


Abstract
Histamine mediates its physiological function through binding to four known histamine receptors. Here, we describe the first selective antagonist of the histamine H4 receptor, the newest member of the histamine receptor family, and provide evidence that such antagonists have anti-inflammatory activity in vivo. 1-[(5-chloro-1H-indol-2-yl)carbonyl]-4-methylpiperazine (JNJ 7777120) has a Ki of 4.5 nM versus the human receptor and a pA2 of 8.1. It is equipotent against the human, mouse, and rat receptors. It exhibits at least 1000-fold selectivity over H1, H2, or H3 receptors and has no cross-reactivity against 50 other targets. This compound has an oral bioavailability of ∼30% in rats and 100% in dogs, with a half-life of ∼3 h in both species. JNJ 7777120 blocks histamine-induced chemotaxis and calcium influx in mouse bone marrow-derived mast cells. In addition, it can block the histamine-induced migration of tracheal mast cells from the connective tissue toward the epithelium in mice. JNJ 7777120 significantly blocks neutrophil infiltration in a mouse zymosan-induced peritonitis model. This model is reported to be mast cell-dependent, which suggests that the compound effect may be mediated by mast cells. These results indicate that the histamine H4 receptor plays a role in the inflammatory process. Selective H4 receptor antagonists like JNJ 7777120 may have the potential to be useful in treating inflammation in humans.
Histamine is a ubiquitous chemical messenger that exerts numerous functions mediated through at least four pharmacologically distinct receptors, which are all members of the G-protein-coupled receptor family (Hill et al., 1997). The H1 receptor is expressed in the brain, endothelial cells, and smooth muscle cells. The most characteristic roles for H1 receptor activation are smooth muscle contraction and increases in vascular permeability (Ash and Schild, 1966). Many of its functions contribute to allergic responses, and H1 receptor antagonists have been very successful drugs for the treatment of allergies. The H2 receptor has been demonstrated to function as a key modulator for gastric acid secretion, and H2 receptor antagonists are widely used for the treatment of gastrointestinal ulcers (Soll and Walsh, 1979). The H3 receptor is predominantly expressed in the human central nervous system (Lovenberg et al., 1999, 2000). Its exact physiological role is unknown, but it is hypothesized to function as a presynaptic release-controlling receptor that may regulate histamine, norepinephrine, serotonin, GABA, acetylcholine, and other neurotransmitters. The histamine receptors couple to different signaling pathways via different G-proteins. Activation of the H1 receptor leads to the mobilization of intracellular Ca2+ by activating the Gq family of G-proteins (Hill, 1990; Leurs et al., 1995). The H2 receptor signals through Gs G-proteins and receptor activation cause increases in cAMP, whereas the H3 receptor couples to Gi/o, leading to decreases in cAMP (Lovenberg et al., 1999).
Recently, a fourth histamine receptor, H4, was identified (Nakamura et al., 2000; Oda et al., 2000; Liu et al., 2001a; Morse et al., 2001; Nguyen et al., 2001; Zhu et al., 2001). The amino acid sequence of the H4 receptor has a 35% amino acid homology with the H3 receptor and a much lower homology to H1 and H2 receptors. This is reflected in the pharmacological profile of known histamine receptor ligands, where most H1 and H2 receptor antagonists and agonists do not bind to the H4 receptor. However, some of the H3 receptor ligands (like thioperamide, clobenpropit, imetit, and R-α-methylhistamine) do bind to the H4 receptor, albeit with affinities different from that of the H3 receptor (see Jablonowski et al., 2004 and Fung-Leung et al., 2004 for reviews). There are also great differences in the expression pattern between H4 and H3 receptors. Expression of the H3 receptor is mainly restricted to cells in the central nervous system (Lovenberg et al., 1999, 2000), whereas the H4 receptor seems to be limited to cells of hematopoietic lineage (Oda et al., 2000; Liu et al., 2001a; Morse et al., 2001; Zhu et al., 2001). In particular, H4 receptor expression has been observed in eosinophils, T cells, dendritic cells, basophils, and mast cells (Liu et al., 2001a; Gantner et al., 2002; O'Reilly et al., 2002; Hofstra et al., 2003).
Very little is known about the actual biological function of the H4 receptor. The H4 receptor can mediate chemotaxis and calcium influx in mast cells and eosinophils (O'Reilly et al., 2002; Hofstra et al., 2003). Gantner et al. (2002) have shown that both the H4 and H2 receptors control histamine-induced interleukin-16 release from CD8+ T cells. These are the only reports of H4 receptor function in the literature.
With the H4 receptor expression pattern limited to hematopoietic cells, a role in inflammation has been postulated. One of the best tools for deciphering the function of a novel receptor is a selective ligand. Here, we describe the first selective histamine H4 receptor antagonist and provide evidence that such antagonists have anti-inflammatory activity in vivo.
Materials and Methods
Materials. Diphenhydramine, ranitidine, and thioperamide were purchased from Sigma-Aldrich (St. Louis, MO). JNJ 7777120 was synthesized as previously described (Jablonowski et al., 2003b). JNJ 5207852 was provided by Dr. Nicholas Carruthers and was synthesized as previously described (Apodaca et al., 2003). Tritiated JNJ 7777120 was synthesized by Amersham Biosciences Inc. (Piscataway, NJ). SK-N-MC cells stably transfected with human, mouse, or rat H4 receptor or the human or rat H3 receptor as previously described (Lovenberg et al., 1999; Liu et al., 2001a,b) were provided by Dr. Timothy Lovenberg. Mouse and human H1 and H2 cell lines were generated as previously described for the H3 and H4 cell lines (Lovenberg et al., 1999; Liu et al., 2001a,b). All of the radiolabeled ligands were purchased from PerkinElmer Life Sciences (Boston, MA).
Binding Assays. Cell pellets from SK-N-MC cells transfected with human, rat, or mouse H4 receptor, human H3 receptor, or human H1 receptor were used. For the mouse H1 receptor binding, fresh mouse brain was used. Cell pellets or mouse brain were homogenized in 50 mM Tris pH 7.5 containing 5 mM EDTA, and supernatants from an 800g spin were collected and recentrifuged at 30,000g for 30 min. Pellets were rehomogenized in 50 mM Tris pH 7.5 containing 5 mM EDTA. For the H4 competition binding studies, human, mouse, and rat cell membranes were incubated with 10, 40, and 150 nM [3H]histamine (specific activity 23 Ci/mmol), respectively, with or without test compounds for 45 min at 25°C. Nonspecific binding was defined using 100 μM unlabeled histamine. The Kd values for the human, mouse, and rat H4 receptor were determined to be 5, 42, and 178 nM, respectively, and the Bmax values were determined to be 1.12, 1.7, and 0.68 pmol/mg protein, respectively. Similarly, the ligand used for the H3 receptor-binding assays was [3H]N-α-methyl histamine (specific activity, 82 Ci/mmol), and the nonspecific binding was defined using 100 μM unlabeled histamine. The Kd values for the human and rat H3 receptor were determined to be 1 and 0.8 nM, respectively, and the Bmax values were 2.13 and 0.4 pmol/mg protein, respectively. The ligand used for the human and mouse H1 receptor binding was [3H]pyrilamine (specific activity, 20 Ci/mmol), and the nonspecific binding was defined using 10 μM unlabeled diphenhydramine. The Kd was 1 nM for human receptor and 3 nM for the mouse receptor. The Bmax was 2.68 pmol/mg protein for the human receptor and 0.2 pmol/mg protein for the mouse receptor. The binding to the guinea pig H1 and H2 receptors was determined by Cerep, Inc. (Redmond, WA). For the H1 receptor-binding assays, guinea pig cerebellum was used. The radioligand was [3H]pyrilamine, and 100 μM triprolidine was used to determine the nonspecific binding. For the H2 receptor-binding assays, guinea pig striatum was used. The radioligand was [3H]aspartate amino-transferase, and 100 μM tiotidine was used to determine the nonspecific binding. Details of both assays can be found on the Cerep website (www.cerep.fr). The Kd of [3H]JNJ 7777120 (specific activity, 84 Ci/mmol) for the human and mouse H4 receptor was determined essentially as described above using increasing concentrations of [3H]JNJ 7777120 instead of [3H]histamine in the presence of 100 μM unlabeled histamine. For all studies, the Ki values were calculated based on an experimentally determined appropriate Kd value according to Cheng and Prusoff (1973).
Cell-Based cAMP Assays. SK-N-MC cell lines were created that express a reporter gene construct and either the human, mouse, or rat H4 receptor; the human or rat H3 receptor; the human or mouse H1 receptor; or the mouse or human H2 receptor full-coding region. The reporter gene was β-galactosidase under the control of cyclic AMP-responsive elements. Cells were plated in 96-well plates the night before the assay. Histamine was used as the agonist for all assays except the mouse and rat H4 assays, where imetit was used. For the H4 and H3 receptor, the inhibition of forskolin-stimulated cAMP production was measured; whereas for the H1 and H2 assays, the direct stimulation of cAMP production was measured. To determine agonist potential, a dose titration of compounds was run from 10-3 to 10-12 M. For determination of antagonist activity, compounds were added 10 min prior to the addition of agonist, which was added directly to the cell medium. In the case of the H4 and H3 receptor assays, forskolin (5 μM final concentration) was added 10 min after the addition of histamine. Cells were returned to the incubator for 6 h at 37°C. The medium was then aspirated, and the cells were washed with 200 ml of phosphate-buffered saline (PBS). Cells were lysed with 25 μl of 0.1× assay buffer (10 mM sodium phosphate, pH 8, 0.2 mM MgSO4, and 0.01 mM MnCl2) and incubated at room temperature for 10 min. Cells were then incubated for 10 min with 100 μl of 1× assay buffer containing 0.5% (v/v) Triton X-100 and 40 mM β-mercaptoethanol. Color was developed using 25 μl of 1 mg/ml substrate solution (chlorophenol red β-d-galactopyranoside; Roche Applied Science, Indianapolis, IN). Color was quantitated on a microplate reader by measuring the absorbance at 570 nm. The data from each concentration-response curve were fitted to a sigmoidal curve to obtain the maximum response, Hill coefficient, and EC50 using Prism (GraphPad Software, San Diego, CA). Dose ratios were calculated from individual concentration-response curves of agonists at three antagonist concentrations. Apparent pA2 values were calculated using a Schild plot.
Selectivity Panel. A panel of 50 different biogenic amine receptors, neuropeptide receptors, ion channel binding sites, and neurotransmitter transporter binding assays were run by Cerep, Inc. (Redmond, WA). The targets run were: human adenosine A1, A2A, and A3 receptors; adrenergic receptors α1 (nonselective), α2 (nonselective), and β1; norepinephrine (NE) transporter; angiotensin II receptor (AT2); rat brain benzodiazepine receptor (BZD); bradykinin receptor 2 (B2); cholecystokinin receptor 1 (CCK1); dopamine D1 and D2 receptors; dopamine transporter (DA); endothelin receptor A (ETA); rat brain GABA receptor; galanin receptor 2 (GAL2); CXCR2; CCR1; vasopressin receptor 1A (V1A); melanocortin receptor 4 (MC4); chicken melatonin receptor 1 (MT1); muscarinic receptors M1, M2, and M3; neurokinin receptors 2 and 3 (NK2, NK3); neuropeptide receptors 1 and 2 (NPY1, NPY2); neurotensin receptor 1 (NT1); opioid receptors δ (DOP), κ (KOP), and μ (MOP); nociceptin receptor (ORL1); serotonin receptors 5-HT1A, 5-HT2A, 5-HT3, 5-HT5A, 5-HT6, 5-HT7, and rat 5-HT1B; rat σ receptor (SST); vasoactive intestinal peptide receptor 1 (VIP1); rat Ca2+ channel verapamil site; rat brain voltage-gated potassium channel (K+V channel); rat brain small-conductance Ca2+-activated K+ channel (SK+Ca channel); rat Na+ channel (site 2); and rat Cl- channel. All assays were run using recombinant human receptors, except where noted. The full methods and references can be found on the Cerep website (www.cerep.fr). The assays were run at 1 μM of JNJ 7777120, and the percentage of inhibition is given as the average of three determinations.
Bone-Marrow Mast Cell Culture. Mast cells were differentiated from bone marrow derived from BALB/c mice as previously described (Hofstra et al., 2003). Briefly, bone marrow cells were cultured in RPMI 1640 medium with 10% (v/v) fetal calf serum, 0.1 mM nonessential amino acids, 50 μg/ml penicillin/streptomycin, and 20% (v/v) conditioned medium from WEHI-3 cells (American Type Culture Collection, Manassas, VA). After a 16-h culture, the nonadherent cells were transferred to a new flask. Four weeks later, the cells were analyzed by flow cytometry for IgE receptor and CD117 expression to confirm the differentiation to mast cells. More than 99% of the cells were IgE receptor- and CD117-positive. Mast cells of 4 to 8 weeks of age were used for the experiments.
Calcium Mobilization in Mast Cells. Calcium mobilization experiments in the mouse bone marrow-derived mast cells were carried out as previously described (Hofstra et al., 2003). Cells were loaded with 4 μM calcium dye Fluo-3 (acetoxymethyl ester) (TEF Labs, Austin, Texas), and calcium mobilization was assayed in fluorometric imaging plate reader 384 (Molecular Devices, Sunnyvale, CA). The fluorescence intensity was calculated as the maximum minus the minimum fluorescence over a 2-min period. All data points were carried out in triplicate, and experiments were repeated at least three times with different batches of mast cells. Histamine and the various histamine receptor antagonists were added to the cells 10 min prior to the calcium measurements.
In Vitro Mast Cell Chemotaxis Assay. Transwells (Costar, Cambridge, MA) of a 8-μm pore size were coated with 100 μl of 100 μg/ml bovine fibronectin (Sigma-Aldrich) for 2 h at room temperature. After removal of the fibronectin, 600 μl of RPMI 1640 medium with 0.5% (w/v) bovine serum albumin in the presence of histamine was added to the bottom chamber, and histamine receptor antagonists were added to both chambers. Mast cells (2 × 105/well) were added to the top chamber. The plates were incubated for 3 h at 37°C. Transwells were removed, and the number of cells in the bottom chamber was counted for 1 min using a flow cytometer.
Histamine-Induced Mast Cell Migration in Vivo. Mice (n = 10 per group) were dosed with either vehicle or JNJ 7777120 15 min before being challenged by a 20-min aerosol inhalation of 0.1 M histamine dihydrochloride or PBS. This was repeated for 2 days. JNJ 7777120 was administered at 20 mg/kg s.c. in 20% (w/v) hydroxypropyl-β-cyclodextrin. Four hours after the last challenge, animals were euthanized by pentobarbital overdose (i.p.) and severing of the abdominal aorta. Trachea were cleared of blood via perfusion of PBS/heparin through the right ventricle and fixed in 10% (w/v) formaldehyde (neutral buffered formalin) for subsequent paraffin cross-sectioning and toluidine blue staining.
Total mast cells were calculated, the location of mast cells was quantified by light microscopy in two sections of trachea from each mouse, and a mean was reported. Mast cells were categorized as subepithelial mast cells if they were located on the luminal side of the tracheal cartilage-smooth muscle cell ring, which is easily visualized. For the analysis of mast cell number, it was deemed relevant to quantify the number of mast cells per section, as the important calculation of subepithelial mast cells was largely based on a histomorphological definition rather than one that could be easily defined by the measurement of area. However, to ensure consistency, care was taken to make sure that the 5-μm sections were of uniform size (90-120 μm) and from uniformly dissected midtracheal tissue.
Histamine-Induced Paw Edema in Mice. Female BALB/c mice (n = 5 per group) were used to test the effect of JNJ 7777120 on histamine-induced paw edema. JNJ 7777120 was administered at 200 mg/kg p.o. in 5% (w/v) dextrose in water 30 min prior to histamine injection. A H1 antagonist, desloratidine, in 5% (w/v) dextrose in water was given at 1 mg/kg i.p. 1 h before histamine injection. Histamine dihydrochloride was dissolved in 0.9% (w/v) NaCl, and 25 μl of a 0.2 mg/ml solution was injected in the left footpad of each mouse under light general gas anesthesia. An injection of 0.9% NaCl in the right footpad served as an internal control. Thirty minutes after histamine challenge, the animals were sacrificed by exsanguination under isoflurane anesthesia. The feet were removed at the ankle joint and placed in preweighed tubes. The total weight of each tube was recorded, and the wet weight of each foot was determined.
Mouse Zymosan-Induced Peritonitis Model. The initial zymosan-induced peritonitis model was carried out at the William Harvey Research Institute (London, United Kingdom). Male outbred Swiss albino mice (n = 8 for each group) were used. JNJ 7777120 was given at 10 mg/kg s.c. in saline (10 ml/kg), and thioperamide in saline was dosed at 5 mg/kg s.c. 15 min prior to i.p. administration of 1 mg of zymosan. A second dose of compound or vehicle was administered 2 h later. Determination of oral activity was run in-house using female CD-1 mice (n = 10 for each group). A single oral dose of compound or vehicle [0.5% (w/v) methocel] was given 15 min prior to i.p. administration of 1 mg of zymosan. After 4 h, the animals were euthanized, and the peritoneal cavities were washed with 3 ml of PBS containing 3 mM EDTA. The number of migrated polymorphonuclear cells (PMN; >95% neutrophils) was determined by taking an aliquot (100 μl) of the lavage fluid and diluting 1:10 in Turk's solution [0.01% (w/v) crystal violet in 3% (v/v) acetic acid]. The samples were then vortexed, and 10 μl of the stained cell solution were placed in a Neubauer hematocymometer. Differential cell counts were performed using a light microscope. PMN cells were easily differentiated by their chromatic characteristics and the shape of the nucleus relative to the cytoplasm.
Mouse Thioglycollate-Induced Peritonitis Model. Male C57/BL6/J mice (n = 3-6 for each group) were used to study the effects of JNJ 7777120 on thioglycollate-induced peritonitis. JNJ 7777120 was given at 10 mg/kg s.c. in saline (10 ml/kg) 15 min prior to i.p. administration of 4% (w/v) thioglycollate in saline. A second dose of compound or vehicle was administered 2 h later. After 4 h, the animals were euthanized, and the peritoneal cavities were washed with 3 ml of PBS containing 3 mM EDTA. The number of migrated PMN (>95% neutrophils) was quantitated using the methods given above.
Pharmacokinetic Analysis. Female CD-1 mice, female Sprague-Dawley rats, and mature male beagle dogs were used in pharmacokinetics studies. Oral dosing was carried out either by oral gavage for mice and rats or via a stomach tube for dogs. I.v. dosing was performed via tail vein for rats or jugular or saphenous veins for dogs. For oral dosing in mice, the compound was prepared in 0.5% (w/v) methocel. For oral and i.v. dosing in rats and dogs, the compound was prepared in 20% (w/v) hydroxypropyl-β-cyclodextran, and the pH value was adjusted to 6. Compound was administered orally at 10 mg/kg to mice, rats, and dogs and intravenously at 1 mg/kg in dogs and 3 mg/kg in rats. After dosing, blood samples were collected via cardiac puncture (mice), tail vein (rats), or jugular or saphenous veins (dogs) into tubes containing heparin at the specified time points. Plasma samples were then immediately prepared, and compound concentrations were determined by LC/MS following acetonitrile extraction. Pharmacokinetic parameters were estimated using WinNonlin (Pharsight Corp., Mountain View, CA).
Statistics. All data are given as the average of several replicates. The error is given as the S.E.M. A Student's t test was used to judge statistical significance where appropriate.
Results
To assist in probing the biological function of the H4 receptor, the identification of potent, selective, nonimidazole histamine H4 ligands is advantageous. A high throughput screen of our corporate compound collection produced several lead compounds that led to the development of JNJ 7777120 (Fig. 1). The structure-activity relationship for this series has been previously described (Jablonowski et al., 2003).

The structure of JNJ 7777120.
The binding of JNJ 7777120 to the human H4 receptor was determined using membranes from SK-N-MC cells transfected with the human H4 receptor in a filter-binding assay. Figure 2 shows that there was a specific and saturatable binding of 3H-labeled JNJ 7777120 to these membranes. The Kd was calculated to be 3.6 nM using a Scatchard analysis and 3.2 nM using a nonlinear curve fit. A competitive radioligand-binding assay was used to measure the binding of JNJ 7777120 to the H4 receptor from other species as well as the selectivity versus H3 or H1 receptors. As shown in Table 1, JNJ 7777120 exhibited high affinity for the human H4 receptor, with a Ki value of 4 nM and at least 1000-fold selectivity over H1 or H3 receptors. The compound had an affinity for the mouse and rat receptors similar to that of the human form.

The binding of JNJ 7777120 to the human histamine H4 receptor. A, saturation binding of [3H]JNJ 7777120 to membranes from SK-N-MC cells expressing the human histamine H4 receptor. The specific binding (▪) was determined by subtracting the total binding (•) from the counts observed when 100 μM unlabeled histamine was used (nonspecific binding, ▾). The results shown are the average of three experiments, and the error bars represent the S.E.M. B, Scatchard plot of the saturation binding data yielding a Kd value of 3.6 nM.
In vitro Ki and pA2 values for JNJ 7777120 interaction with various histamine receptors
Experimental details are given under Materials and Methods. n > 3 for all. A less than or greater than symbol indicates that this was the highest concentration run.
The functional antagonism of the human H4 receptor signaling by JNJ 7777120 was determined using SK-N-MC cells expressing either the recombinant human or mouse receptor. Histamine dose-dependently inhibits adenylate cyclase in these cells as measured by a decrease in the forskolin-dependent activation of a cAMP reporter construct. The presence of increasing concentrations of JNJ 7777120 produced rightward shifts in the histamine concentration-response curves, indicating that it was a competitive antagonist (Fig. 3). Schild regression analysis (Fig. 3, inset) of the EC50 concentration ratios produced a pA2 of the apparent antagonist effect of 8.1 for both the human (Fig. 3A) and mouse (Fig. 3B) receptors. This same analysis was carried out for the rat H4 receptor, as well as the other histamine receptors, using cells transfected with the proper receptors; and the results are tabulated in Table 1. As seen in the binding assays, JNJ 7777120 had a similar potency against the mouse and rat H4 receptors. In addition, no antagonism of H1, H2, or H3 receptors was detected.
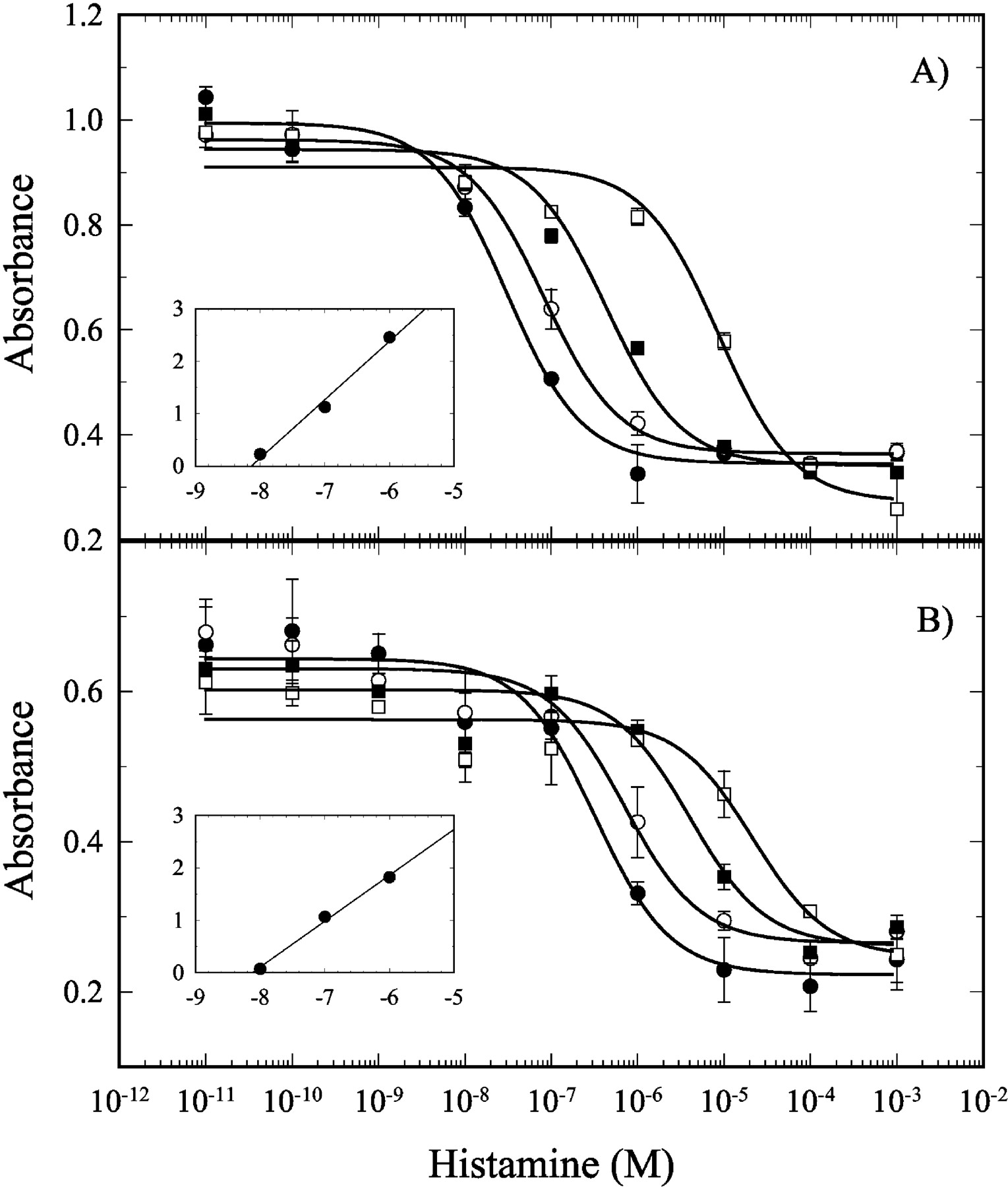
Functional antagonism of the human histamine H4 receptor by JNJ 7777120. Antagonism of the histamine inhibition of forskolin-stimulated cAMP-mediated reporter gene activity in SK-N-MC cells expressing the human (A) or mouse (B) histamine H4 receptor by JNJ 7777120. A titration of histamine was carried out in the presence of 0 (•), 10 (○), 100 (▪), and 1000 (□) nM JNJ 7777120. Inset, Schild plot analysis. The results shown are the average of two experiments, and the error bars represent the S.E.M.
To determine the selectivity of JNJ 7777120 for the H4 receptor, it was tested in radioligand-binding assays for 50 other targets. These targets represent major classes of biogenic amine receptors, neuropeptide receptors, ion channel binding sites, and neurotransmitter transporters. Only serotonin receptor 5-HT2A (34%) and norepinephrine transporter (27%) showed greater than 20% inhibition at 1 μM (Table 2). These results, coupled with the results for the different histamine receptors, indicate that JNJ 7777120 is a selective and potent H4 receptor antagonist with little or no affinity for over 50 other targets.
Inhibition of radioligand binding to various targets by JNJ 7777120
Recently, we described a role for the H4 receptor in mouse bone marrow-derived mast cells (Hofstra et al., 2003). It was shown that activating the H4 receptor induces a calcium influx in mast cells that results in chemotaxis toward histamine. These conclusions were made using a combination of cells from H4 receptor-deficient mice and inhibition by thioperamide, which is a dual H4/H3 inhibitor. Since JNJ 7777120 is a selective antagonist for the H4 receptor, it can be used to verify that these responses are indeed due to activation of the H4 receptor. Figure 4A shows the effects of different histamine receptor antagonists on the histamine-mediated increase in cellular calcium as measured by an increase in fluorescence. At 10 μM, the H1-, H2-, or H3-specific antagonists (diphenhydramine, ranitidine, or JNJ 5207852, respectively) had no effect, whereas JNJ 7777120 and thioperamide completely blocked the calcium response. Histamine has also been shown to induce chemotaxis of these cells (Hofstra et al., 2003). As for the calcium response, this effect is mediated by the H4 receptor, since only JNJ 7777120 and thioperamide blocked the migration (Fig. 4B). The IC50 for inhibition (Fig. 4C) of the histamine-induced chemotaxis assay was 40 nM using 10 μM histamine, showing that JNJ 7777120 is a very potent antagonist of the H4 receptor in primary cells, as seen in transfected cells. The use of JNJ 7777120 provides definitive evidence that the H4 receptor is responsible for histamine-induced calcium increase and chemotaxis in mouse mast cells.

Effects of JNJ 7777120 on mast cell function in vitro. A, effects of histamine receptor antagonists on the histamine-mediated calcium increase. Mast cells were loaded with 4 μM calcium dye Fluo-3, and calcium mobilization was assayed in a fluorometric imaging plate reader. The relative fluorescence intensity was calculated as the maximum minus the minimum fluorescence over a 2-min period. Histamine (black bars) and the various histamine receptor antagonists (all 10 μM) were added to the cells 10 min prior to the calcium measurements. The antagonists used were H1, diphenhydramine (white diagonal bars); H2, ranitidine (gray bars); H3, JNJ 5207852 (white horizontal bars); H4, JNJ 7777120 (white bars); and H3/H4, thioperamide (gray diagonal bars). B, effects of histamine receptor antagonists on histamine-induced mast cell chemotaxis. Histamine (HA) was added to the bottom chamber at a concentration of 10 μM, and various histamine receptor antagonists were added to both the top and bottom chambers at concentrations of 10 μM. The antagonists used were the same as in panel A. The migration is given as relative migration, where the number of migrating cells is normalized to that seen with 10 μM histamine. C, IC50 for JNJ 7777120 inhibition of histamine-induced mast cell chemotaxis. Histamine (10 μM) was added to the bottom chamber, and various concentrations of JNJ 777120 were added to both the top and bottom chambers. In all panels, the results shown are the average of three experiments, and the error bars represent the S.E.M. In panels A and B, the statistical significance relative to histamine-only treatment was judged using a Student's t test (★★★, p < 0.001).
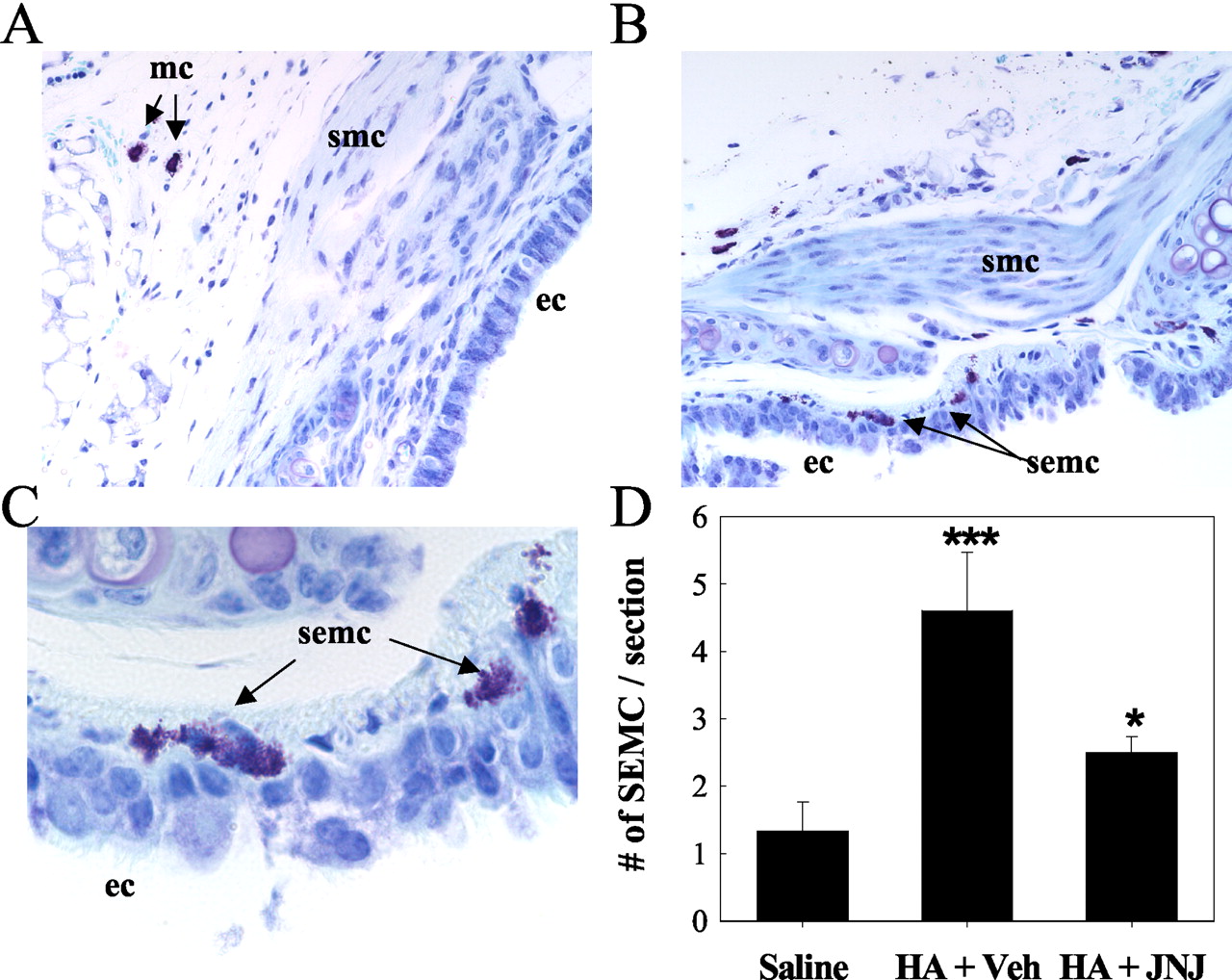
The in vivo migration of mast cells to histamine has been investigated. Mice (n = 10 per group) were given a 20-min aerosol administration of histamine or PBS once a day for 2 days. JNJ 7777120 or vehicle was given at 20 mg/kg s.c. 15 min before a 20-min aerosol administration of histamine or PBS. This was repeated for 2 days. Sections of the trachea were stained for mast cells, and the number of mast cells in the subepithelial space per tracheal section was determined. With aerosol administration of PBS, most of the tracheal mast cells remained in the surrounding connective tissue with only a very few resident cells in the subepithelial region (Fig. 5A). Upon administration of histamine, there was an increase in the number of mast cells per tracheal section and an apparent migration of the mast cells toward the tracheal epithelium (Fig. 5, B and C). This effect was blocked by s.c. administration of JNJ 7777120, indicating that, in this model, the mast cells are migrating along the histamine concentration gradient created by the aerosol and that this is mediated by the H4 receptor (Fig. 5D). This conclusion is supported by the fact that the migration was not significantly affected by H1-, H2-, or H3-selective antagonists (data not shown). There was also an H4 receptor antagonist-mediated reduction in the number of total mast cells per tracheal section that had been enhanced by histamine aerosol (data not shown).

Histamine-induced mast cell migration in vivo. Aerosolized histamine-induced accumulation of subepithelial mast cells (semc) in the trachea of mice. Representative tracheal sections stained with toluidine blue are shown. In animals that received saline-only aerosol (A), the mast cells (arrows, mc) were found mainly outside of the smooth muscle cell layer (smc). When the mice received aerosolized histamine (B, C), there was an increase in the total number of mast cells and in the number of subepithelial mast cells (semc). C, magnification of the epithelia cell (ec) layer from panel B showing that upon histamine treatment, mast cells were found in this layer. The increase in subepithelial mast cells was blocked by JNJ 7777120 (20 mg/kg s.c.). The total number of subepithelial mast cells per tracheal section was quantitated (D). The average number of subepithelial mast cells per tracheal section (n = 10 animals, with two sections analyzed for each animal) is shown, and the error bars represent the S.E.M. Statistical significance was judged using a Student's t test (★★★, p < 0.001 versus saline-only administration; ★, p < 0.01 versus vehicle treatment).
One of the most characteristic H1 receptor responses is increases in vascular permeability. A model for this response is the edema induced upon histamine injection into the paws of mice. This is thought to be mediated via the H1 receptor, but discovery of the H4 receptor led us to revisit this issue. The histamine-induced edema, which was measured as an increase in paw weight, was blocked by the H1 receptor antagonist, desloratidine, but not by JNJ 7777120 (Fig. 6). This suggests that the H4 receptor has little role in this traditional initial inflammatory response associated with histamine and that JNJ 7777120 does not significantly cross-react with the H1 receptor in vivo.

Histamine-induced paw edema in mice. A histamine H1 receptor antagonist, desloratidine, blocks histamine-induced paw swelling in mice, but JNJ 7777120 has no effect. The average difference in weight between the left and right paw was quantitated after the injection of histamine into the left footpad. The mice received vehicle, a H1 antagonist (desloratidine, i.p. 1 mg/kg), or JNJ 777120 (p.o. 200 mg/kg). The results shown are the average of several replicates (n = 5), and the error bars represent the S.E.M. Statistical significance was judged using a Student's t test (★★, p < 0.005 versus vehicle treatment).
A peritonitis model in mice was used to probe the role of the H4 receptor in general inflammation. If mice are injected intraperitoneally with zymosan, there is a dramatic extravasation of polymorphonuclear cells, which peaks at 4 h. Figure 7 shows that when these mice were treated s.c. with 10 mg/kg of JNJ 7777120 or 5 mg/kg thioperamide 15 min prior to injection of zymosan, there was a significant reduction in PMN accumulation 4 h after treatment. A dose titration of JNJ 7777120 in the zymosan-induced peritonitis model was studied. The test compound was administered as a single oral dose 15 min before zymosan injection. There was a statistically significant reduction of neutrophil infiltration for all doses at or above 30 mg/kg (Table 3). However, it is worth noting that the dose titration is somewhat shallow, and in no case can complete inhibition be obtained. The thioglycollate-induced peritonitis model was also run. The infiltrating cells were measured at 4 h, and, with the addition of thioglycollate, there was an accumulation of neutrophils [7.8 ± 0.2 × 105 cells per mouse (n = 6) versus 0.1 ± 0.1 × 105 (n = 3) when no thioglycollate was added]. In contrast to the results with the zymosan-induced peritonitis model, treatment with 10 mg/kg s.c. JNJ 7777120 had no significant effect on the PMN accumulation [5.8 ± 0.1 × 105 cells per mouse (p = 0.37; n = 5)]. This may be due to differences in the cell type involved in initiating the inflammation (see Discussion).

JNJ 7777120 reduces the neutrophil influx in mouse peritonitis models. A, mice (n = 7 per group) were treated with JNJ 7777120 (10 mg/kg s.c.), thioperamide (5 mg/kg s.c.), or PBS. Fifteen minutes later, zymosan was injected i.p. At 4 h, the peritoneal cells were harvested by lavage, and polymorphonuclear cells were quantitated. The symbols indicate results for individual mice, and the bar represents the average. The inhibition by both JNJ 7777120 and thioperamide was judged to be significant using a Student's t test (p < 0.001).
Inhibition of PMN influx at 4 h by JNJ 7777120 in the mouse zymosan-induced peritonitis model
Experimental details are given under Materials and Methods.
JNJ 7777120 shows oral activity in mouse peritonitis models, which implies that it is orally bioavailable. To probe this, single-dose pharmacokinetics were evaluated in mice (n = 4), rats (n = 3), and dogs (n = 3). The compound was administered orally at 10 mg/kg to mice, rats, and dogs and intravenously at 1 mg/kg in dogs and 3 mg/kg in rats. LC/MS analysis was used to quantitate the plasma levels. After oral administration, JNJ 7777120 had an absolute oral availability of 22 to 100% and an oral half-life of 1 to 2 h depending on the species (Table 4).
In vivo pharmacokinetic parameters for JNJ 7777120a
Discussion
The recent discovery of the histamine H4 receptor has lead to a re-evaluation of the biological function of histamine. Indeed, with its expression pattern limited to hematopoietic cells, a role for the H4 receptor in inflammation has been postulated. One of the best tools for probing the pharmacology and in vivo function of a novel receptor is a selective receptor ligand. In some cases, the endogenous agonists are selective and can be used to validate the biology. However, histamine binds to at least four receptors, and therefore a pharmacological approach using the native ligand is complicated. Selective synthetic ligands have been used for many years to elucidate the function of the various histamine receptions. However, to date, the only described H4 ligands are not selective and have affinities for the H3 receptor as well as H4. Here, we describe the pharmacological characterization of the first selective H4 antagonist, JNJ 7777120, which should prove invaluable in helping to decipher the biological role of the H4 receptor.
JNJ 7777120 binds with high affinity to the H4 receptor (Kd, ∼4 nM) and has a greater than 1000-fold selectivity over the other histamine receptors. Several other proteins (∼50) have been studied for inhibition by JNJ 7777120, and little or no activity was found at ∼300× the Kd for the H4 receptor. As with any compound, inhibition of other targets can never completely be ruled out, but it appears that JNJ 7777120 is highly selective for the H4 receptor.
JNJ 7777120 has an affinity for the mouse and rat H4 receptors that is similar to the affinity versus the human H4 receptor. This is not a trivial observation, since there is significant sequence variation between the species (Liu et al., 2001b). Indeed, even histamine itself is a log less potent for the mouse and rat H4 receptors than for the human receptor. This is also true for most of the ligands previously tested, with the exception of thioperamide (Liu et al., 2001b). Potency in mice and rats, as well as reasonable pharmacokinetic parameters, make JNJ 7777120 an extremely useful tool for probing the physiological role of the H4 receptor.
The transfected cells used in this study to measure functional antagonism contain the H4 receptor and a cAMP responding reporter construct. The addition of histamine to these cells causes an inhibition of forskolin-stimulated cAMP increases as previously described (Oda et al., 2000; Liu et al., 2001a; Zhu et al., 2001). However, the cAMP-inhibitory effect is low in comparison with that mediated by the H3 receptor. This may be due to the fact that the cell line used is of neuronal origin and may express a different set of G-proteins than those found in leukocytes. It has also been reported that an increase in calcium mobilization can be observed in cells cotransfected with the H4 receptor and chimeric or promiscuous G-proteins (Oda et al., 2000; Liu et al., 2001b; Morse et al., 2001; Zhu et al., 2001). In primary mouse mast cells, we have previously observed that the H4 receptor signals through increases in calcium, whereas no effect on cAMP levels could be observed (Hofstra et al., 2003). This effect was blocked by pertussis toxin. This is also likely to be true in eosinophils, since the original hint of the existence of the H4 receptor came from the findings of Raible et al. (1992, 1994) showing a histamine-induced calcium influx in eosinophils that was not blocked by H1 or H2 antagonists but could be blocked by thioperamide. All of these results are consistent with coupling of the H4 receptor to Gαi/o G-proteins in mast cells, as well as eosinophils, that can trigger calcium mobilization.
Previously, a role has been suggested for the H4 receptor in mediating histamine-induced chemotaxis of mast cells (Hofstra et al., 2003). This has been characterized using a dual H4/H3 antagonist and mast cells from H4 receptor knockout mice. The use of the selective H4 receptor antagonist described here, JNJ 7777120, allows for more definitive evidence for the role of the H4 receptor. The IC50 calculated for JNJ 7777120 inhibition of mast cell chemotaxis is consistent with the affinity for the H4 receptor, since the amount of histamine used was 10-fold over the EC50, and hence, a log shift in the potency of the compound would be expected. These results conclusively prove that the H4 receptor mediates mast cell histamine-induced chemotaxis.
What is the physiological role for histamine induced-chemotaxis of mast cells? Increases in mast cell numbers are found in conditions such as asthma, rhinitis, rheumatoid arthritis, psoriasis, and cardiovascular disease (Marone et al., 1997; Peterson et al., 1998; Krishnaswamy et al., 2001; Woolley, 2003). For mast cells, it is known that progenitor cells from the bone marrow migrate to connective or mucosal tissue, where they differentiate into mature cells. The major chemoattractant for this localization is thought to be stem cell factor. In humans, there is a redistribution of mast cells to the epithelial lining of the nasal mucosa in response to antigens (Fokkens et al., 1992; Slater et al., 1996), and it is conceivable that this chemotaxis is mediated by histamine via the H4 receptor. To mimic this effect, we carried out aerosol administration of histamine in mice. In response to histamine, there was redistribution of mast cells to the tracheal epithelium that could be blocked by systemic administration of the H4 receptor antagonist. This may mimic what happens in allergic conditions, where the presence of allergens in the airways leads to release of histamine either by the resident mast cells or by other cells. This, in turn, recruits additional mast cells, resulting in an amplification of the allergic symptoms and inflammation. In support of this, increases in mast cell numbers are found in allergic rhinitis and allergy in humans (Kirby et al., 1987; Crimi et al., 1991; Amin et al., 2000; Gauvreau et al., 2000; Kassel et al., 2001).
Peritonitis models in mice have been used for many years as a readout of acute inflammation. Here, we show that a H4 receptor antagonist, JNJ 7777120, can partially block inflammation in a zymosan-induced peritonitis model. This model is characterized by the influx of neutrophils in response to an irritant. This response includes the release of histamine and other proinflammatory cytokines and chemokines. Histamine is known to play a role in these models, since it has been shown that both H1 and H2 receptor antagonists can partially block zymosan-induced plasma exudation and that H1 receptor antagonists can partially block the neutrophil influx (Kolaczkowska et al., 2001). The effects on neutrophil influx are similar for both the H4 and H1 receptor antagonists, indicating that they may both be playing a role. In both cases, only partial inhibition can be obtained. This may indicate that histamine is only one of the factors driving the neutrophil influx or that both receptors need to be blocked simultaneously. We have not, as of yet, tried to treat animals with both antagonists to see whether the effects are additive.
This model is somewhat complicated, and several different cell types are involved. It is possible that JNJ 7777120 exerts its effect by acting directly on the neutrophils. The actions of histamine via the H2 receptor on neutrophils have been well documented, and there is also some evidence for the presence of H1 receptors. To date, there has been no described H4 activity on neutrophils. H4 receptor expression has been reported on human neutrophils (Oda et al., 2000; Morse et al., 2001; Zhu et al., 2001), but we have not been able to confirm this and have found no evidence for H4 receptor expression on mouse neutrophils (data not shown). Furthermore, although peritoneal lavage fluid from zymosan-treated animals is chemotactic for neutrophils in vitro, the addition of JNJ 7777120 had no effect on this chemotaxis (data not shown). Peritoneal macrophages may also play a major role in initiating the response, but, once again, it does not appear that they express the H4 receptor (Hofstra et al., 2003). A more likely target is the mast cell. The zymosan-induced peritonitis model is reported to be mast cell-dependent, since the neutrophil influx is reduced in mast cell-deficient or -depleted mice (Ajuebor et al., 1999). The fact that JNJ 7777120 is not effective in the thioglycollate-induced peritonitis model, which is reported not to be mast cell-dependent (Ajuebor et al., 1999), is consistent with the conclusion that JNJ 7777120 is acting on mast cells. We do not believe that JNJ 777120 is acting as a stabilizer of mast cells, since the H4 receptor does not seem to play a role in degranulation (Hofstra et al., 2003). We are currently investigating the role of the H4 receptor in the release of other mast cell mediators. The inflammatory response in the zymosan model is only partially mediated by mast cells, since a lack of mast cells only reduces, but does not abolish, the response. This is thought to be due to additional contributions of a macrophage response. A partial response is also seen when using the H4 receptor antagonists, which may be consistent with mast cells being their target, since the H4 receptor is not expressed on macrophages.
The role of the H4 receptor in mast cell, eosinophil, and T cell function, as well as the effects of the H4 receptor antagonist, JNJ 7777120, in a mouse peritonitis model point to a more general role for the H4 receptor in inflammation. Selective H4 receptor antagonists like JNJ 7777120 may have the potential to be useful in treating inflammation in humans. Allergic rhinitis, asthma, and rheumatoid arthritis are just a few of the conditions where mast cells and eosinophils are involved and where H4 receptor antagonists may have therapeutic utility.
Acknowledgments
We thank Tim Lovenberg, Changlu Liu, and Sandy Wilson for providing the H4, H3, and H1 cell lines as well as advice on running the binding and functional assays. Sherry Baker developed the human and mouse H2 receptor cell lines and provided the H2 receptor functional data. We are indebted to Kiev Ly, Jill Jablonowski, Nicholas Carruthers, Curt Dvorak, and Cheryl Grice for the synthesis of JNJ 7777120. Druie Cavender and Gill Olini ran the thioglycollate-induced peritonitis models. We are also indebted to Bill Hageman and Jefferey Hall for pharmacokinetics studies in dogs as well as Kenway Hoey and Mark Feinstein for LC/MS analysis of pharmacokinetic plasma samples.
Footnotes
-
↵1 Current address: NV Organon, Molenstraat 110, P.O. Box 20, 5340 BH Oss, The Netherlands.
-
DOI: 10.1124/jpet.103.061754.
-
ABBREVIATIONS: JNJ 7777120, 1-[(5-chloro-1H-indol-2-yl)carbonyl]-4-methylpiperazine; JNJ 5207852, 1-[3-(4-((piperidin-1-yl)methyl)phenoxy)propyl]piperidine; PBS, phosphate-buffered saline; PMN, polymorphonuclear cells; LC/MS, liquid chromatography/mass spectrometry.
- Received October 16, 2003.
- Accepted December 16, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}