


Abstract
We have previously reported that CYP3A cross-links with polyubiquitinated proteins in microsomes from nicardipine-treated rats in a process that is distinct from classical polyubiquitination. To further examine the role of the proteasome in CYP3A degradation, we investigated the effects of proteasome inhibitors lactacystin, MG132, proteasome inhibitor 1, and hemin in primary cultures of rat and human hepatocytes. With the exception of hemin, these agents increased the total pool of ubiquitinated proteins in microsomes isolated from rat hepatocytes, indicating that lactacystin, MG132, and proteasome inhibitor 1 effectively inhibited the proteasome in these cells. All four agents caused a reduction in the amount of the major ∼55-kDa CYP3A band, opposite to what would be expected if the ubiquitin-proteasome pathway degraded CYP3A. Only hemin treatment caused an increase in high molecular mass (HMM) CYP3A bands. Because hemin treatment did not alter levels of ubiquitin in CYP3A immunoprecipitates, the HMM CYP3A bands formed in response to hemin treatment clearly were not due to proteasome inhibition. Rather, because hemin treatment also caused an increase in HMM CYP3A in the detergent-insoluble fraction of the 10,000g pellet, the HMM CYP3A seems to represent a large protein complex that is unlikely to primarily represent ubiquitination.
Cytochrome P450 (P450) 3A (CYP3A) is present at high levels in the liver and intestine of humans. Relative to other P450s, the CYP3A subfamily has an unusually large substrate binding pocket, allowing it to metabolize a diverse variety of lipophilic compounds, including approximately 50% of all prescribed drugs (Guengerich, 1999). CYP3A protein has a relatively short half-life compared with most microsomal proteins, including other P450s. Substrates stabilize CYP3A in a process that may be dependent upon protein conformational changes associated with substrate binding (Watkins et al., 1986). In contrast, treatment with “suicide substrates” accelerates CYP3A degradation (Correia et al., 1992). The evidence to date has suggested that the ubiquitin-proteasome system may be responsible for the accelerated degradation of CYP3A observed after treatment with suicide substrates. In one study, animals were cotreated with 3,5-dicarbethoxy-4-ethyl-2,6-dimethyl-1,4-dihydropyridine (DDEP), a CYP3A suicide substrate, and hemin, a proteasome inhibitor (Correia et al., 1992). Treatment with only DDEP increased the amount of ubiquitin detected in hepatic microsomes and in immunoprecipitates of CYP3A. Cotreatment with hemin further increased the amount of polyubiquitinated proteins detected in microsomes from DDEP animals, although the effects of hemin treatment on ubiquitin conjugated to immunoprecipitated CYP3A were not reported. More recently, the proteasome inhibitors aclarubicin and MG132 were used to examine the effects of DDEP on CYP3A degradation in primary cultured rat hepatocytes and were found to decrease the DDEP-mediated loss of both the 55-kDa and HMM CYP3A forms (Wang et al., 1999). Studies examining the degradation of CYP3A expressed in yeast have provided strong evidence that CYP3A is degraded by the proteasome in this system, although it is unclear whether the expressed CYP3A was folded normally or was catalytically active (Murray and Correia, 2001).
We recently developed an in vitro model of CYP3A degradation that involves the conversion of CYP3A to high molecular mass (HMM) conjugates in hepatic microsomes incubated at 37°C (Zangar et al., 2002). Because CYP3A substrates prevented the formation of the HMM CYP3A conjugates in the incubated microsomes (Zangar et al., 2002), this in vitro system provides a mechanistic model for studying substrate-mediated stabilization of CYP3A. Although HMM ubiquitin could be detected in CYP3A immunoprecipitates, it was clear that ubiquitin was conjugated to CYP3A in a process that was distinct from the “classical” ubiquitination process. That is, this reaction did not require cytosol, ubiquitin-activating enzyme E1, ATP, Mg2+, or free monoubiquitin. In addition, CYP3A was apparently in large molar excess to ubiquitin units in these complexes, and the cytosol-mediated degradation of these HMM bands was not affected by proteasome inhibitors. These results indicated that CYP3A was conjugated to a pool of microsomal proteins that are already polyubiquitinated, rather than CYP3A itself being directly ubiquitinated (Zangar et al., 2002). In turn, this conclusion suggests that the presence of ubiquitin in CYP3A immunoprecipitates provides insufficient evidence to determine that classical polyubiquitination of CYP3A has occurred and that processes other than proteasome-mediated degradation may be important in the relatively short half-life of CYP3A.
To further evaluate the role of proteasome-mediated degradation in defining CYP3A protein stability, we examined the effects of proteasome inhibitors on CYP3A protein levels in primary cultured rat hepatocytes. These inhibitors consistently decreased CYP3A protein in microsomal fractions, a result opposite of what would be predicted if CYP3A were degraded by the 26S proteasome. In addition, we found that treatment of cells with hemin resulted in the formation of HMM CYP3A bands in a process that seemed to be unrelated to proteasome inhibition. These HMM CYP3A bands were found to be concentrated in the “detergent insoluble” portion of the 10,000g (S10) pellet, as is typical of large protein aggregates. This last result suggests that the CYP3A complexes potentially may be degraded by a mechanism that is independent of the classical ubiquitin-proteasome system.
Materials and Methods
Chemicals. MG132, proteasome inhibitor 1 [PI1 or Z-Ile-Glu-(OtBu)-Ala-Leu-CHO], and lactacystin were from Calbiochem (San Diego, CA). Monoclonal anti-CYP3A1 was a generous gift from Dr. C. Roland Wolf (Ninewells Hospital and Medical School, Dundee, UK). The polyclonal anti-CYP3A2 antibody used in the immunoprecipitation studies was raised in goat and was obtained from BD Gentest (Bedford, MA). The monoclonal anticystic fibrosis transmembrane conductance regulator (CFTR), anti-ubiquitin, and horseradish peroxidase-conjugated secondary antibody were from Affinity Biore-agents (Golden, CO), Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), and Jackson Immunoresearch Laboratories (West Grove, PA), respectively. SuperSignal West Pico was from Pierce Chemical (Rockford, IL). Other reagents were from Sigma-Aldrich (St. Louis, MO).
Primary Hepatocytes. Primary rat hepatocytes were isolated and cultured on collagen-coated dishes as described previously (Zangar et al., 1995). Three 100-mm dishes were pooled for each sample. Primary human hepatocytes were prepared essentially as described previously (Duanmu et al., 2002), except that they were plated onto Matrigel-coated dishes (1.5 mg of Matrigel/60-mm dish). Nine 60-mm dishes were pooled for each protein sample for the human hepatocyte study. Hepatocytes were cultured without treatment for 3 days before initiation of a 30-h treatment with 2 mM phenobarbital. For the last 6 h of phenobarbital treatment, some cells were cotreated with 20 μM lactacystin, 200 μM MG132, 200 μM proteasome inhibitor 1, 100 μM hemin, or 0.2% ethanol (vehicle for MG132 and proteasome inhibitor 1). The proteasome inhibitors, lactacystin, MG132, and proteasome inhibitor 1, were prepared from a previously unopened manufacturer's vial immediately before cell treatment. This step seemed to be necessary to achieve a reproducibly high level of proteasome inhibition.
Subcellular Fractionation and Western and Northern Blots. Subcellular fractions were prepared essentially as described previously (Zangar et al., 1992, 1995). Briefly, cultured cells were harvested, washed, and homogenized in 50 mM KPO4, 250 mM sucrose, 1 mM EDTA, 100 μg of phenylmethylsulfonyl fluoride/ml, 0.2 units of aprotinin/ml, 10 μg of leupeptin/ml, and 10 mM N-ethylmaleimide (homogenization buffer). Cell homogenates were centrifuged at 10,000g for 10 min at 4°C to prepare the S10 pellet and supernatant. The S10 pellet was washed by suspending in 1 ml of homogenization buffer and repeating the centrifugation step. The washed pellet was suspended in 200 μl of homogenization buffer containing 0.5% NP40 and vortexed vigorously at 4°C for 30 min. The samples were then centrifuged at 10,000g for 10 min, 4°C and the supernatants saved as the “NP40-soluble” fraction. The remaining pellets were washed with 500 μl of homogenization buffer containing 0.5% NP40, collected by centrifugation, and suspended by sonication in 1% lauryl sulfate, 62.5 mM Tris-Cl, 1.5 M glycerol, and saved as the “NP40-insoluble” S10 pellet fraction. Samples were stored at –80° and were typically analyzed by Western blot analysis within 1 to 7 days of cell harvest.
The S10 supernatant was spun at 100,000g for 1 h. The supernatant was saved as the cytosol fraction. The pellet was first gently washed and then resuspended by sonication in 50 mM KPO4, 1 mM EDTA. Samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and analyzed by Western blotting, as described previously (Kim et al., 2001; Zangar et al., 2002). Typically, samples were diluted in an equal volume of loading buffer [62.5 mM Tris, pH 6.8, 1% lauryl sulfate, 11% glycerol, 370 μM bromphenol blue, and 700 mM (5% by volume) 2-mercaptoethanol] and then heated to 65°C for 5 min. Alternatively, to determine whether increased detergent might alter the levels of HMM CYP3A, samples were also diluted in 62.5 mM Tris, pH 6.8, 11% glycerol, 370 μM bromphenol blue, 10% lauryl sulfate, 40 mM dithiothreitol, and 560 mM (4%) 2-mercaptoethanol before heating. Microsomal and NP40-insoluble fractions were loaded at 20 μg of protein/lane, whereas other fractions were loaded at 30 μg/lane. Protein was then transferred to nitrocellulose membrane (Bio-Rad, Hercules, CA) using a Bio-Rad Transblot apparatus operated at 50 V for 2 h, with 4°C circulating water for cooling. Blots were blocked in 5% milk powder and probed with 1:10,000 dilution of primary antibody in buffered saline (pH 7), 0.1% bovine serum albumin, and 0.05% Tween 20. Blots were then probed with a 1:5000 dilution of goat anti-mouse secondary antibody in saline (pH 7), 0.05% Tween 20, and 5% powdered milk. Protein levels were determined using SuperSignal chemiluminescent reagents and were imaged using a Lumi-Imager F1 (Roche Diagnostics, Indianapolis, IN).
Total RNA was isolated from individual 60-mm dishes of human hepatocytes (two dishes per treatment group) as described previously (Xie and Rothblum, 1991). Northern blot analyses for CYP3A and 7S RNA (used to control for RNA loading) were performed as described previously (Kocarek et al., 2002).
Microsomal Incubations. Hepatic microsomes were prepared from rats that had been treated with 100 mg of nicardipine/kg/day for 7 days, as described previously (Zangar et al., 1999) and were stored as aliquots at –70°C for 1 year or more before use. Treatment of rats with nicardipine induces high levels of microsomal CYP3A, and microsomes from these animals spontaneously form HMM CYP3A conjugates when incubated at 37°C (Zangar et al., 1999, 2002). Microsomes were incubated at 37°C using 75 μg of microsomal protein, 50 mM Tris, pH 7.5, 25 mM sucrose, 0.154 mM KCl, 2 mM CaCl2, and 3 μM ZnCl2 in a total volume of 50 μl. Reactions were terminated by addition of 50 μl of loading buffer [62.5 mM Tris, pH 6.8, 1% lauryl sulfate, 11% glycerol, 370 μM bromphenol blue, and 700 mM (5% by volume) 2-mercaptoethanol] and heating to 65°C for 5 min. Samples were then analyzed by Western blotting, as described above. Twelve micrograms of protein was loaded per lane for these samples.
CYP3A catalytic activity was measured using erythromycin N-demethylase activity, as described previously (Wrighton et al., 1985). Proteasome inhibitors were added to the microsomes at concentrations that were found to stimulate an increase in polyubiquitinated proteins in primary cultured rat hepatocytes in this study (see below) or, in the case of aclarubicin, were used by others for proteasome inhibition (Wang et al., 1999).
CYP3A Immunoprecipitation Analyses. The immunoprecipitation studies were undertaken essentially as described previously (Zangar et al., 2002). Each microsome sample (100 μg of protein) was suspended in 400 μl of immunoprecipitation buffer (RIPA; final concentration was 0.9% NaCl, 0.1 M sodium phosphate, pH 7.4, 1% NP40, 0.5% sodium deoxycholate, 0.1% sodium lauryl sulfate, 0.1 mg/ml phenylmethylsulfonyl fluoride, 0.07 mg/ml aprotinin, 1 mM orthovanadate, and 100 μM butylated hydroxytoluene) and added to 100 μl of beads containing covalently attached anti-CYP3A2. Samples were gently mixed at 4°C for 4 h. The beads were collected in Micro Bio-Spin columns (Bio-Rad), drained by gravity flow, and washed with 2 ml of RIPA. CYP3A was gently eluted at room temperature using 1 ml of 0.1 M glycine, 1% NP40, and 1% CHAPS, pH 2.2. The protein was precipitated by adding trichloroacetic acid to a final concentration of 20% (w/v), pelleted by centrifugation, washed in 70% ethanol, and repelleted. The pellets were suspended directly in loading buffer and analyzed by Western procedures, as described above.
Statistics. Data were initially analyzed using a one-way analysis of variance. Multiple comparisons were undertaken using Tukey's test. P < 0.05 was used to define significant differences on all tests.
Results
Proteasome inhibitors have been reported to maintain CYP3A levels in primary cultured rat hepatocytes when added before treatment with DDEP (Wang et al., 1999), but the effects of these agents on CYP3A levels in the absence of treatment with suicide substrates have not been reported. Therefore, we treated cells with lactacystin, hemin, and MG132 for 6 h. Lactacystin and MG132 inhibited the proteasome in primary cultured rat hepatocytes, as demonstrated by respective ∼122 and 79% increases in the amounts of polyubiquitinated proteins presence in microsome samples (Fig. 1). However, treatment with hemin did not significantly alter levels of ubiquitinated proteins.

Effects of treatment with proteasome inhibitors on microsomal CYP3A levels in primary cultured rat hepatocytes. Cells were cultured for 3 days before 30-h treatment with 2 mM phenobarbital alone (Con) or with cotreatment for the last 6 h with 20 μM lactacystin (Lac), 100 μM hemin (Hem), 200 μM MG132 (MG), or 0.2% ethanol (EtOH). Microsomal fractions were prepared and analyzed by Western blot techniques using CYP3A-, ubiquitin-, or CFTR-specific antibodies. Lactacystin and hemin stocks were prepared in water. MG132 was prepared and added such that 0.2% ethanol was present in the medium. A, representative Western blots from a single hepatocyte preparation. The approximate mass of the protein bands is shown on the right of the Western blots and was determined using prestained protein standards. B, graph showing combined results from two (CFTR) or three (all others) primary hepatocyte preparations, with two or three separate microsomal samples prepared and analyzed for each hepatocyte preparation (total sample numbers vary from four to seven). As such, each column and crossbar represent the mean and S.E., respectively, of four to seven analyses. The arrowhead and bracket in the CYP3A blot in A represent the band or range of bands used to quantitate the 55-kDa CYP3A and HMM CYP3A, respectively. Values are statistically significant from both the Con and EtOH treatment groups (*, P < 0.05).
Opposite to their effects on ubiquitinated protein levels, treatment with the proteasome inhibitors consistently reduced microsomal CYP3A protein levels (Fig. 1). Specifically, mean levels of the 55-kDa CYP3A band were decreased ∼37, 30, or 53% in cells treated with lactacystin, hemin, or MG132, respectively, relative to control levels. CFTR is known to be degraded by the proteasome when improperly folded after synthesis (Gelman et al., 2002) and was included as a control here. Western blot analysis of CFTR indicated that levels of this protein were not altered after treatment with lactacystin, hemin, or MG132 (Fig. 1), suggesting that CFTR is efficiently folded in the primary cultured hepatocytes and therefore not degraded by the proteasome. More importantly, this result suggests that the down-regulation of CYP3A observed after treatment with proteasome inhibitors is selective for CYP3A. Additional studies were undertaken using proteasome inhibitor 1 and results were similar to those observed with lactacystin and MG132. That is, treatment with proteasome inhibitor 1 significantly increased microsomal levels of ubiquitinated proteins by 63%, decreased levels of the ∼55-kDa CYP3A band by 52%, but did not alter HMM CYP3A levels (proteasome inhibitor 1 results are from two separate primary hepatocyte preparations, with two individual microsomal samples prepared for each cell preparation, for four samples total). Overall, these data demonstrate that proteasome inhibition decreased microsomal CYP3A levels in primary cultured rat hepatocytes and that this effect was selective for CYP3A.
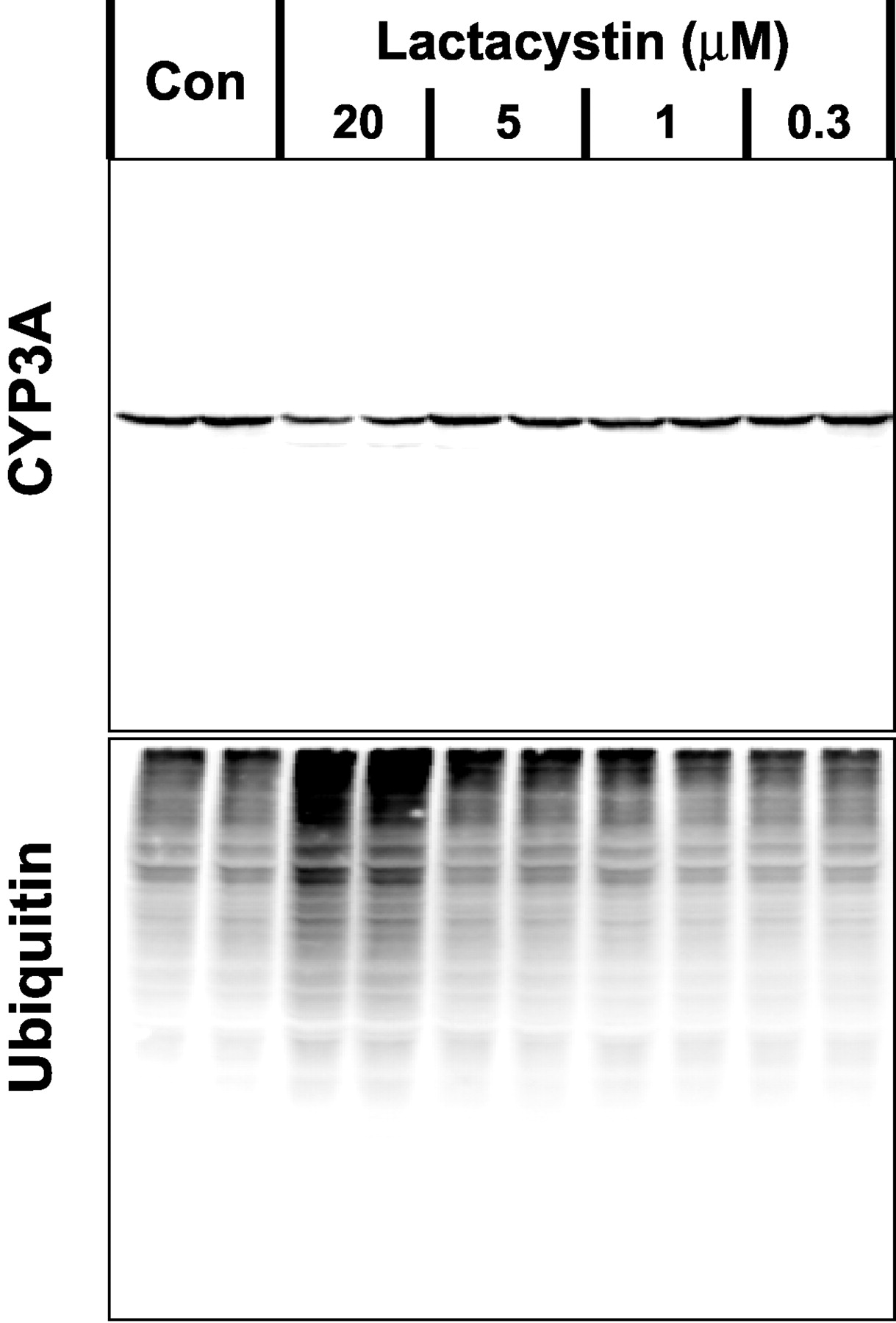
To determine whether there was an inverse correlation between microsomal CYP3A levels and inhibition of the proteasome, the concentration response of lactacystin was determined. We found that 20 μM lactacystin caused a marked increase in ubiquitinated proteins and a clear suppression of microsomal CYP3A levels (Fig. 2). Lower concentrations of lactacystin had negligible effects on either CYP3A levels or ubiquitinated protein levels.

Concentration-dependent effects of lactacystin on microsomal CYP3A levels in primary cultured rat hepatocytes. Cells were cultured for 3 days before 30-h treatment with 2 mM phenobarbital alone (Con) or were cotreated for the last 6 h of culture with 0.3 to 20 μM lactacystin. Microsomal fractions were prepared and analyzed by Western blot techniques using CYP3A- and ubiquitin-specific antibodies. Data are representative of two replicate analyses from different preparations of primary cultured rat hepatocytes.
If CYP3A is degraded by the proteasome in primary cultured hepatocytes, then treatment with proteasome inhibitors would be predicted to increase the amount of HMM CYP3A. Hemin was the only agent we studied that resulted in an increase in HMM CYP3A bands (Fig. 1). To determine whether these HMM CYP3A bands might be disrupted by increasing amount of detergent, microsomes from control, lactacystin-treated, or hemin-treated cells were diluted 1:1 in loading buffer containing either 1% (our usual concentration) or 10% lauryl sulfate. After heating, paired samples were then loaded side by side on a single gel, and CYP3A levels were analyzed by Western blot analysis. Paired samples seemed identical regardless of the loading buffer used (data not shown), indicating that the HMM CYP3A could not be dissociated with increased amounts of detergent. Such a result is consistent with other studies on HMM CYP3A bands we have undertaken with the microsome samples, where samples that were prepared in loading buffer containing a final concentration of 8 M urea or 1 M dithiothreitol did not alter the levels of HMM CYP3A detected in Western blot analyses (A. L. Kimzey, N. Bollinger, K. K. Weitz, and R. C. Zangar, unpublished data).
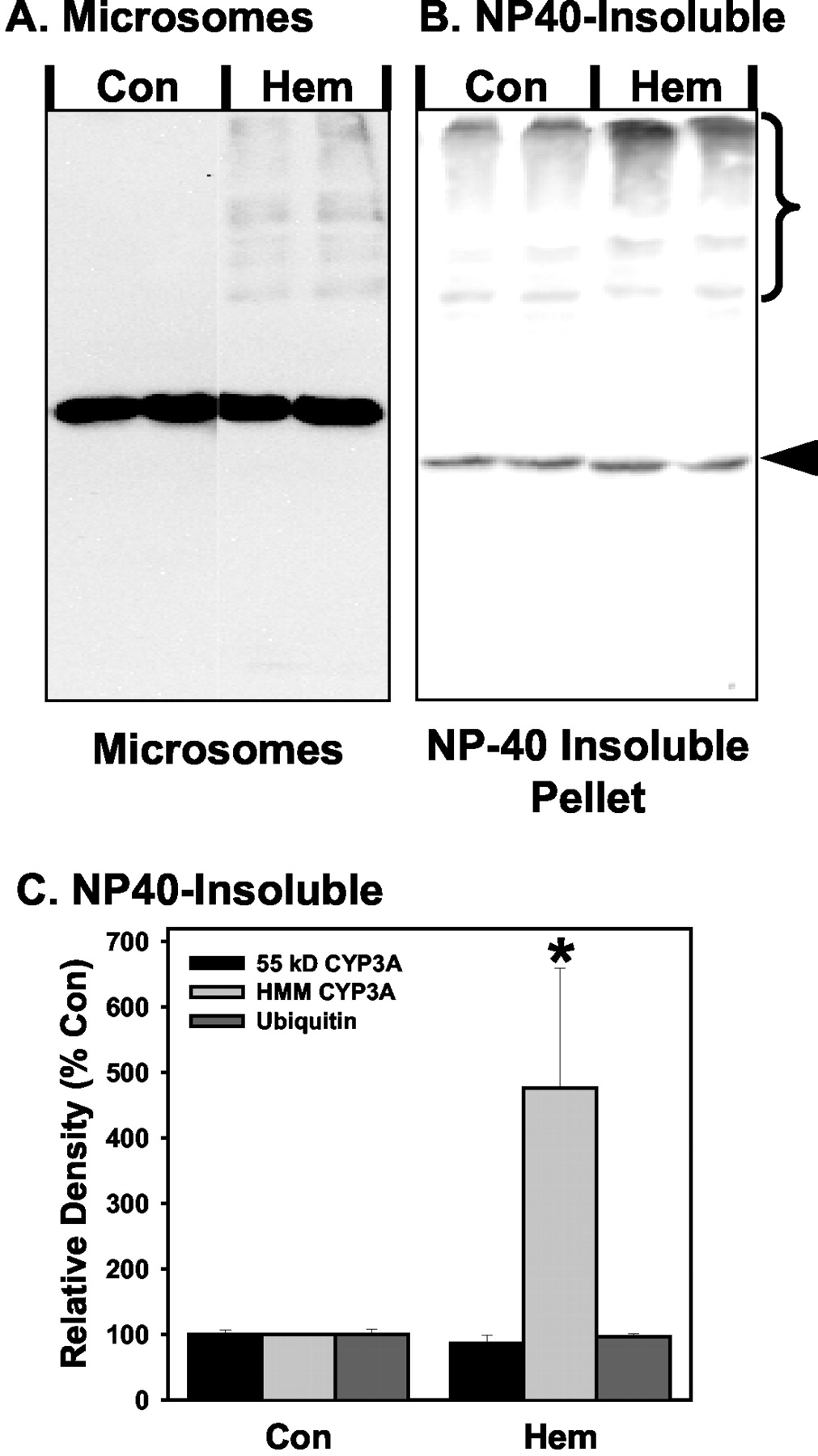
We were unable to detect any CYP3A in cytosolic fractions by Western blot analysis (data not shown). Analysis of the NP40-soluble portion of the S10 pellet only demonstrated a weak signal that otherwise looked identical to the strong signal in the microsome fraction (data not shown), suggesting there may be a low-level contamination of microsomal protein in the NP40-soluble fraction. In contrast, in the NP40-insoluble fraction obtained from the S10 pellet, significant amounts of CYP3A were detected (Fig. 3). Similar to microsomes, hemin treatment resulted in an increase in the amount of HMM CYP3A present in the NP40-insoluble fraction.

Effect of treatment with hemin on CYP3A levels in NP40-insoluble fractions from primary cultured rat hepatocytes. Cells were cultured for 3 days before 30-h treatment with 2 mM phenobarbital alone (Con) or cotreated for the last 6 h with 100 μM hemin (Hem). Microsomal fractions and the NP40-insoluble fractions were prepared and analyzed by Western blot techniques using CYP3A-specific antibodies. Representative Western blots of microsomal (A) and NP40-insoluble (B) fractions from the same hepatocyte preparation are shown. C, graph of combined results from two separate primary hepatocyte preparations, with each preparation having subcellular samples prepared in duplicate from separate dishes of cells. As such, each column and crossbar represent the mean and S.E., respectively, of four analyses. The arrowhead and bracket in the CYP3A blot in A represent the band or range of bands used to quantitate the 55-kDa CYP3A and HMM CYP3A, respectively. Values are statistically significant from the Con group (*, P < 0.05).
Immunoprecipitation studies were performed to examine the role of ubiquitination in the formation of HMM CYP3A bands. As we have noted previously, levels of HMM CYP3A do not immunoprecipitate as efficiently as the ∼55-kDa band, presumably due to steric hindrance (Zangar et al., 2002). Even so, consistent with results observed in microsomes, levels of HMM CYP3A were increased over 50% in immunoprecipitates from microsomes from hemin-treated cells relative to control cells or cells treated with lactacystin (Fig. 4). Low levels of HMM ubiquitin were also detected by Western blot analysis in the CYP3A immunoprecipitates, although no increase of ubiquitinated proteins was observed in the microsomes from hemin-treated cells compared with controls (Fig. 4). In contrast, there was a clear increase in HMM ubiquitin in the immunoprecipitated sample from the lactacystin-treated cells (Fig. 4).

Identification of HMM ubiquitin in CYP3A immunoprecipitates. CYP3A was immunoprecipitated from microsomes from primary cultured rat hepatocytes, as described under Materials and Methods. Identical Western blots were probed using antibodies specific for CYP3A or ubiquitin. Ten micrograms of microsomal protein per lane (Micros.) that represents the samples before immunoprecipitation, or equal amounts of immunoprecipitated samples (IP) that were loaded and analyzed on a single blot are shown. Sample codes are beads-only mock immunoprecipitation (M), microsomes from cells that were untreated (C) or treated with lactacystin (L) or hemin (H). The asterisk (*) indicates nonspecific bands detected in immunoprecipitated samples that comigrate with the heavy chain of the antibody.
To determine whether proteasome inhibitors had similar effects on CYP3A levels in human cells, we exposed primary cultured human hepatocytes to lactacystin and hemin. Six-hour treatment with lactacystin or hemin reduced microsomal CYP3A protein levels by 26 or 41%, respectively (Fig. 5). In contrast to rat hepatocytes, hemin treatment did not result in an increase in detectable levels of HMM CYP3A in any subcellular fraction. Similar to changes in microsomal protein levels, lactacystin and hemin treatment decreased CYP3A mRNA levels by 20 and 24%, respectively (Fig. 5).

Effects of proteasome inhibitors on CYP3A protein and mRNA levels in primary cultured human hepatocytes. Human hepatocytes were cultured for 3 days before 30-h treatment with 2 mM phenobarbital alone (Con) or were cotreated for the last 6 h with 20 μM lactacystin (Lac) or 100 μM hemin (Hem). Microsomal fractions or total RNA were analyzed by Western blot or Northern blot analysis, respectively, as described under Materials and Methods. The 7S RNA band intensity was used to normalize for loading differences between lanes.
CYP3A substrates are known to stabilize CYP3A protein in vivo, in primary cultured rat hepatocytes and in vitro (Watkins et al., 1986; Eliasson et al., 1994; Zangar et al., 2002). Given that approximately half of all prescribed drugs are believed to be metabolized by CYP3A (Guengerich, 1999), care must be taken when selecting pharmacological tools to ensure that these agents are not CYP3A substrates that may affect protein levels by substrate-mediated stabilization. For this reason, we examined the effects of a series of proteasome inhibitors, used in this study or by others (Wang et al., 1999), on a model CYP3A-catalyzed reaction, erythromycin N-demethylase activity. Clotrimazole, a high-affinity CYP3A substrate, was also included in this analysis as a positive control. Clotrimazole nearly completely blocked CYP3A catalytic activity (Fig. 6). When at the same concentration used to inhibit proteasome activity, we found that aclarubicin decreased CYP3A activity by about 20% (Fig. 6). Because erythromycin is present at a 20-fold greater concentration than aclarubicin, these results suggest that CYP3A may have a significant affinity for aclarubicin when there is not an excess of competitive substrate. MG132 and lactacystin seemed to have no effect on CYP3A activity.

Effects of proteasome inhibitors on CYP3A catalytic activity. Erythromycin N-demethylase activity in hepatic microsomes from rats treated with nicardipine was assayed in the presence of 0.1% acetonitrile alone (Con) or cotreated with 50 μM aclarubicin (ACL), 200 μM MG132, 20 μM lactacystin (LAC), or 10 μM clotrimazole (CTZ).
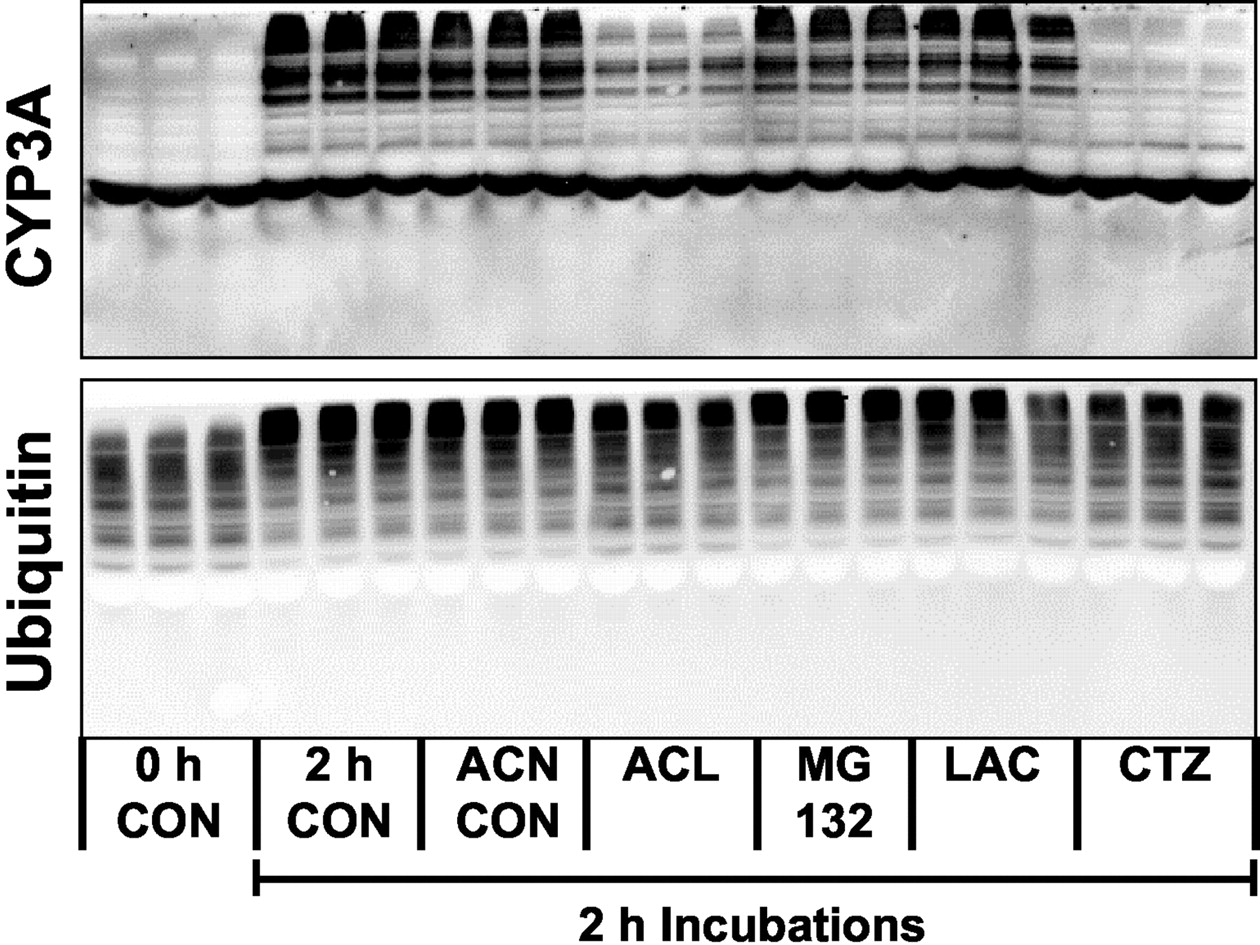
We have previously described an in vitro incubation system in which CYP3A is converted to HMM bands that are degraded in the presence of cytosol (Zangar et al., 2002). Because CYP3A substrates block this process, it is likely that this system is a mechanistic model of the processes associated with substrate-mediated stabilization of CYP3A. Therefore, the ability of drugs to block the formation of HMM CYP3A in this system is likely to be a good predictor of substrate-mediated stabilization effects in living cells. Consistent with previous results, HMM CYP3A was formed in hepatic microsomes from nicardipine-treated rats when the microsomes were incubated for 2 h at 37°C (Fig. 7). Addition of acetonitrile, a solvent control, was without effect on this reaction, whereas clotrimazole effectively blocked the formation of HMM CYP3A. Aclarubicin was also effective in blocking this reaction. MG132 and lactacystin did not seem to have any effect. None of the agents examined blocked the upward migration of ubiquitin that is also observed in the incubated microsomes (Zangar et al., 2002), suggesting that aclarubicin's inhibition of the formation of the HMM CYP3A bands were due to specific interactions with the CYP3A protein.

Effects of proteasome inhibitors on the formation of HMM CYP3A bands in incubated microsomes. Hepatic microsomes from nicardipine-treated rats were incubated at 37°C for 0 or 2 h in the presence of 0.1% acetonitrile (ACN, solvent control for aclarubicin, MG132, and clotrimazole), 50 μM aclarubicin (ACL), 200 μM MG132, 20 μM lactacystin (LAC), or 10 μM clotrimazole (CTZ).
Discussion
Evidence has been presented that the ubiquitin-proteasome system is responsible for the degradation of CYP3A, at least after inactivation with DDEP (Correia et al., 1992; Wang et al., 1999). However, in this study, we found that treatment with proteasome inhibitors resulted in the loss of microsomal CYP3A in primary cultured rat hepatocytes, a result opposite of what would be expected if the ubiquitin-proteasome system contributed to the relatively short half-life of CYP3A protein that is observed even in the absence of suicide substrates. Because proteasome inhibitors also decreased CYP3A mRNA levels, it seems that these agents act at a pretranslational level to suppress CYP3A expression. Possibly the likeliest explanation for this effect is that there is a protein that inhibits CYP3A transcription that is degraded by the proteasome. When the proteasome is inhibited, levels of this suppressor protein increase, resulting in decreased expression of CYP3A. However, we cannot rule out that changes in protein stability may have occurred in this study. We observe an increase in HMM ubiquitin in CYP3A immunoprecipitates from lactacystin-treated cells (Fig. 4), suggesting that some CYP3A may be degraded by the proteasome. In this light, it may be that the reason that lactacystin treatment does not increase HMM CYP3A is due to suppression of the precursor ∼55-kDa CYP3A. However, we have previously observed that CYP3A can cross-link to other ubiquitinated proteins in incubated microsomes independent of classical ubiquitination processes (Zangar et al., 2002). Therefore, an alternative explanation is that the increased levels of ubiquitin in the immunoprecipitated CYP3A samples simply reflected an increase in the pool of ubiquitinated microsomal proteins available for cross-linking and was not related to classical ubiquitination of CYP3A.
We examined several proteasome inhibitors that had previously been reported to affect CYP3A degradation. Notably, a recent study reported that CYP3A was stabilized in the presence of aclarubicin (Wang et al., 1999). Aclarubicin is an antineoplastic agent with a mass of 812 Da, too big for most P450s to metabolize but typical of CYP3A substrates. We found that aclarubicin inhibited CYP3A catalytic activity, as would be expected of a CYP3A substrate. More importantly, we found that aclarubicin blocked the formation of HMM CYP3A conjugates in incubated microsomes. Because the formation of these HMM CYP3A conjugates seems to precede CYP3A proteolysis but is independent of classical ubiquitination processes (Zangar et al., 2002), the ability of aclarubicin to interfere with this process suggests that it has an effect on CYP3A stability that is independent of proteasomal inhibition. That is, the inhibition of the CYP3A conjugation in incubated microsomes suggests that aclarubicin is likely to stabilize CYP3A in living cells at concentrations used for proteasome inhibition.
Wang et al. (1999) found that MG132 and lactacystin were ineffective in freshly isolated primary rat hepatocytes unless glutathione levels were suppressed. However, we found these agents to be effective proteasome inhibitors in cultured primary rat hepatocytes without suppression of glutathione. The differences between these two studies may reflect differences between cultured and freshly isolated hepatocytes. However, because primary rat hepatocytes cultured for 1 to 6 days have levels of intracellular glutathione that are at least as great as those in freshly isolated cells (Lii et al., 1996), the differences between these studies are unlikely to be due to higher glutathione levels in freshly isolated cells.
Increases in HMM CYP3A that were observed after hemin and DDEP cotreatment have been used as evidence that inactivated CYP3A is degraded by the proteasome (Correia et al., 1992). We also found that hemin treatment of primary cultured hepatocytes increased HMM CYP3A. However, it seems unlikely that this effect was the result of proteasome inhibition, because hemin did not seem to inhibit the proteasome in this study and because the three active proteasome inhibitors we examined did not increase HMM CYP3A. This conclusion is further supported by evidence that levels of ubiquitin were not increased in CYP3A samples immunoprecipitated from hemin-treated cells (Fig. 4). Although hemin has been reported to inhibit proteasomal activity by ∼50% at 25 μM in in vitro experiments, hemin concentrations of 500 μM typically have been used in studies on living cells (Etlinger and Goldberg, 1980; Vierstra and Sullivan, 1988). Therefore, it seems likely that the 100 μM hemin used in this study was insufficient to block proteasome activity in the cultured hepatocytes. We have observed that oxidative stress is likely to be an important factor in the formation of the HMM CYP3A conjugates (A. L. Kimzey, N. Bollinger, K. K. Weitz, and R. C. Zangar, unpublished observations). Therefore, one possible explanation for the formation of the HMM CYP3A bands is that hemin released free iron, which could have acted as a pro-oxidant.
Overall, our data suggest that previous studies examining CYP3A degradation have suffered from the use of proteasome inhibitors with nonspecific properties. That is, these studies have relied primarily on aclarubicin and hemin, which seem to have effects on CYP3A protein independent of proteasome inhibition. Furthermore, because proteasome inhibitors may suppress CYP3A mRNA levels, it seems that any data on CYP3A protein degradation obtained with these inhibitors must be interpreted with care. Based on these conclusions and previous evidence that CYP3A-ubiquitin conjugates can be formed independent of classical ubiquitination processes and are not degraded by the proteasome (Zangar et al., 2002), mechanisms in addition to the ubiquitin-proteasome pathway must be considered as potentially contributing to the relatively short half-life of CYP3A.
Aggregates of endoplasmic reticulum proteins that are not degraded by the proteasome have been described previously. A mutant form of urate oxidase was found to form large aggregates that contained ubiquitin but seemed to be degraded by processes other than the proteasome (Yokota et al., 2000). Similar complexes called “aggresomes” have been reported (Kopito, 2000). These aggresomes may contain ubiquitin but are potentially degraded by an autophagic route as well as by the proteasome (Kopito, 2000). One of the characteristics of aggresomes is that they tend to be dense particles that are poorly soluble in detergents (Kopito, 2000). For this reason, we examined the S10 pellet for NP40-insoluble CYP3A complexes. In contrast to the decrease in the ∼55-kDa CYP3A band observed in microsomes (Fig. 1), there was no change in the ∼55-kDa CYP3A band in the NP40-insoluble fraction with hemin treatment (Fig. 3) or in response to lactacystin or MG132 (data not shown). In addition, CFTR was readily detectable in microsome samples (Fig. 1) but was undetectable in the NP40 insoluble fraction (data not shown). Therefore, it is clear that the NP40-insoluble and microsomal fractions represent distinct pools of protein. We found that hemin treatment of primary hepatocytes not only stimulated the formation of HMM CYP3A bands in microsomes but also in the NP40-insoluble fraction of the S10 pellet (Fig. 3). However, in the NP40-insoluble fraction, the amount of HMM CYP3A was greater than the ∼55-kDa CYP3A band that represents the intact apoprotein. In contrast, levels of the ∼55-kDa CYP3A band were in large excess to the HMM CYP3A in microsomes. Even across the range of HMM CYP3A bands, the HMM CYP3A were predominately of a greater mass in the NP40-insoluble fraction than in microsomes (Fig. 3). That is, the proportion of the HMM CYP3A that is above 200 kDa compared with the proportion that is in the 80- to 200-kDa range is greater in the NP40-insoluble fraction compared with the microsomes. These results suggest that hemin treatment not only stimulated the formation of HMM CYP3A but that, once formed, the HMM CYP3A was primarily recovered in the NP40-insoluble fraction.
Although we cannot rule out a role for the proteasome in degradation of the HMM CYP3A complexes from the current studies, the results of this study suggest that alternative routes of degradation may be important in the absence of exposure to CYP3A suicide substrates. Taken together, the results presented here and in a previous report (Zangar et al., 2002) suggest a novel hypothesis about how CYP3A protein may be degraded. That is, CYP3A is initially cross-linked to ubiquitinated proteins and forms a large protein complex in a process that is increased by hemin treatment. This complex may be degraded by either an autophagic mechanism or by the proteasome. The presence of these HMM complexes has apparently been overlooked in previous studies on P450 degradation, because previous studies did not examine the detergent-insoluble, 10,000g pellet that contains most of the HMM CYP3A complexes.
Footnotes
-
This work was supported by National Institutes of Health Grants DK54812 (to R.C.Z., S.S., N.B.), HL50710, and P30-ES06639 (to T.A.K.). D.W.L. was supported by the Community College Initiative in Science and Technology, funded through U.S. Department of Energy's Office of Science.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
DOI: 10.1124/jpet.102.044628.
-
ABBREVIATIONS: P450, cytochrome P450; DDEP, 3,5-dicarbethoxy-2,6-dimethyl-4-ethyl-1,4-dihydropyridine; HMM, high molecular mass; PI1, proteasome inhibitor 1 or Z-Ile-Glu(OtBu)-Ala-Leu-CHO; CFTR, cystic fibrosis transmembrane conductance regulator; S10, pellet and supernatant fractions formed from the 10,000g centrifugation of hepatocyte homogenates; CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]propanesulfonate; RIPA, radioimmunoprecipitation assay.
- Received September 19, 2002.
- Accepted February 19, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}