be inhibited.
Discussion
Protein Phosphatase PP2A as a Target of cAMP Regulation.
We observed a robust decrease in phosphorylation of many proteins after forskolin/IBMX was used to elevate intracellular cAMP. Substrates for various kinases were affected, including cdks, EF-2 kinase, PKC, and the unidentified kinase that phosphorylates protein no. 2. This points to a common phosphatase rather than any specific kinase as the primary target for regulation through the cAMP-stimulated pathway, and inhibitor studies narrowed it to PP2A or a related phosphatase. The obligatory involvement of a phosphatase was supported by the observation that preincubation with calyculin A abolished the forskolin effect. Furthermore, if calyculin A was removed, forskolin/IBMX significantly accelerated net dephosphorylation. Activated PP2A probably directly dephosphorylates proteins such as EF-2, but it may also dephosphorylate various protein kinases, down-regulating their activity (Millward et al., 1999). The final level of phosphorylation would then depend either exclusively on PP2A activation or on coordinated phosphatase/kinase effects, depending on the protein substrate.
In agreement with our data, EF-2 is rapidly dephosphorylated by PP2A during 8-Br-cAMP treatment in permeabilized neuroblastoma cells (Li et al., 1995). PP2A has been shown to dephosphorylate several cdk substrates [high mobility group protein-I(Y), caldesmon, and histone H1] in tissue extracts (Ferrigno et al., 1993). Preincubation of striatal slices with forskolin or dopamine decreased the phosphorylation state of DARPP-32 at Thr75, which is phosphorylated by cdk5, while increasing phosphorylation at Thr34 by PKA (Nishi et al., 2000). In striatal slices, cdk5 activity was not altered, but okadaic acid blocked the effect, implicating PP2A activation. Others have observed forskolin-induced reduction in 32P incorporation into proteins of rat sciatic nerve while investigating Na,K-ATPase regulation in normal and diabetic nerve (Borghini et al., 1994). A number of other reports have also indicated that increased cAMP results in protein dephosphorylation (Christoffersen et al., 1994;Begum and Ragolia, 1996; Mukhopadhyay et al., 1998). The generality of the effect of cAMP on phosphatase activity has not been previously recognized as a common phenomenon, however, perhaps because it does not fit the prevailing paradigm that cAMP acts via PKA.
Decrease in concentration of ATP in forskolin/IBMX-treated cells was not sufficient to prevent kinase action. EF-2, for example, was fully dephosphorylated after 10 to 15 min of forskolin/IBMX, but addition of calyculin A restored its phosphorylation, implying that the protein kinase was still active (Fig. 6A). Phosphorylation of ARPP-19 protein, a known PKA substrate (Dulubova et al., 2001), was increased by forskolin/IBMX in the same experiments (see Fig. 6, A and B). This demonstrates that cAMP can stimulate both phosphorylation and dephosphorylation processes apparently through different pathways.
The Novel cAMP-Stimulated Pathway for PP2A Regulation Is PKA-Independent.
Similar observations were made with L6 myoblast cells, which exhibit a 500-fold increase in cAMP, but dephosphorylation was not seen in C6 glioma cells, which exhibit only a 5-fold cAMP increase with forskolin/IBMX (Feschenko et al., 2000). This suggests that the phenomenon has a high enough threshold that its contribution may depend on the strength of the cAMP signal, and vary from cell to cell depending on the activity of adenylyl cyclase and phosphodiesterase. A high threshold may also distinguish it from activation of PKA. In agreement with our data, concentration-dependent effects of cAMP were reported in the regulation of insect neuronal acetylcholine receptors (Courjaret and Lapied, 2001). cAMP at up to 0.1 mM increased the nicotinic response, and 1 μM forskolin mimicked it. In contrast, internal cAMP concentration higher than 0.1 mM (or 100 μM forskolin) significantly decreased the nicotinic response, and the effect was reversed by 1 μM okadaic acid, implicating a protein phosphatase in the pathway. We observed that treatment of NRK cells with 1 mM dibutyryl-cAMP did not activate PP2A, but it raised intracellular cAMP only 3- to 4-fold, and activated PKA only 40% or less (M. S. Feschenko, unpublished observations). Our data imply that similar to activation of cAMP-GEFs, activation of cAMP-stimulated phosphatase requires higher cAMP levels than activation of PKA.
The cAMP-dependent effects here were not blocked by H89, an inhibitor of PKA catalytic activity, although the drug readily penetrated the cells and inhibited PKA phosphorylation of ARPP-19, a known PKA substrate. Moreover, PKA was inhibited in extracts obtained from H89-pretreated cells. Although H89 may inhibit protein kinases other than PKA, this inhibitor is still regarded as very useful for excluding PKA involvement in cellular processes (Davies et al., 2000). Because it directly inhibits the catalytic subunit activity, rather than antagonizing the RI or RII regulatory and anchoring subunits, unexpected alternative mechanisms for catalytic subunit release and activation are ruled out.
The view that all effects of cAMP are mediated by PKA in eukaryotic tissues has evolved with the discovery of other cAMP-binding proteins, such as cyclic nucleotide-gated ion channels (Li et al., 1997) and the cAMP-binding guanine nucleotide exchange factors (cAMP-GEFs Epac1 and 2; CNrasGEF) (de Rooij et al., 1998; Kawasaki et al., 1998). There are numerous other recent reports in which cell-specific cAMP effects were shown to be PKA-independent (Li et al., 2000; Yusta et al., 2000;Martin et al., 2001; Robert et al., 2001), some implicating cAMP-GEFs (Ozaki et al., 2000; Schmidt et al., 2001). There are apparently also cAMP-dependent events independent of either PKA or known cAMP-GEFs (Wang et al., 2001).
Further investigation will be required to understand how cAMP activates PP2A. cAMP did not activate phosphatase activity directly in disrupted cell extracts, so it must act through a regulatory protein. The mechanism could be as direct as a cyclic nucleotide-binding domain in an undiscovered regulatory subunit of the phosphatase itself. PP2A is a multimeric protein. Its catalytic subunit (C) is usually found tightly associated with subunit A, which serves as a scaffold for the association of one of a variety of interchangeable regulatory B subunits (Janssens and Goris, 2001). Alternatively, PP2A may interact with a regulatory subunit that is a target of one of the other cAMP-activated pathways, activated by a cyclic nucleotide-regulated ion channel or a cyclic nucleotide-binding GEF. A formal possibility is that PP2A may be required to remain active only to maintain some critical pathway protein in a dephosphorylated state, but this seems unlikely because of the evidence that PP2A is the most important phosphatase acting directly on the affected phosphoproteins. Whatever the mechanism, the impact of the phenomenon is likely to be considerable. Thus, cAMP-regulated, PKA-independent PP2A activation may be a major regulatory mechanism involved in cell cycle control and other cellular processes. The ability of particular cells to activate protein phosphatase activity through this cAMP pathway seems to depend on the levels of intracellular cAMP achieved, and may vary in different cell types depending on the activity of adenylate cyclase and phosphodiesterase.
Footnotes
-
This work was supported by National Institutes of Health Grant NS27653 (to K.J.S.), by American Cancer Association Grant IRG-173-J (to M.S.F.), and by National Institutes of Health Grant GM50402 (to A.C.N.).
- Abbreviations:
- PKA
- cAMP-dependent protein kinase
- PKC
- protein kinase C
- PP1 and PP2A
- protein phosphatases 1 and 2A
- cdk
- cyclin-dependent kinase
- EF-2
- elongation factor 2
- Rb
- retinoblastoma protein
- IBMX
- 3-isobutyl-1-methylxanthine
- DMEM
- Dulbecco's modified Eagle's medium
- cAMP-GEF
- cyclic nucleotide-regulated guanine exchange factors
- Received January 28, 2002.
- Accepted March 25, 2002.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}