


Abstract
About one-third of epilepsy patients are resistant to treatment with antiepileptic drugs (AEDs). This refractoriness is not fully understood, but is thought to be attributed to overexpression of multidrug transporters at the blood-brain barrier, particularly P-glycoprotein (Pgp). In certain cases pharmacoresistance can be overcome by add-on therapy, raising the question of whether the coadministered drugs act as inhibitors of Pgp. Indeed, several AEDs are substrates and to some extent also inducers of Pgp. To date nothing is known about possible Pgp inhibitory activities of AEDs. Therefore, we investigated whether AEDs commonly used in mono or add-on therapy inhibit Pgp using a calcein acetoxymethylester uptake assay with L-MDR1 cells and primary porcine brain capillary endothelial cells, as well as confocal laser-scanning microscopy with L-MDR1 cells and bodipy-verapamil as Pgp substrate. In the calcein assay only carbamazepine inhibited Pgp, which was confirmed in confocal laser-scanning microscopy. In this assay, also phenytoin, lamotrigine, and valproate revealed Pgp inhibitory potency. Because Pgp inhibition by AEDs appeared at significantly higher concentrations than that of well known inhibitors and because the effective concentrations exceeded therapeutic AED plasma concentrations, Pgp inhibition appears not to be of clinical relevance, at least in antiepileptic monotherapy. However, it is possible that this inhibition may contribute to the effectiveness of certain add-on therapies.
About 30% of epileptic patients do not respond to the usual antiepileptic drugs (AEDs), representing a major health problem associated with increased morbidity and mortality (Regesta and Tanganelli, 1999). An important characteristic of pharmacoresistant epilepsy is that these patients do not respond to most, and often all, AEDs, pointing to nonspecific and possibly adaptive mechanisms. The underlying cause is not completely understood; mechanisms leading to AED resistance are most likely complex network phenomena and may include development of tolerance to AED action or alterations in drug targets (including genetic factors). A currently favored hypothesis is the overexpression of multidrug transporters, especially P-glycoprotein (Pgp), in the blood-brain barrier (BBB) (Abbott et al., 2002; Löscher and Potschka, 2002), restricting the access of these drugs to their site of action. Pgp (gene product of the multidrug resistance 1 (MDR1) gene) contributes to renal, biliary, and intestinal elimination of drugs and is expressed at the BBB (Schinkel et al., 1994; van Asperen et al., 1997, 1998; Kusuhara et al., 1998; Drescher et al., 2003). At the BBB Pgp is localized in the apical membrane of brain capillary endothelial cells (BCECs), where it transports substrates toward the blood compartment (Cordon-Cardo et al., 1989; van Asperen et al., 1997). Therefore, this transporter can limit the penetration into and retention within the brain and thus modulate effectiveness and central nervous system toxicity of numerous compounds (Schinkel et al., 1994, 1996).
The brain expression of Pgp is markedly increased in the majority of patients with intractable partial epilepsy (Tishler et al., 1995), suggesting that at least in some resistant patients the overexpression of drug transporters might contribute to AED refractoriness. This assumption was further supported by other studies demonstrating an overexpression of multidrug transporters in BCECs and brain tissue from treatment-resistant patients (Dombrowski et al., 2001; Sisodiya et al., 2002). Such an overexpression may be of importance because AEDs such as phenytoin, carbamazepine, phenobarbital, and lamotrigine are Pgp substrates (Schinkel et al., 1996; Potschka and Löscher, 2001a; Potschka et al., 2001, 2002) and phenytoin, carbamazepine, and valproate are substrates of the multidrug resistance-associated protein (MRP) (Huai-Yun et al., 1998; Potschka et al., 2001; Potschka and Löscher, 2001b). Furthermore, recent evidence suggests a possible role of the C3435T polymorphism of the MDR1 gene in pharmacoresistant epilepsy (Siddiqui et al., 2003).
Interestingly, in certain cases pharmacoresistance can be overcome by add-on therapy with a second AED (Marson et al., 2000; Ramaratnam et al., 2001; Jette et al., 2002), raising the question of whether the coadministered drug acts by inhibition of Pgp. We therefore investigated the Pgp inhibitor potency of the most prevalent AEDs using the well established in vitro calcein assay and confocal laser-scanning microscopy with bodipy-verapamil as Pgp substrate.
Materials and Methods
Materials. Culture media, fetal calf serum, medium supplements, antibiotics, and Hanks' balanced salt solution (HBSS) were purchased from Invitrogen (Karlsruhe, Germany), collagenase/dispase and dispase were from Roche Applied Science (Mannheim, Germany), collagen-R from Serva (Heidelberg, Germany), DMSO and Triton X-100 were from AppliChem (Darmstadt, Germany), dextran and poly-d-lysine from Sigma-Aldrich (Taufkirchen, Germany), Percoll from Amersham Biosciences (Freiburg, Germany), calcein-AM and bodipy-verapamil from MoBiTec (Göttingen, Germany), vincristine from Merck (Darmstadt, Germany), 96-well microtiter plates and culturing bottles were from NUNC GmbH & Co. KG (Wiesbaden, Germany), and the coverslips for confocal laser-scanning microscopy from H. Saur (Reutlingen, Germany).
Drugs. Gabapentin was a kind gift from Gödecke/Parke Davis AG (Freiburg, Germany), lamotrigine from GlaxoSmithKline (Uxbridge, Middlesex, UK), LY335979 was obtained from Eli Lilly Company (Bad Homburg, Germany), topiramate from Janssen-Cilag (Neuss, Germany), and vigabatrin from Aventis (Bad Soden, Germany). Carbamazepine was purchased from ICN (Eschwege, Germany), sodium phenytoin, sodium phenobarbital, sodium valproate, and verapamil were from Sigma-Aldrich.
LLC-PK1 and L-MDR1 Cells. As a model for human Pgp we used L-MDR1 cells, a cell line generated by transfection of the porcine kidney epithelial cell line LLC-PK1 with the human MDR1 gene (Schinkel et al., 1996) and the parental cell line LLC-PK1 (American Type Culture Collection, Manassas, VA) as a control. The L-MDR1 cell line was kindly provided by Dr. A. H. Schinkel (The Netherlands Cancer Institute, Division of Experimental Therapy, Amsterdam, The Netherlands). The cells were cultured under standard cell culture conditions with medium M199 supplemented with 10% heat-inactivated fetal calf serum, 2 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin sulfate. To maintain Pgp expression, the culture medium for L-MDR1 was supplemented with 0.64 μM vincristine. For the calcein assay, cells were seeded on collagen-coated microtiter plates in a density of 10,000 cells/cm2 and cultured for 3 days. For confocal laser-scanning microscopy, about 1 × 106 cells were seeded onto poly-d-lysine and collagen-coated coverslips (diameter 30 mm) and cultured for 4 days. One day before the assays both cell lines were fed with vincristine-free culture medium.
Porcine Brain Capillary Endothelial Cells (pBCECs). As a model for the BBB we used pBCECs. The isolation of these cells was essentially based on the method described by Audus and coworkers (Audus et al., 1996) with minor changes and after thorough validation as described previously (Weiss et al., 2003). Cells were seeded on collagen-coated microtiter plates in a density of 100,000 cells/cm2 and cultured under standard cell culture conditions with medium M199 containing 0.7 mM l-glutamine, 100 μg/ml streptomycin sulfate, 100 U/ml penicillin G,10 mM HEPES, and 10% heat-inactivated horse serum. Eight days after seeding the confluent monolayers were used for the calcein assay.
Stock Solutions. Stock solutions of test compounds were prepared strictly following the manufacturers instructions. Gabapentin, sodium valproate, topiramate, and vigabatrin were soluble in aqua bidest. Carbamazepine, lamotrigine, sodium phenobarbital, sodium phenytoin, quinidine, verapamil hydrochloride, and LY335979 were dissolved in DMSO. The DMSO concentration in the assays never exceeded 1% (v/v), a concentration that was found not to influence the results of the assay in pilot experiments.
Calcein Uptake Assay. The assay was used to determine the Pgp inhibitor potency of the test compounds. Calcein-AM is a fluorogenic, highly lipid-soluble dye that rapidly penetrates the plasma membrane. Inside the cell endogenous esterases cleave the ester bonds, producing the hydrophilic and fluorescent dye calcein, which cannot leave the cell via the plasma membrane. Whereas calcein-AM is a substrate of Pgp, calcein is not (Homolya et al., 1993). Cells expressing high levels of Pgp rapidly extrude nonfluorescent calcein-AM from the plasma membrane, thus preventing accumulation of fluorescent calcein in the cytosol. Because the transport capacity of Pgp is inversely proportional to the accumulation of intracellular calcein fluorescence, inhibition of Pgp will lead to intracellular calcein accumulation.
The calcein-AM uptake assay was performed in 96-well plates. All incubation steps and the cell lysis were conducted at 37°C on a rotary shaker at 450 rpm. Before the uptake assay the cells were washed with prewarmed HBSS supplemented with 10 mM HEPES (HHBSS) and preincubated with HHBSS for 30 min and subsequently with the test compound for 10 min in octuplet. After preincubation, calcein-AM was added (final concentration 1 μM) and the cells were incubated for 60 min. The uptake was stopped by transferring the plates on ice and washing the cells twice with HHBSS precooled to 4°C. Subsequently, cells were lysed in 1% Triton X-100 for 15 min. Calcein fluorescence generated within the cells was analyzed in a Fluoroskan Ascent fluorimeter (Labsystems, Frankfurt, Germany) with 485-nm excitation and 535-nm emission filters. Each experiment was performed at least in duplicate (if no effect was observed) or in triplicate on different days.
Confocal Laser-Scanning Microscopy. Intracellular accumulation of the Pgp substrate bodipy-verapamil (Lelong et al., 1991) in LLC-PK1 and L-MDR1 cells was analyzed with a DM IRE 2 TCS SP II confocal laser-scanning microscope from Leica (Bensheim, Germany). For excitation a 488 nm argon laser line was used and a 500 to 550 nm band-pass filter to detect emission. The objective used was a Leica HCX PL APO CS 63x with a numerical aperture of 1.2. Confluent monolayers cultured on coverslips were preincubated with the test compound for 20 min in darkness at 37°C in 1 ml HHBSS. Subsequently, the fluorescent labeled bodipy-verapamil (final concentration 1 μM) was added and incubated again for 20 min. Afterward, the coverslip was transferred into a closed miniperfusion chamber (H. Saur, Reutlingen, Germany) and three series of 20 sections in z-plane through the cells were acquired for each coverslip. The thickness of each optical section was about 0.25 μm. The mean amplitude of fluorescence intensity was compared between L-MDR1 with and without preincubated test compound. In each series fluorescence was quantified along three lines of 168 ± 0.1 μm drawn in the image (Chen and Simon, 2000). Only areas with confluent cell monolayers were used for quantification. The experiment was performed at least in triplicate on different days and the values were averaged. LLC-PK1 cells were used as a control.
Quenching Test. Each test compound was analyzed for possible quenching effects on calcein fluorescence. To simulate the conditions of the calcein assay we generated calcein by incubating LLC-PK1 cells with 1 μM calcein-AM for 60 min at 37°C on a rotary shaker at 450 rpm. Increasing concentrations of the test compounds were added to aliquots of the cell lysate and the fluorescence was compared with control wells without test compounds.
All compounds were also tested for quenching effects on bodipyverapamil fluorescence. To exclude possible self-quenching effects of calcein and bodipy-verapamil we ascertained that the fluorescence of these dyes was linear up to the respective concentration applied in the assays.
Cytotoxicity Assay. Each test compound was screened for possible cytotoxic effects with the Cytotoxicity Detection Kit (Roche Diagnostics, Mannheim, Germany), a colorimetric assay for the quantification of lactate dehydrogenase activity released from the cytosol of damaged cells into the supernatant.
Statistical Analysis. The p values were calculated by analysis of variance with Dunnett's multiple comparison test for post hoc pairwise comparison with the control value or by a two-tailed unpaired t test (comparison of only two values obtained with confocal laser-scanning microscopy in LLC-PK1 cells). All statistical analyses were performed with GraphPad InStat, version 3.05, GraphPad Software (San Diego, CA). Data are expressed as mean ± S.D. unless indicated otherwise. A p value of ≤0.05 was considered significant.
Results
Method Validation. Expression of Pgp in the cell lines used and exclusion of possible involvement of MRP1 and MRP2, which may also transport calcein-AM, were demonstrated as described previously (Weiss et al., 2003). The suitability of the confocal laser-scanning microscopy for detecting Pgp inhibition in L-MDR cells was demonstrated with the well known Pgp inhibitor LY335979, which showed a significant increase in basal bodipy-verapamil fluorescence already at a concentration of 1 nM (Table 1). Moreover, the fact that all compounds with Pgp inhibitor activity in the confocal laser-scanning microscope assay only enhanced the bodipyverapamil fluorescence in the overexpressing cell line L-MDR1 and not in the parental LLC-PK1 cells without human Pgp (Table 1) strongly indicates that the observed effects are related to Pgp activity.
Increase of baseline bodipy-verapamil fluorescence (where baseline values were set to 1.0) by AEDs and LY335979 in L-MDR1 and LLC-PK1 cells measured by confocal laser-scanning microscopy
LLC-PK1 cells, porcine kidney epithelial cell line (control without human Pgp); L-MDR1 cells, LLC-PK1 cells transfected with the human MDR1 gene; n = number of experiments. Values represent mean ± S.D. of at least three independent assays. The p values are determined with analysis of variance with Dunnett's multiple comparison test for post hoc pairwise comparison of the results with the control value (basal fluorescence without compound = 1; L-MDR1 cells) and with unpaired t test (LLC-PK1 cells).
None of the test compounds was quenching calcein or bodipy-verapamil fluorescence and no autofluorescence at the excitation wavelength used to measure calcein fluorescence (485 nm) was observed. Moreover, no self-quenching of calcein or bodipy-verapamil was present. None of the test compounds was cytotoxic in any of the cell lines, even at the highest concentrations used.
Evaluation of the Pgp Inhibitory Potency of AEDs. Of all AEDs tested, only carbamazepine significantly increased the intracellular calcein concentration in pBCECs at concentrations >10 μM and in L-MDR1 cells at concentrations >200 μM, but not in the control cell line LLC-PK1 (Fig. 1); 500 μM carbamazepine enhanced basal fluorescence by a factor of 1.8 in pBCECs and 1.4 in L-MDR1 cells. In comparison to the potent and efficient Pgp inhibitor verapamil, which doubles baseline fluorescence at a concentration of 1.9 μM in pBCECs and 3.9 μM in L-MDR1 cells (Weiss et al., 2003), the inhibitor potency of carbamazepine was low.

Concentration-dependent effect of carbamazepine on the basal calcein fluorescence in pBCECs, LLC-PK1, and L-MDR1 cells. Data are presented as mean ± S.D. for n = 32 (4 experiments on different days in octuplet) in pBCECs and LLC-PK1 cells or n = 24 (3 experiments on different days in octuplet) in L-MDR1 cells. The p values are determined by analysis of variance with Dunnett's multiple comparison test for post hoc pairwise comparison of the results with the control value (basal fluorescence without carbamazepine). *, p < 0.05; **, p < 0.01.
Using confocal laser-scanning microscopy, carbamazepine-induced inhibition was confirmed (Table 1, Fig. 2) and phenytoin, lamotrigine, and valproate also inhibited Pgp (Table 1, Fig. 3). Carbamazepine, lamotrigine, phenytoin, and valproate in concentrations >100 μM significantly increased the uptake of bodipy-verapamil. Carbamazepine and phenytoin even doubled the basal fluorescence at 500 μM (Table 1). There was no significant effect of 500 μM carbamazepine, lamotrigine, phenytoin, and valproate on bodipy-verapamil accumulation in LLC-PK1 cells. All other AEDs had no significant effect either in L-MDR1 or in LLC-PK1 cells (Table 1, Fig. 3).
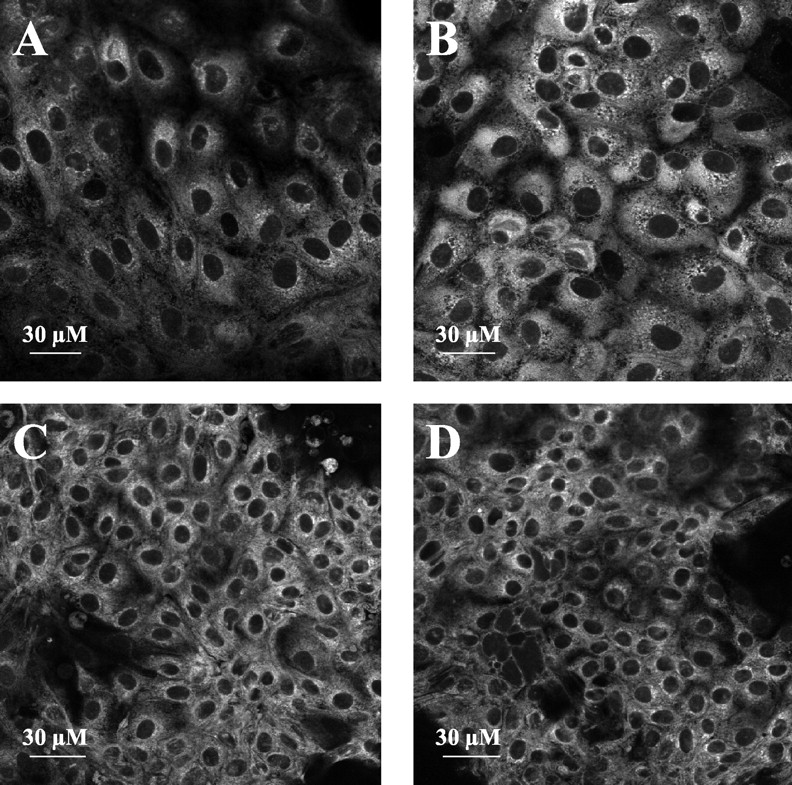
Confocal laser-scanning microscopy: bodipy-verapamil uptake into L-MDR1 (A, B) and LLC-PK1 (C, D) cells. On the left: without carbamazepine; on the right: after exposure for 20 min with 500 μM carbamazepine as Pgp inhibitor.

Confocal laser-scanning microscopy: net change in basal bodipyverapamil fluorescence induced by AEDs (500 μM). Values are expressed as ratio of effect in L-MDR1/LLC-PK1 (see also Table 1).
Discussion
Resistance to AEDs represents a common problem in epilepsy treatment (Regesta and Tanganelli, 1999) but the phenomenon is not well understood and cannot be attributed to low serum concentrations. One of the currently favored hypotheses assumes that overexpression of multidrug transporters at the BBB leads to ineffective brain concentrations of the AEDs (Abbott et al., 2002; Löscher and Potschka, 2002). Other mechanisms are less well defined and include alterations in drug targets as well as inherent risk factors associated with certain seizure etiologies (Regesta and Tanganelli, 1999; Löscher and Potschka, 2002).
AEDs are frequently associated with clinically important drug interactions that often affect safety and effectiveness of drug treatment in epilepsy patients (Tanaka, 1999; Hachad et al., 2002; Patsalos et al., 2002). However, certain interactions have a favorable impact, as has been shown for a series of add-on therapies, which are often more effective than monotherapies at similar plasma concentrations (Deckers et al., 2000; Marson et al., 2000; Ramaratnam et al., 2001; Jette et al., 2002). These studies thus raise the question of whether their superiority might be caused by changes in drug distribution to the site of action.
Pgp is an efflux transporter that may confer drug resistance in cancer chemotherapy and anti-HIV therapy. Indirect evidence suggests that this might also be a cause for impaired drug responses in epilepsy (Tishler et al., 1995; Dombrowski et al., 2001; Sisodiya et al., 2002). Moreover, Pgp activity may substantially affect the kinetics of many drugs and may contribute to numerous pharmacokinetic drug-drug interactions (Yu, 1999). Thus far the impact of Pgp on bio-availability, distribution, and excretion of AEDs and its role in the interaction with coadministered drugs (including AEDs in combination therapy) has not been thoroughly investigated.
While for some AEDs such as carbamazepine, lamotrigine, phenobarbital, and phenytoin Pgp-mediated transport and induction of the MDR1 gene (not for lamotrigine) has already been shown (Schinkel et al., 1996; Potschka and Löscher, 2001a; Potschka et al., 2001, 2002), little is known about potential inhibitor characteristics of AEDs on Pgp. This is of particular interest because inhibition of Pgp around an epileptic focus would facilitate tissue penetration of its substrates, increase anticonvulsant concentrations at the site of action, and thus modulate both adverse drug reactions and efficacy.
Thus far, only carbamazepine has been tested for inhibition and was found not to affect the rhodamine-123 efflux in lymphocytes as measured by flow cytometry (Owen et al., 2001). Because carbamazepine was applied only up to 100 μM, a concentration that was in the lower effective concentration range in this study, carbamazepine-induced inhibition may have escaped detection. Our series of in vitro experiments for the first time shows that carbamazepine, lamotrigine, phenytoin, and valproate all inhibit Pgp, whereas topiramate, vigabatrin, phenobarbital, and gabapentin do not. Compared with potent Pgp inhibitors like verapamil and LY335979, inhibition by the AEDs was weak-appearing only in concentrations higher than 100 μM.
As expected, carbamazepine was less effective in the overexpressing L-MDR1 cells compared with pBCECs, in which less inhibitor is necessary to reach similar effects. Confocal laser-scanning microscopy was more sensitive in detecting Pgp inhibition than the calcein assay. The differences observed in this series of experiments could have several reasons. 1) The most obvious is that the confocal laser-scanning microscope method is indeed more sensitive due to a higher accumulation of bodipy-verapamil compared with calcein within the cells or to a more sensitive detection system of the microscope compared with a fluorimeter; 2) a further explanation could be a lower affinity of bodipy-verapamil to Pgp compared with calcein-AM enabling competitive inhibition of the transport by substrates with lower affinity. Thus far, there is no published information on the affinity of bodipyverapamil to Pgp; and 3) generally, it has been shown that the inhibition of Pgp transport is unique to a given pair of substrate and inhibitor (Wang et al., 2001), therefore application of different substrates commonly leads to different results (Yasuda et al., 2002). Because calcein fluorescence exceeds the fluorescence of bodipy-verapamil when compared at equimolar concentrations (Weiss et al., unpublished data), differences in fluorescence intensity of the two marker substrates cannot explain the results. Moreover, because the two dyes were used at identical concentrations, differences in substrate concentrations, which may also modulate the results, can be excluded.
Whereas not all inhibitors of Pgp are also transported by Pgp, compounds that are transported (substrates) are usually also inhibitors because Pgp substrates generally share a specific binding site for which they can compete. Hence concentration and affinity of the simultaneously present competitor will ultimately determine which compound will act as an inhibitor in a given drug combination and lead to accumulation of the competitor. In this regard our results agree well with the findings by others demonstrating that those AEDs that are known Pgp substrates (carbamazepine, lamotrigine, and phenytoin) (Potschka and Löscher, 2001a; Potschka et al., 2001, 2002) are indeed also inhibitors. For valproate there is no experimental evidence to date for being a Pgp substrate, but it is an inhibitor of MRP (Huai-Yun et al., 1998) and may also be transported by this multidrug transporter because probenecid inhibits the transport of valproate out of the cerebrospinal fluid (Frey and Löscher, 1978). Interestingly, the Pgp substrate phenobarbital (Potschka et al., 2002) revealed no Pgp inhibitory activity. This might indicate a weak affinity for Pgp, which enables the transport of phenobarbital itself but not the inhibition of calcein-AM and bodipy-verapamil transport by Pgp.
Effective concentrations in the in vitro assays were clearly higher than therapeutic plasma concentrations present after usual therapeutic doses (lying between 20 and 100 μM for all studied compounds except valproate (300–600 μM)). Even when considering the fact that carbamazepine, phenytoin, and lamotrigine may accumulate in the human brain by a factor of 1.2 to 2.8 (Sironi et al., 1980; Meyer et al., 1999), it seems unlikely that the weak inhibition found for these AEDs is of clinical relevance in the monotherapy with AEDs. However, our study does not exclude that inhibition might be potentiated in add-on therapies, when two AEDs with Pgp substrate and inhibitor characteristics are given in combination. Combinations of AEDs can improve effectiveness by improving efficacy, tolerability, or both (Deckers et al., 2000). In many cases the underlying mechanisms are not elucidated yet. Effectiveness of AEDs results from a multifactorial process and increases in efficacy of antiepileptic therapy may, therefore, have several reasons. First, the pharmacokinetics of an AED may be altered to augment the availability of the compound at the site of action and thus result in a rightward shift on the concentration-response curve toward higher responses. This may be caused by modulation of any pharmacokinetic process (i.e., absorption, distribution, metabolism, or excretion). In the case of substantial Pgp inhibition, drug distribution may be facilitated and the AEDs may have more ready access to the brain. Moreover, it is also possible that the coordinate effect of AEDs acting through different mechanisms of action will improve therapeutic responses by interacting with distinct pathways that may not have sufficiently suppressed seizure activity when stimulated alone (Deckers et al., 2000). Indeed, valproate and lamotrigine have recently been shown to synergistically increase antiepileptic effects of each other in a seizure model in mice (Cuadrado et al., 2002). This interaction appeared largely pharmacodynamic, but there was also a significant increase in lamotrigine brain concentrations when low doses were administered. However, the determination of AED tissue concentrations in acute epilepsy models may not be a suitable surrogate for the chronic, treatment-resistant situation in which changes in Pgp expression may have occurred (Tishler et al., 1995; Dombrowski et al., 2001; Sisodiya et al., 2002).
The superiority of antiepileptic combination therapy has been shown in numerous experimental and human studies for combinations of older AEDs (e.g., phenobarbital/phenytoin, phenytoin/valproate, valproate/ethosuximide, and carbamazepine/valproate; Deckers et al., 2000) and add-on therapy with newer antiepileptics (e.g., valproate/lamotrigine, carbamazepine/vigabatrin, combinations with topiramate, lamotrigine, or gabapentin; Deckers et al., 2000; Marson et al., 2000; Ramaratnam et al., 2001; Jette et al., 2002). In a clinical setting it is, however, rather difficult to distinguish between the contribution of a potential pharmacodynamic interaction in the presence of pharmacokinetic interactions. The evaluation of the interactions of the known Pgp substrates phenytoin and carbamazepine with lamotrigine is complex because both phenytoin and carbamazepine are potent enzyme-inducing agents. However, although in combination therapy with lamotrigine carbamazepine concentrations are unchanged and lamotrigine concentrations are lowered, there is a remarkable increase in adverse events typically associated with carbamazepine (Besag et al., 1998). Although this could result from a pharmacodynamic interaction, it could obviously also indicate a facilitated distribution of carbamazepine to the brain caused through Pgp inhibition by lamotrigine.
In summary, this study demonstrates an inhibitory interaction of several AEDs with Pgp in vitro at concentrations exceeding therapeutic plasma concentrations, suggesting that modulation of Pgp is not a major determinant of AED action in monotherapy. It is, however, possible that additive or even synergistic inhibition of transport may occur when several AEDs that compete for Pgp binding sites are given concomitantly.
Acknowledgments
The authors thank Stephanie Fuchs for excellent technical assistance, Aventis (Bad Soden, Germany), Eli Lilly Company (Bad Homburg, Germany), GlaxoSmithKline (Uxbridge, Middlesex, UK), and Janssen-Cilag (Neuss, Germany) for providing the test substances, and Dr. Alfred H. Schinkel (The Netherlands Cancer Institute, Division of Experimental Therapy, Amsterdam, The Netherlands) for generously providing the cell line L-MDR1.
Footnotes
-
This work was supported by Grant 01EC9902 from the German Ministry for Education and Research (BMBF, Bundesministerium für Bildung und Forschung).
-
DOI: 10.1124/jpet.103.054197.
-
ABBREVIATIONS: AED, antiepileptic drug; Pgp, P-glycoprotein; BBB, blood-brain barrier; BCEC, brain capillary endothelial cell; MRP, multidrug resistance-associated protein; HBSS, Hanks' balanced salt solution; calcein-AM, calcein-acetoxymethylester; DMSO, dimethyl sulfoxide; LY335979; 1-piperazinethanol,4-(1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa-[c]cyclohepten-6-yl)-α-[(5-quinolinoxy)methyl]-trihydrochloride; pBCECs, porcine BCECs; HHBSS, HBSS supplemented with 10 mM HEPES; MDR, multidrug resistance.
- Received May 8, 2003.
- Accepted July 1, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}