


Abstract
The effects of prolonged exposure of M2 muscarinic acetylcholine receptors (mAChRs), stably expressed in Chinese hamster ovary cells, to the allosteric modulators gallamine, alcuronium, and heptane-1,7-bis (dimethyl-3′-phthalimidopropyl)-ammonium bromide (C7/3′-phth) were compared with the effects of the agonist carbachol (CCh) and antagonists atropine and N-methylscopolamine (NMS). Intact cell saturation binding assays using [3H]NMS found that pretreatment of the cells for 24 h with CCh caused a significant down-regulation of receptor number, whereas atropine, NMS, and all three allosteric modulators caused receptor up-regulation. Functional assays using a cytosensor microphysiometer to measure whole-cell metabolic rate found no acute effects of gallamine on receptor signaling, whereas atropine seemed to behave as an inverse agonist. Pretreatment of the cells with gallamine (20 μM) or atropine (20 nM) resulted in a significant enhancement of the maximal effect evoked by CCh. In contrast, CCh (100 μM) pretreatment resulted in a significant reduction in maximal receptor signaling capacity. Time-course experiments revealed that the effects of atropine and gallamine on receptor up-regulation are only visualized after at least 12-h ligand exposure, compared with the more rapid effects of CCh, which achieve steady-state down-regulation within 90 min. Additional experiments monitoring CCh-mediated M2 mAChR internalization in the presence of gallamine revealed that part of the mechanism underlying the effects of the modulator on receptor expression may involve a change in receptor internalization properties. These findings suggest that, like orthosteric ligands, G protein-coupled receptor allosteric modulators also are able to mediate long-term effects on receptor regulation.
The majority of all known drugs mediate their therapeutic effects by targeting cell-surface receptors, of which the G protein-coupled receptor (GPCR) superfamily constitutes that largest group (Drews, 2000). The ability to selectively target drugs to various GPCRs has traditionally involved the design of ligands that act at the receptor's orthosteric binding site, that is, the binding site recognized by the endogenous ligand for that receptor (Neubig et al., 2003). However, it is now becoming accepted that many GPCRs can also possess at least one allosteric binding site, that is, a site topographically distinct from the orthosteric site that, when occupied by ligand, can modulate the binding and/or functional properties of orthosteric ligands (Christopoulos and Kenakin, 2002).
Allosteric modulators possess a number of theoretical advantages over orthosteric drugs. For instance, they can either inhibit or potentiate ligand binding affinity and/or function. This is in contrast to orthosteric ligands, which can only act competitively (May and Christopoulos, 2003). Allosteric modulators also have the possibility of displaying greater subtype selectivity for some receptors by recognizing binding domains that show high sequence divergence between receptor subtypes (Christopoulos, 2002), or by selectively exerting a cooperative effect on the binding of an endogenous orthosteric agonist at a single receptor to the exclusion of other subtypes (Lazareno et al., 2004). Many of these properties of allosteric modulators have been investigated in some detail using the using muscarinic acetylcholine receptor (mAChR) family as a model GPCR system (Lee and El-Fakahany, 1991; Tucek and Proska, 1995; Birdsall et al., 1996; Ellis, 1997; Christopoulos et al., 1998; Holzgrabe and Mohr, 1998). In particular, the M2 mAChR is well established as possessing a high affinity for a variety of structurally diverse modulators that, nevertheless, seem to interact at a common allosteric binding site (Ellis and Seidenberg, 1992; Lanzafame et al., 1997; Tränkle et al., 1998).
In addition to the acute effects of both orthosteric and allosteric drugs on mAChR function, the cellular host system itself can exert a profound effect on the function of the receptor when the latter is exposed to a drug for prolonged periods of time. For example, it is widely accepted that upon prolonged agonist exposure, mAChRs, like most other GPCRs, undergo signal attenuation by mechanisms commonly referred to as desensitization, sequestration, and/or down-regulation (Bunemann et al., 1999). More recently, inverse agonist ligands have also been demonstrated to exert profound effects on GPCR regulation and expression after prolonged exposure, although in a manner opposite to that seen with agonists; a common observation upon prolonged inverse agonist treatment is an up-regulation in GPCR expression levels (Milligan and Bond, 1997). Interestingly, although allosteric modulators of GPCRs are now being recognized as potentially novel therapeutic agents, surprisingly little is known about their long-term effects on receptor function, even though this information is vital if these drugs are to be used clinically.
Given that allosteric modulators engender potentially unique receptor conformations by binding to domains that are distinct from those of orthosteric agonists and inverse agonists, the aim of the present study was to use the human M2 mAChR as a model system to investigate the effects of long-term exposure of allosteric modulators on the cell-surface expression and the signaling properties of this receptor, and to gain some insight into the mechanisms underlying these effects. We report, for the first time, that prolonged exposure to three different allosteric modulators of M2 mAChRs results in a significant enhancement of cell-surface receptor expression that may reflect the ability of the modulators to promote a receptor conformation that displays modified internalization properties.
Materials and Methods
Materials. Dulbecco's modified Eagle's medium (DMEM), geneticin, and trypsin were obtained from Invitrogen (Carlsbad, CA). (-)[N-methyl-3H]scopolamine methyl chloride (70-87Ci/mmol) was from PerkinElmer Life and Analytical Sciences (Boston, MA). Bio-Rad protein assay kit was obtained from Bio-Rad (Hercules, CA). Ultima Gold was from Packard (Greningen, The Netherlands). Alcuronium was a generous gift from F. Hoffmann-La Roche (Basel, Switzerland), and heptane-1,7-bis-(3′-phthalimidopropyl)-ammonium bromide (C7/3-phth) was synthesized at the Institute of Drug Technology (Boronia, Victoria, Australia). All other chemicals and reagents were from Sigma-Aldrich (St. Louis, MO).
Cell Culture. CHO-K1 cell lines stably transfected with the human M2 mAChR (CHO M2 cells; provided by Dr. Mark Brann, University of Vermont College of Medicine, Burlington, VT), were cultured in DMEM supplemented with 10% fetal bovine serum, 20 mM HEPES, and 50 μg/ml geneticin. Cells were grown at 37°C in 5% CO2, 95% O2. At approximately 90% confluence, cells were harvested by trypsinization followed by centrifugation (400g, 3 min) and resuspension (three times) in HEPES buffer (110 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 1 mM MgSO4, 25 mM glucose, 20 mM HEPES, and 58 mM sucrose; pH 7.4 with NaOH). Cells between passages 6 to 20 were used in all experiments, unless otherwise indicated.
Saturation Binding Assays. Two different types of saturation binding assays were performed: 1) Complete saturation binding isotherms, using increasing concentrations of the radiolabeled muscarinic antagonist, [3H]NMS (0.02 nM-5 nM), were constructed in intact CHO M2 cells. This assay used approximately 105 cells per assay tube, made up in a final volume of 1 ml of HEPES buffer, with 10 μM atropine used to determine nonspecific binding. For initial control experiments, cells were incubated at 37°C for 1 h in a shaking water bath. In subsequent experiments, cells were pretreated with a fixed concentration (approximately 10 × KB; see Results) of carbachol (CCh), atropine, or gallamine, at 37°C for 24 h (unless otherwise indicated under Results), before extensive washing on ice to remove pretreatment ligand while preventing any receptor cycling. The system was then allowed to equilibrate with radioligand for 3 h at 4°C, and the reaction was then terminated by rapid vacuum filtration through Whatman GF/C filters using a Brandel cell harvester, followed by three washes of ice-cold 0.9% sodium chloride buffer. Control experiments (not shown) indicated that 3 h was sufficient for binding equilibrium to be achieved at 4°C. Filters were then dried, placed in scintillation vials, 4 ml/vial of Packard Ultima Gold scintillant was added, and the vials were left to sit for at least 3 h. Radioactivity was then determined by scintillation counting. 2) A “two-point” saturation binding assay was also performed on CHO M2 cells. This assay is a modified saturation binding assay where only two concentrations, 0.1 and 1 nM, of [3H]NMS are used, allowing a calculation of cell surface receptor density (Bmax; see also “Data Analysis”). This assay was performed in triplicate and used 2 × 105 cells per tube in a total volume of 1 ml of HEPES buffer. Cells were initially pretreated with a fixed concentration (approximately 10 × KB; see Results) of orthosteric ligand (CCh, atropine, or NMS), allosteric modulator (gallamine, C7/3-phth, or alcuronium), or a combination of NMS and alcuronium, at 37°C for 24 h (unless otherwise indicated under Results), before extensive washing on ice. Due to a loss of activity over time (presumably due to breakdown), the C7/3-phth and alcuronium were replaced every 6 h. All other details are as described above for the complete saturation binding experiments.
Inhibition Binding Assays. In initial experiments, approximately 2 × 105 CHO M2 cells in 1 ml of HEPES buffer/tube were incubated with a fixed concentration of [3H]NMS (0.2 nM) in the absence or presence of increasing concentrations of CCh (10 nM-3 mM), atropine (3 pM-3 μM), or NMS (0.1 pM-0.1 μM). Reactions were allowed to proceed for 1 h at 37°C, with nonspecific binding defined using 10 μM atropine. Reaction termination and determination of radioactivity were as described in the preceding section.
To determine the equilibrium binding parameters describing the interaction between the agonist CCh, the allosteric modulator gallamine, and the radioligand [3H]NMS, additional combination inhibition binding experiments were undertaken. Cells were incubated with a fixed concentration of [3H]NMS (0.2 nM) in the absence or presence of increasing concentrations of CCh alone (0.1 μM-10 mM), gallamine alone (10 nM-1 mM), or gallamine (10 nM-1 mM) together with fixed concentrations of CCh (30 or 200 μM for three experiments; 100 or 1 μM for three experiments). Incubations (105 cells per tube; 1-ml total volume) were for 3 h at 4°C. Reactions were terminated and radioactivity was determined as described above.
Receptor Internalization Assay. The experimental protocol used to monitor the internalization of cell surface M2 mAChRs was modified from that of Pals-Rylaarsdam et al. (1997). Approximately 105 cells/well were distributed in 24-well plates and allowed to reach confluence over 24 h. Subsequently, cells were exposed to CCh (100 μM), gallamine (20 μM), atropine (20 nM), or a combination of CCh and gallamine for various time points (see Results) at 37°C before being washed five times with ice-cold HEPES buffer. Cells were then incubated at 4°C, to arrest receptor cycling, with a near-saturating concentration of [3H]NMS (2 nM) for 3 h to measure the remaining accessible cell surface receptors. Nonspecific binding was determined using 10 μM atropine. At the end of the 3-h incubation period, buffer and drugs were aspirated and the wells were washed three times. Cells were solubilized from the wells with 2 × 0.5 ml of 0.2 M NaOH, and both fractions were then combined and radioactivity was determined by scintillation counting as described above.
Cytosensor Microphysiometer Assay. Approximately 2 × 105 cells were plated into sterile capsule cups (12 mm in diameter, 3.0-μm pore) in a 12-well plate 24 h before use. At the same time as plating, the cells were exposed to various drug or vehicle pretreatment conditions as indicated under Results. At the end of the 24-h pretreatment, cells were loaded into the sensor chambers of a cytosensor microphysiometer (Molecular Devices, Monlo Park, CA) and superfused with medium (bicarbonate-free DMEM, pH 7.4) until the cellular acidification rate was steady (∼30 min). Increasing concentrations of CCh (1 nM-1 mM) were then superfused across each chamber at a pump speed of 100 μl/min to construct a cumulative concentration-response curve. Each pump cycle was of 1-min 30-s duration, with drug being perfused for 1 min and then the pump being switched off for the remaining 30 s. Recordings of extracellular pH were made from 1 min 8 s to 1 min 28 s of each cycle, and the extracellular rate of acidification (ECAR; mV s-1/change in pH units) was calculated using the Cytosoft program (Molecular Devices).
Data Analysis. Both total and nonspecific saturation binding data sets were globally fitted to the following equation via nonlinear regression using Prism 4.0 (GraphPad Software Inc., San Diego, CA): 
where Y denotes the total binding, [A] is the concentration of [3H]NMS, Bmax is the maximum number of binding sites, KA is the equilibrium dissociation constant of [3H]NMS, and NS is the fraction of the total binding that represents nonspecific binding. This latter parameter was shared between both total and nonspecific binding data sets. Bmax values from the two-point saturation binding experiments were calculated using the following equation (Lazareno and Birdsall, 1995; Christopoulos, 2000): 
where Bmax is as described previously, [A1] and [A2] are the low and high concentrations of [3H]NMS used, and [B1] and [B2] are the corresponding specific binding counts. It should be noted that this approach provides reliable estimates of the Bmax relative to estimates obtained from complete saturation binding curve analysis, provided that the experimental error is ≤10% CV (A. Christopoulos, unpublished data), as is normally the case for [3H]NMS binding at mAChRs. These experiments also allowed for a calculation of the equilibrium dissociation constant KA of [3H]NMS after the various treatment conditions using the following equation (Lazareno and Birdsall, 1999): 
Inhibition Binding Assays. Normalized orthosteric ligand competition binding data were fitted to the following equation according to a simple mass-action model for competition for one binding site: 
where [B] denotes the concentration of orthosteric ligand present, KB is the equilibrium dissociation constant for the orthosteric ligand, and Y, [A], and KA are as defined previously.
For the combination experiments between the radioligand [3H]NMS, allosteric modulator gallamine, and unlabeled orthosteric ligand CCh, data sets were globally fitted to the following extended allosteric ternary complex model (Fig. 1; see Christopoulos, 2000; Christopoulos and Kenakin, 2002): 

An extended allosteric ternary complex model. A, B, and I denote the radioligand, allosteric modulator, and unlabeled orthosteric ligand, respectively, and KA, KB, and KI denote their respective equilibrium dissociation constants; α denotes the cooperativity factor for the allosteric interaction between the radioligand and allosteric modulator, and β denotes the cooperativity factor for the allosteric interaction between the unlabeled orthosteric ligand. Values of α and β greater than 1 denote positive cooperativity, whereas values less than 1 denote negative cooperativity.
with 
where [A], [B], and [I] denote the concentrations, and KA, KB, and KI denote the equilibrium dissociation constants, of the radioligand, allosteric modulator, and unlabeled orthosteric ligand, respectively; α denotes the cooperativity factor for the allosteric interaction between the radioligand and allosteric modulator; β denotes the cooperativity factor for the allosteric interaction between the unlabeled orthosteric ligand and allosteric modulator; and the parameter slope denotes a logistic slope factor for the binding of the unlabeled orthosteric ligand. According to the allosteric ternary complex model (Ehlert, 1988; Lazareno and Birdsall, 1995; Christopoulos, 2002), the cooperativity factor is a measure of the extent by which the affinity of one ligand is modified by the concomitant binding of another ligand on the same receptor. In this model, values of α and β greater than 1 denote positive cooperativity, whereas values less than 1 denote negative cooperativity. For this analysis, the KA parameter was fixed to a constant value, as determined separately in [3H]NMS saturation experiments.
For the receptor internalization experiments, the kinetics of loss of cell surface [3H]NMS binding were analyzed according to the following two-phase exponential decay model: 
where Span1 and Span2 denote the percentage of each phase, Plateau denotes the minimal asymptotic value, and k1 and k2 denote the rate constants for the components defined by Span1 and Span2, respectively. An extra-sum-of-squares (F-test) was used to determine whether the data were significantly better fitted to this model compared with a simpler model characterized by a single Span and k value.
Agonist concentration-response data from the cytosensor assays were consistently characterized by a marked decline in maximal agonist response at high agonist concentrations (see Results). Thus, to obtain a close model fit to the data and derive estimates of agonist potency and maximal response range, the following Gaussian curve equation was used (Christopoulos et al., 2001; Motulsky and Christopoulos, 2004): 
with 
where E denotes effect, Basal denotes the minimum asymptotic effect in the absence of agonist, Log[A] is the logarithm of the concentration of agonist, slope is a slope factor, Log EC50 is the logarithm of the midpoint location parameter, and Range denotes the maximal response range over the Basal value.
In practice, all affinity, potency, and cooperativity parameters were determined as logarithms (Christopoulos, 1998). In all instances, results are expressed as mean ± S.E.M. Statistical analyses were performed by one-way ANOVA, with p < 0.05 indicating statistical significance.
Results
Determination of Ligand Binding Properties in Intact CHO M2 Cells. Initial saturation binding experiments were performed to determine the affinity of [3H]NMS for the human M2 mAChR in intact CHO M2 cells. The data were well described by a standard one-site hyperbolic binding model, with a calculated value of Log KA being -9.60 ± 0.01 and Bmax being 5.50 ± 1.10 fmol/105 cells (n = 3). Subsequent inhibition binding experiments were performed to determine the affinity (Log KB) of a variety of test compounds for the M2 mAChR, because this information was required to inform the design of experiments investigating the effects of prolonged ligand exposure. The results of these characterization experiments for the orthosteric agonist CCh and the orthosteric antagonists atropine and NMS are shown in Table 1, together with affinity and cooperativity values for the allosteric modulators gallamine, alcuronium, and C7/3-phth derived from a recent study by our group using the same cells under identical experimental conditions (Avlani et al., 2004). It should be noted that the Log KB value for CCh is only an apparent measure of agonist affinity, because CCh can cause internalization under these assay conditions (see below). In all instances of competition binding, the curve slope factors were not significantly different from unity.
Binding parameters for orthosteric ligands and allosteric modulators against [3H]NMS at human M2 mAChRs expressed in intact CHO cells
Values are mean ± S.E.M. of three experiments conducted in triplicate at 37°C for 60 min. Log KB is the logarithm of the ligand equilibrium dissociation constant at the free receptor, determined using nonlinear regression as outlined under Materials and Methods. Log α is the logarithm of the cooperativity factor. Antilogarithm (geometric mean) is given in parentheses.
Effects of Prolonged Ligand Exposure on M2 mAChR Cell Surface Expression.Figure 2A illustrates the effects of 24-h pretreatment of CHO M2 cells with CCh, atropine, or gallamine (at approximately 10× KB values from Table 1) on the binding properties of [3H]NMS. In comparison with vehicle controls, pretreatment of CHO M2 cells with the agonist CCh led to a significant decrease in cell surface expression of the M2 mAChR (Table 2). In contrast, the orthosteric antagonist atropine, and interestingly, the allosteric modulator gallamine each caused a significant increase in the [3H]NMS Bmax value after 24-h exposure (Table 2). No effects were observed on radioligand affinity (Table 2). Furthermore, the Log KA values determined for [3H]NMS from these experiments were not significantly different (p > 0.05) between any of the groups and the vehicle controls, indicating that the washing protocol used was sufficient to fully remove the pretreatment ligands from the receptor compartment.

Cell surface binding of [3H]NMS to M2 mAChRs, expressed in intact CHO cells that had been pre-exposed to a variety of ligands for 24 h at 37°C, before extensive washout on ice. A, saturation binding isotherms after pretreatment with vehicle (•), 100 μM CCh (□), 20 nM atropine (▿), or 20 μM gallamine (○). Data points represent the mean ± S.E.M of three to six experiments performed in triplicate at 4°C for 3 h. B, [3H]NMS two-point Bmax determinations after pretreatment with the orthosteric ligands CCh (100 μM), atropine (20 nM), or NMS (2.5 nM); the allosteric modulators gallamine (20 μM), C7/3-phth (20 μM), or alcuronium (10 μM); or a combination of 2.5 nM NMS and 10 μM alcuronium. Bmax values were normalized to those obtained using vehicle pretreated controls (mean = 4.95 ± 0.65 fmol/105 cells). Values of [3H]NMS Log KA were also calculated (see Materials and Methods) for each of the treatments and were as follows: vehicle (-9.69 ± 0.28), CCh (-9.77 ± 0.23), atropine (-9.66 ± 0.25), NMS (-9.69 ± 0.28), gallamine (-9.54 ± 0.26), C7/3-phth (-9.79 ± 0.18), alcuronium (-9.83 ± 0.25), and NMS + alcuronium (-9.86 ± 0.15). Data represent the mean ± S.E.M of four to eight experiments performed in triplicate at 4°C for 3 h.
Saturation binding parameters for [3H]NMS at human M2 mAChRs expressed in intact CHO cells after pretreatment with the indicated ligand for 24 h
Values are mean ± S.E.M. Cells were treated as indicated at 37°C, followed by extensive washout on ice prior to assay.
Specificity of Ligand Pretreatment Effects on M2 mAChR Cell Surface Expression. To investigate the effect of prolonged pretreatment of CHO M2 cells with a wider variety of allosteric and orthosteric ligands, we used a two-point saturation binding assay, which specifically allows for the calculation of radioligand Bmax values without the need to construct complete saturation binding curves. The results of these experiments are shown in Fig. 2B, where significant (p < 0.05) increases in receptor cell surface expression were revealed after pretreatment with the potent antagonist NMS, the allosteric inhibitor C7/3′-phth, or the allosteric enhancer alcuronium, as well as with atropine or gallamine. In addition, the combination of NMS and alcuronium, which is normally characterized by positive binding cooperativity at M2 mAChRs (Avlani et al., 2004), also resulted in a significant enhancement of receptor cell surface expression, although this was no greater than that observed with either ligand used alone. As before, pretreatment with CCh led to a significant decrease in cell surface M2 mAChR expression. In all instances, there was no significant effect of pretreatment on calculated [3H]NMS Log KA (see Fig. 2B legend).
Effects of Prolonged Ligand Exposure on M2 mAChR Function. The functional consequence of M2 mAChR activation after various pretreatment conditions was determined via microphysiometric measurements of ligand-mediated changes on whole cell ECAR. The presence of increasing concentrations of gallamine did not cause any significant change in ECAR, in contrast to the clear stimulation observed with CCh (Fig. 3A). Interestingly, atropine caused a small, but significant (p < 0.05), concentration-dependent decline in basal ECAR, with a Log EC50 of -7.3 ± 0.4; this was not due to a pH change in the highest concentrations of antagonist used, because this was routinely adjusted to pH 7.4 for all concentrations. In contrast to the effects of acute exposure of the CHO cells to gallamine, 24-h pretreatment with the modulator before washout caused a significant effect on cellular responsiveness, as evidenced by changes in the concentration-response profile for CCh between the various pretreatment groups. These experiments are summarized in Fig. 3B and Table 3. Although no drug pretreatment resulted in a change in CCh potency relative to vehicle-pretreated cells, significant differences were noted for the maximal agonist response in cells pretreated with CCh, atropine, or gallamine. These effects on response range were in accordance with the effects seen on receptor cell surface expression in the binding assays, with atropine and gallamine causing an increase in agonist responsiveness, whereas pretreatment with CCh caused a significant decrease in subsequent responsiveness.
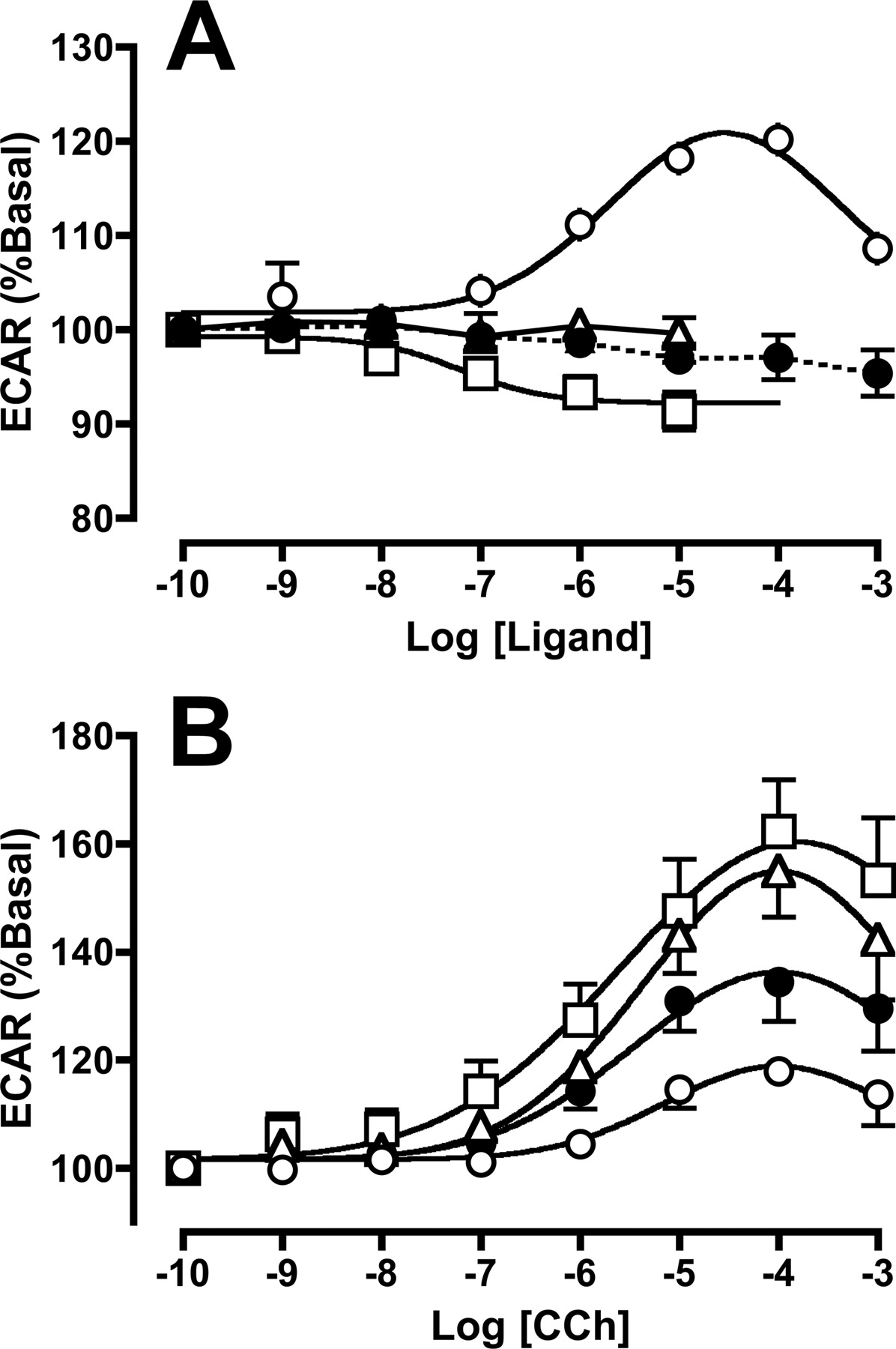
A, ECARs of CHO M2 cells exposed to vehicle (•) or increasing concentrations of CCh (○), atropine (□), or gallamine (▵). Individual prestimulated ECAR responses were normalized to 100% and are shown as the mean ± S.E.M of four experiments. B, effects of CCh on ECAR in CHO M2 cells that had been pretreated for 24 h with vehicle (•), 100 μM CCh (○), 20 nM atropine (□), or 20 μM gallamine (▵) at 37°C, followed by extensive washout on ice. Data points represent ± S.E.M of 13 to 17 experiments.
Estimates of CCh potency and maximal response range for M2 mAChR-mediated increases in whole-cell extracellular acidification rate in CHO cells under varying pretreatment conditions
Values are mean ± S.E.M. Cells were treated as indicated at 37°C, followed by extensive washout prior to assay.
Time Course of Ligand Effects on M2 mAChR Cell Surface Expression. Further experiments were performed to investigate the time course of the effect of pretreatment with CCh, atropine, or gallamine on cell-surface M2 mAChR expression. As shown in Fig. 4, CCh clearly caused maximal reduction of receptor expression within 3 h, whereas atropine and gallamine only began to cause an enhancement in cell surface expression from 12 h, which was statistically significant (p < 0.05) at 24 h.

Time course for change in M2 mAChR cell surface expression in intact CHO cells. CHO M2 cells were pretreated with either 100 μM CCh (•), 20 nM atropine (▾), or 20 μM gallamine (▴) for the indicated times at 37°C, followed by extensive washing on ice. Bmax values were calculated using two-point [3H]NMS saturation binding performed at 4°C for 3 h. Nonspecific binding was defined using 10 μM atropine. Bmax values were normalized to those obtained using vehicle pretreated controls (mean = 2580 ± 258 dpm). Each point represents the mean ± S.E.M of 7 to 10 experiments conducted in triplicate.
It is possible that the effects of prolonged exposure to gallamine lead to a long-term stabilization of a conformation of the M2 mAChR on the cell surface that is less prone to (agonist-independent) internalization, as has previously been shown for inverse agonist ligands (Gether et al., 1997). The fact that gallamine occupies a topographically distinct binding site on the M2 mAChR to that used by CCh provided us with a unique opportunity to further investigate the effects of the modulator on actual agonist-driven internalization, which is not possible when studying classic orthosteric antagonist/inverse agonist effects on receptor trafficking. To perform such experiments, however, it was first necessary to determine the occupancy binding relationships for CCh and gallamine, both alone and combined, at the M2 mAChR, to account for the negative cooperativity between the two ligands when they concomitantly occupy the receptor. Figure 5 shows a family of inhibition binding curves that are representative of this type of experiment. The curves of the inhibition of [3H]NMS binding by CCh alone, gallamine alone, and gallamine in the presence of two different concentrations of CCh were simultaneously fitted to an extended ternary complex model of allosteric interaction (eqs. 3-5) as described in Materials and Methods. The results from these experiments and accompanying analysis are summarized in Table 4. Although the estimates of both CCh and gallamine dissociation constants are approximately half a log unit lower than those shown in Table 1, this is most likely due to the fact that the combination experiments were performed at 4°C, to ensure that the estimate of CCh affinity, in particular, is not influenced by any concomitant internalization that the agonist may cause at 37°C. Importantly, these inhibition binding experiments allowed for the determination of β, the cooperativity factor describing the allosteric interaction between CCh and gallamine, which indicated that there was an approx. 200-fold reciprocal reduction in ligand affinity when both CCh and gallamine bound to the M2 mAChR at the same time.

Determination of the negative cooperativity between gallamine and CCh. CHO M2 cells were incubated with a fixed concentration of [3H]NMS (0.2 nM) for 3 h at 4°C in the presence of increasing concentrations of CCh alone (•), gallamine alone (○), or gallamine together with fixed concentrations of CCh (100 μM □ or 1 mM fl). Data represent one of six such experiments, with each point representing the mean ± S.E.M of triplicate determinations.
Binding parameters for interaction according to an extended allosteric ternary complex model between [3H]NMS, gallamine, and CCh at human M2 mAChRS expressed in intact CHO cells
Estimates are the mean ± S.E.M. for six binding assays conducted in triplicate at 4°C for 3 h.
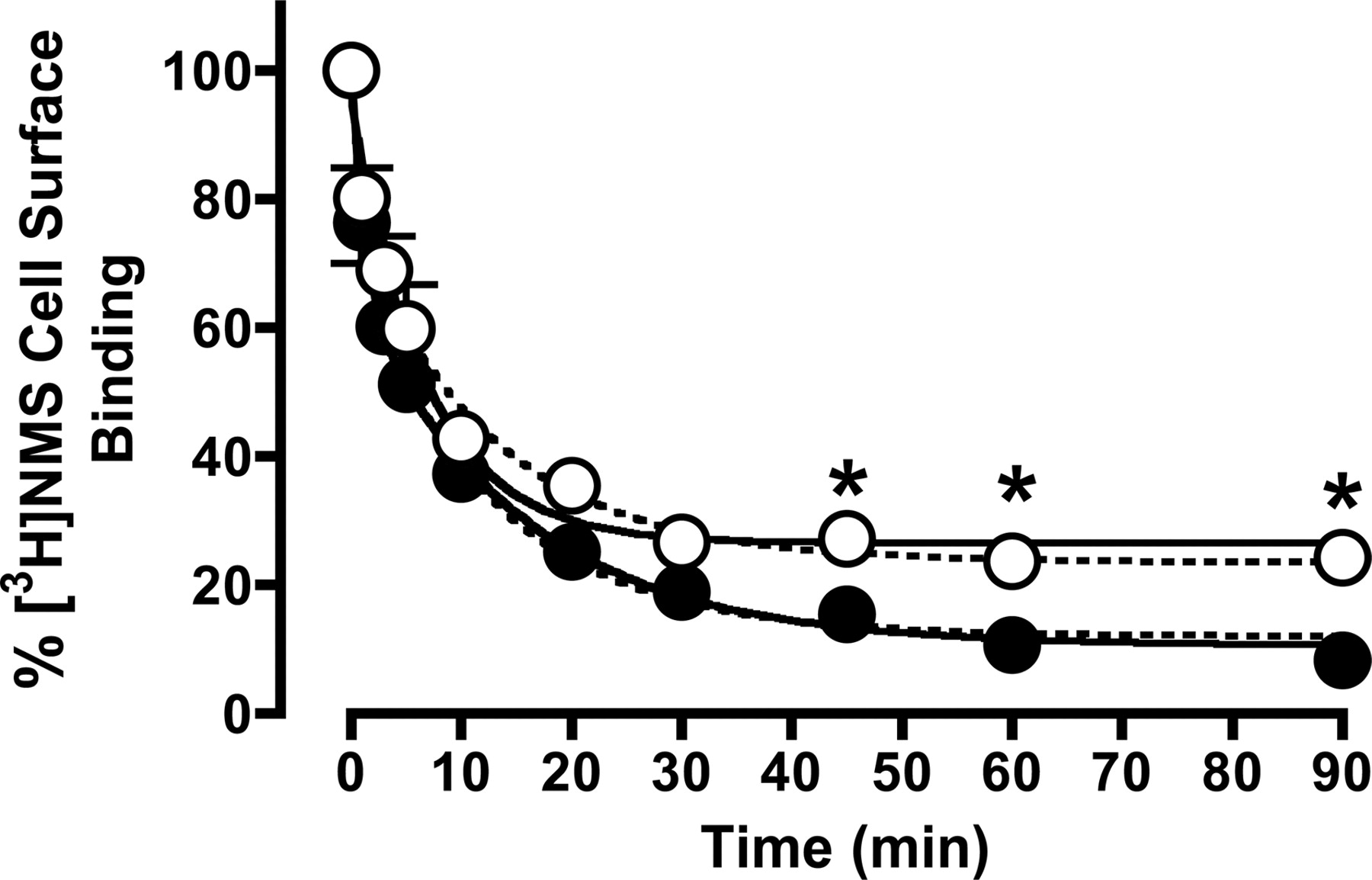
Based on the results shown in Table 4, 100 μM CCh was calculated to occupy approximately 80% of the M2 mAChRs, and this concentration was chosen for further receptor internalization studies. To measure the effect on internalization of the same level of receptor occupancy by CCh in the presence of gallamine, however, the concentration of CCh that was needed was calculated to be 250 μM when combined with 10 μM gallamine. Under these conditions, the modulator will occupy approximately 13% of the receptors, which is equivalent to the receptor occupancy of 1 μM gallamine in the absence of CCh. The results of these internalization experiments are shown in Fig. 6. In the presence of 100 μM CCh alone, internalization resulted in an approximately 90% loss of cell-surface [3H]NMS binding within 90 min and was characterized by two exponential phases (k1 = 0.82 ± 0.30 min-1; 34 ± 8%; k2 = 0.07 ± 0.01 min-1; 56 ± 7%; n = 4), as determined by F-test (F = 8.4; p < 0.001). Repeating these experiments using 1 mM CCh resulted in almost identical observations (data not shown), indicating that maximal internalization is attained at 100 μM CCh. In the presence of both CCh and gallamine, internalization seemed to be better described by a monophasic function (0.15 ± 0.02 min-1; n = 4), as determined by F-test (F = 2.9; p > 0.05); this finding would indicate a significant reduction in the extent of internalized receptors being observed at 45 min onward.
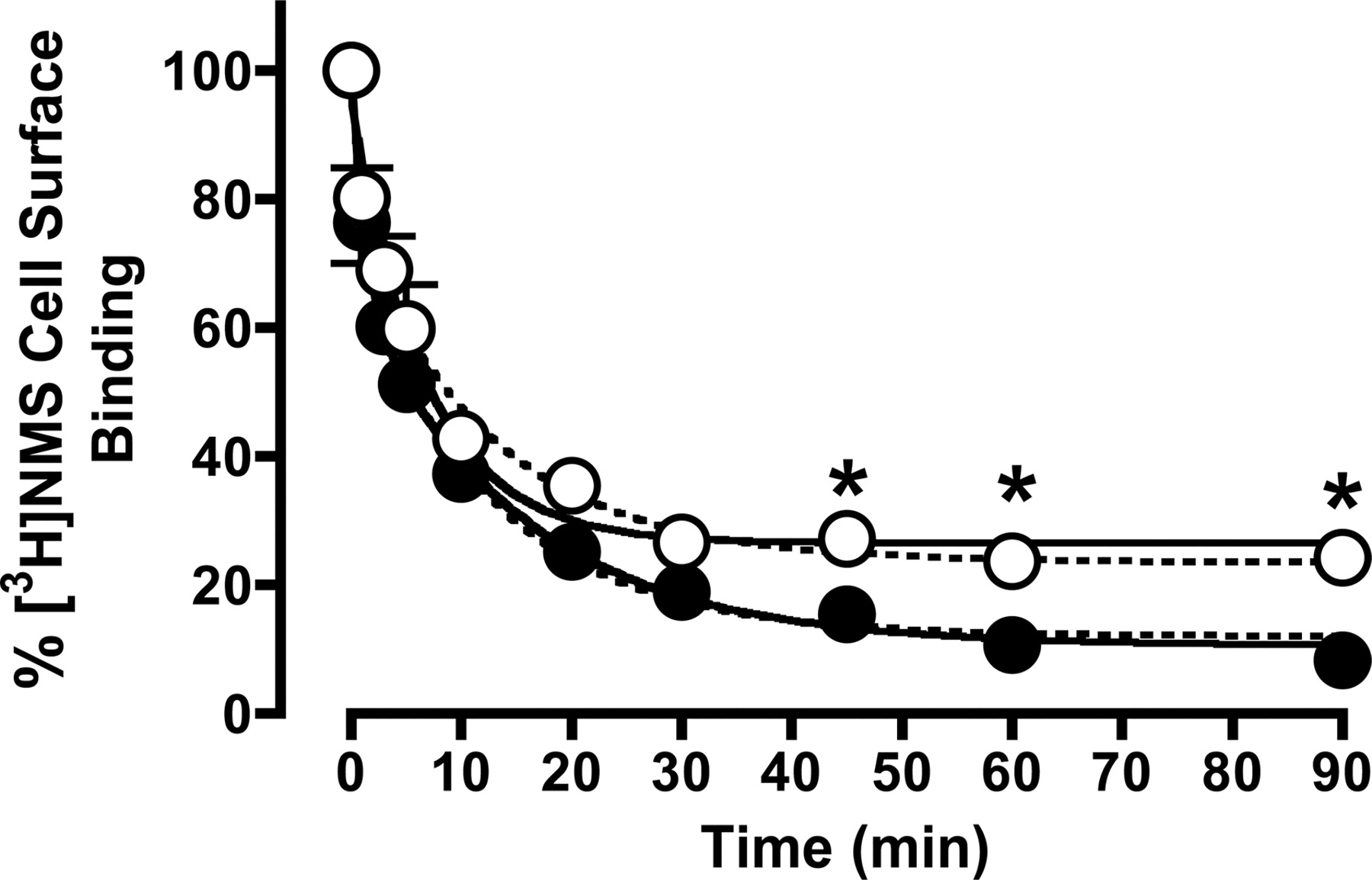
Time course of internalization of M2 mAChRs in CHO cells after exposure to 100 μM CCh (•) or 250 μM CCh + 10 μM gallamine (○) for various time intervals as shown, before extensive washing and assessment of surface receptor density with 2 nM [3H]NMS at 4°C for 3 h. The increased concentration of CCh in the presence of gallamine was chosen based on the estimated negative cooperativity in binding between the two ligands (Table 4), thus ensuring equivalent levels of receptor occupancy to those observed in the presence of 100 μM CCh alone. Solid curves represent the best fit of either a two-phase (CCh alone) or one-phase (CCh plus gallamine) exponential model, as determined by F-test. Dashed curves represent the best global fit of a two-phase exponential model to both data sets, with the rate constants for each phase shared between the data sets. Data are mean ± S.E.M. of four experiments, with incubations performed in duplicates. *, p < 0.05 for the difference between percentage of internalization between the two treatments.
It is also possible, however, that the two phases of internalization observed in the absence of gallamine are retained in the presence of the modulator, but that one or other of the amplitudes is reduced such that the F-test cannot statistically resolve a biphasic fit over a (simpler) monophasic fit. To explore this possibility, the internalization data were refitted with the two rate constants constrained to be shared between data sets, whereas the amplitudes (Span values) were allowed to vary; this represents a conservative hypothesis that assumes gallamine has no effect on the rate of CCh internalization. The results of this analysis are also shown in Fig. 6 (dashed lines) and were characterized by the following parameters: k1 = 0.83 ± 0.37 min-1; k2 = 0.07 ± 0.02 min-1; Span 1 (Control) = 33 ± 8%; Span 1 (+ Gallamine) = 16 ± 8%; Span 2 (Control) = 56 ± 7%; Span 2 (+ Gallamine) = 59 ± 7%. Thus, in contrast to the initial analysis, the data can also be adequately described by assuming that gallamine had no effect on the rate of CCh internalization, but rather reduced the amplitude of the fast phase (Span 1). A comparison of the two analytical methods by F-test found a statistical preference for the initial model (biphasic internalization converted to a monophasic internalization by gallamine), with F = 2.9 and p > 0.05, but it can be seen from visual inspection of Fig. 6 that the data do not allow for an unambiguous discrimination between the two models.
Discussion
The major finding of this study is that prolonged exposure to allosteric modulators can cause up-regulation of cell surface M2 mAChR expression and cellular responsiveness. These effects are independent of the well documented actions of acute modulator administration on orthosteric ligand binding affinity, since both allosteric antagonists (gallamine and C7/3-phth) and an enhancer (alcuronium) of NMS binding affinity have the same effect on cell surface receptor expression. Investigation of the potential mechanism underlying these effects, using gallamine as a prototypical example of an mAChR modulator, suggests that the modulators are able to promote a receptor conformation that possesses modified internalization properties.
The initial experiments described in this study directly assessed cell surface receptor expression using saturation binding with the hydrophilic antagonist, [3H]NMS. In agreement with previous reports (Pitcher et al., 1998; Tsuga et al., 1998; Bunemann et al., 1999), pretreatment of CHO M2 cells with the agonist CCh for 24 h significantly down-regulated the maximal number of cell-surface receptors. In contrast, pretreatment with the antagonists atropine or NMS, or the modulators gallamine, alcuronium, or C7/3-phth had the opposite effect, resulting in a significant up-regulation of M2 mAChRs after 24 h. Interestingly, combination of NMS and alcuronium, which are known to allosterically enhance each other's binding to the M2 mAChR, did not produce any greater effect than either compound alone; this may reflect a “ceiling” to the up-regulation that can be achieved at high degrees of receptor occupancy. Since both atropine and NMS have previously been shown to possess inverse agonist activity at M2 mAChRs (Jakubík et al., 1995), the effects of these ligands in our study can most readily be reconciled with the fact that inverse agonists are known to cause GPCR up-regulation, presumably by stabilizing a conformational state of the receptor that is less prone to down-regulation (Gether et al., 1997; Milligan and Bond, 1997). The effect of the allosteric modulators, however, is novel. By their very nature, these ligands bind to a common site on the M2 mAChR that is topographically distinct from that recognized by orthosteric ligands and engender a conformation that is negatively cooperative toward the binding of the endogenous agonist acetylcholine (Lazareno and Birdsall, 1995; Christopoulos 2000). However, gallamine and alcuronium may also show positive cooperativity toward the binding of G proteins under certain experimental conditions (Jakubík et al., 1996, 1998), although we have not observed any evidence for such an effect in our studies (Fig. 3A). Our current findings now also suggest that these modulator-induced conformations can also be “inverse agonist-like” with respect to their effects on regulation of receptor number. Indeed, Zahn et al. (2002) have previously demonstrated inverse agonism for alcuronium in an assay of M2 mAChR-mediated guanosine 5′-O-(3-thio)triphosphate turnover. Overall, our observations provide further support to the concept that efficacy at GPCRs encompasses a broader spectrum of receptor behaviors than the classic activation-deactivation paradigm (Kenakin, 2002).
To our knowledge, only one other study has investigated the potential for long-term effects of allosteric modulators on family A GPCRs. Bhattacharya and Linden (1996) found that 24-h pretreatment of A1 adenosine receptors with the allosteric enhancer PD 81,723 had no effect on the maximal density of binding sites recognized by either agonist or antagonist radioligands in membrane binding assays. However, they did note a small down-regulation of antagonist-labeled sites in intact cells, which was more pronounced when an agonist radioligand was used. These findings were reconciled with the fact that PD 81,723 seems to have a small propensity to mediate receptor-G protein coupling in its own right, and thus produce “agonist-like” effects on receptor regulation. Since we have found no evidence for acute effects of these allosteric modulators on signaling under our experimental conditions, we have no reason to expect a similar effect to that observed by Bhattacharya and Linden (1996) at the A1 receptor.
The cytosensor microphysiometer was used in our study to measure integrated whole-cell functional responses as an index of receptor activation, both acutely and after prolonged ligand exposure. As expected, CCh caused an increase in ECAR, although this was characterized by a decline at the highest concentrations of agonist used, presumably due to acute desensitization. Interestingly, atropine was also able to mediate a response, but opposite to that observed with CCh. This observation may be indicative of atropine behaving as an inverse agonist, in agreement with previous studies (Jakubík et al., 1995; Zahn et al., 2002). However, the potency of atropine as an inverse agonist in the cytosenor experiments was approx. 25-fold weaker than its Log KB value shown in Table 1. This is at odds with the classic expectation that an inverse agonist's potency should approximate its receptor affinity. It is possible that the reduced potency of atropine as an inverse agonist in our hands reflects nonlinear stimulus-response coupling in the CHO cells, which would be expected to increase agonist potency while decreasing inverse agonist potency.
The cytosensor experiments also addressed the consequences of prolonged cellular exposure to different mAChR ligands on the subsequent ability of the receptor to signal. Pretreatment with CCh resulted in a marked attenuation of receptor signaling, consistent with the findings of the saturation assays and indicative of receptor down-regulation. In contrast, pretreatment of the cells with gallamine and atropine caused a significant enhancement in the maximal agonist-mediated response. These experiments thus provided functional evidence for an up-regulation of M2 mAChRs in response to atropine, an inverse agonist, and gallamine, an allosteric modulator.
Collectively, the effects of gallamine, C7/3-phth, or alcuronium pretreatment on saturation binding, and gallamine pretreatment on cellular function, suggested a role for allosteric modulators in regulating receptor trafficking. The final series of experiments in our study were performed to gain insight into potential mechanisms mediating the phenomenon. The time for achieving the maximal effect of CCh on receptor expression was much faster than the effects of either atropine or gallamine, which required at least 12 to 24 h (Fig. 5). This may suggest that the maximal increase in receptor cell surface expression in the presence of atropine or gallamine reflects a slowing of agonist-independent receptor cycling such that a new steady-state cell surface expression is achieved, with/without additional de novo protein synthesis.
In addition to studying the effects of gallamine alone on receptor regulation, the ability to monitor CCh-mediated internalization in the concomitant presence of gallamine was a particularly advantageous experimental manipulation. This was possible due to the lack of mutual exclusivity in binding between orthosteric and allosteric sites. However, because internalization is occupancy-driven, it was first necessary to account for the negative allosteric effect that gallamine and CCh exert on each other's binding affinities to ensure that the occupancy of the orthosteric site by the agonist remained the same in internalization assays conducted in the presence of gallamine to those conducted in its absence. The relevant binding parameters for determining these occupancy relationships were obtained from separate inhibition binding experiments. At equilibrium, 100 μM CCh was calculated to occupy approximately 80% of the receptors; in the presence of 10 μM gallamine, the concentration of CCh thus needed to be increased to 250 μM for equivalent occupancy. Similarly, 10 μM gallamine in presence of 100 μM CCh gave the same occupancy (13%) as that calculated for 1 μM gallamine alone. However, there are two caveats to this approach. First, these latter binding experiments were performed at 4°C, whereas the internalization was performed at 37°C. This is an unavoidable consequence of radioligand binding assays using intact CHO cells, since the receptor will internalize to agonist during the time course of the assay if it were performed at 37°C. Second, it would have been desirable to have attained a higher degree of receptor occupancy by the modulator in the internalization experiments, but the high negative cooperativity between gallamine and CCh made this impracticable due to the high concentrations of CCh that would have been required to maintain 80% receptor occupancy under such conditions. Nevertheless, the results from our internalization experiments are consistent with previous observations that M2 mAChRs display a biphasic mode of loss of cell surface receptors in response to agonist. Koenig and Edwardson (1996) have demonstrated that this biphasic response reflects the composite effects of receptor internalization and recycling. Our analysis suggests that the ability of gallamine to up-regulate cell surface receptor expression possibly involved a loss of the initial fast component of agonist-driven internalization, although we are currently unable to conclude whether this is also accompanied by a significant change in the internalization rate.
In conclusion, our results suggest that allosteric modulators change the trafficking properties of M2 mAChRs by a mechanism that may involve a modification in receptor internalization properties. These changes in receptor trafficking can be manifested at both the level of ligand binding as well as the level of cellular responsiveness. Since allosteric modulators of GPCRs are now being targeted as novel therapeutic entities (Christopoulos, 2002), the present study has provided important insight into some of the consequences of prolonged exposure to these types of agents, as would be expected, for example, in a chronic dosing regimen.
Footnotes
-
This work was supported by Project Grant 251538 of the National Health and Medical Research Council of Australia. A.C. and P.S. are Senior Research Fellows of the National Health and Medical Research Council of Australia. L.M. is a recipient of a Melbourne Research Scholarship.
-
doi:10.1124/jpet.104.073767.
-
ABBREVIATIONS: GPCR, G protein-coupled receptor; mAChR, muscarinic acetylcholine receptor; DMEM, Dulbecco's modified Eagle's medium; C7/3-phth, heptane-1,7-bis (dimethyl-3′-phthalimidopropyl) ammonium bromide; CHO, Chinese hamster ovary; CCh, carbachol; NMS, N-methylscopolamine; ANOVA, analysis of variance; ECAR, extracellular acidification rate; PD 81,723, (2-amino-4,5-dimethyl-3-thienyl)-[3(trifluoromethyl)-phenyl]methanone.
- Received July 5, 2004.
- Accepted August 26, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}