


Abstract
We found that 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide (CDPPB) is a potent and selective positive allosteric modulator of the metabotropic glutamate receptor subtype 5 (mGluR5). In Chinese hamster ovary cells expressing human mGluR5, CDPPB potentiated threshold responses to glutamate in fluorometric Ca2+ assays more than 7-fold with an EC50 value of approximately 27 nM. At 1 μM, CDPPB shifted mGluR5 agonist concentration response curves to glutamate, quisqualate, and (R,S)-3,5-dihydroxyphenylglycine 3- to 9-fold to the left. At higher concentrations, CDPPB exhibited agonist-like activity on cells expressing mGluR5. No other activity was observed on any other mGluR or cell type at concentrations up to 10 μM. CDPPB had no effect on [3H]quisqualate binding to mGluR5 but did compete for binding of [3H]methoxyPEPy, an analog of the selective mGluR5 negative allosteric modulator MPEP. CDPPB was found to be brain penetrant and reversed amphetamine-induced locomotor activity and amphetamine-induced deficits in prepulse inhibition in rats, two models sensitive to antipsychotic drug treatment. These results demonstrate that positive allosteric modulation of mGluR5 produces behavioral effects, suggesting that such modulation serves as a viable approach to increasing mGluR5 activity in vivo. These effects are consistent with the hypothesis that allosteric potentiation of mGluR5 may provide a novel approach for development of antipsychotic agents.
Glutamate, the predominant excitatory neurotransmitter in the mammalian central nervous system (CNS), exerts its effects through two classes of receptors. The first class of receptor, ionotropic glutamate receptors, are postsynaptic, multimeric ligand-gated ion channels classified into three groups named for group-selective agonists: N-methyl-d-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazopropionic acid, and kainate receptors. The NMDA receptor (NMDAR) is known to play an important role in processes related to schizophrenia. NMDAR antagonists, such as phencyclidine and ketamine, induce positive, negative, and cognitive symptoms reminiscent of schizophrenia in human volunteers and worsen existing symptoms in schizophrenic patients. This observation has led to the hypothesis that changes in CNS circuits induced by NMDAR hypofunction may play a key role in the development and/or in the underlying symptoms of schizophrenia (Olney et al., 1999). Therefore, development of compounds that selectively increase NMDAR function could be used to test this hypothesis.
The metabotropic glutamate receptors (mGluRs), the second class of glutamate receptor, are members of family C of the G-protein-coupled receptors and are characterized by a large extracellular agonist binding domain on the amino-terminal end of the receptor that is distinct from the seven-transmembrane domain characteristic of all G-protein-coupled receptors. Eight mGluR subtypes have been identified and cloned, and these have been classified into three groups based on sequence similarity, effector coupling, and pharmacology. The group I mGluRs (mGluR1 and 5) are mainly located postsynaptically and are coupled to Gαq and its associated effectors, including activation of phospholipase C, increases in phosphoinositide hydrolysis, and increases in intracellular calcium levels. Activation of group I mGluRs often results in an increase in neuronal excitability. The groups II (mGluR2 and 3) and III (mGluR4, 6, 7, and 8) mGluRs are located mainly presynaptically and are coupled to Gαi/o and its associated effectors, including decreases in cAMP levels. Activation of groups II and III mGluRs results in a decrease in neurotransmitter release. In native systems, groups II and III mGluRs can be distinguished from each other pharmacologically (for review, see Conn and Pin, 1997).
We have been interested in the function of mGluR5 because numerous reports indicate that this receptor may also play a role in schizophrenia, as well as in Parkinson's disease, anxiety, and cocaine addiction (Spooren et al., 2000; Chiamulera et al., 2001; Tatarczynska et al., 2001; Chavez-Noriega et al., 2002; Moghaddam, 2004). We have recently reported that the novel mGluR5 positive allosteric modulator N-{4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl] phenyl}-2-hydroxybenzamide (CPPHA) potentiates responses to mGluR5 agonists in cell lines and increases potentiation of NMDA receptor currents induced by subthreshold concentrations of an mGluR5 agonist (O'Brien et al., 2004). However, CPPHA does not have the properties needed for use in behavioral and other in vivo studies. We here report discovery of a novel allosteric potentiator of mGluR5, 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide (CDPPB). This compound is a selective positive allosteric modulator of mGluR5 from a new structural series and has pharmacokinetic properties and brain penetration that makes it suitable for in vivo studies. Interestingly, this compound is active in rodent locomotor and prepulse inhibition models, providing the first in vivo evidence that positive allosteric modulation of mGluR5 has behavioral effects in two models that have been used to predict antipsychotic efficacy.
Materials and Methods
Structure and Synthesis of CDPPB. The structure of CDPPB is shown in Fig. 1. The synthetic route for CDPPB is detailed by Lindsley et al. (2004).

Structure of CDPPB.
Stable Cell Lines. Methods describing the development of the mGluR-expressing stable cell lines used in these studies have been detailed previously (O'Brien et al., 2004). CHO cells stably expressing human and rat mGluR5 receptors are referred to as human mGluR5 CHO cells and rat mGluR5 CHO cells, respectively.
Fluorometric Imaging Plate Reader (FLIPR). Methods used for Ca2+ flux measurements using the FLIPR384 fluorometric imaging plate reader (Molecular Devices, Sunnyvale, CA) have been detailed previously (O'Brien et al., 2003). The peak of the calcium response was used to construct agonist concentration-response curves.
Radioligand Binding Assays. Methods used for binding studies using the agonist binding site radioligand [3H]quisqualate and the 2-methyl-6-(phenylethynyl)-pyridine (MPEP) analog radioligand [3H]3-methoxy-5-(2-pyridinylethynyl)pyridine have been detailed previously (O'Brien et al., 2003).
Mathematical Modeling. Data were compared with the model of Hall (2000) using Prism 4.0 (GraphPad Software Inc., San Diego, CA), as described previously (O'Brien et al., 2004).
Pharmacokinetics Studies. Rats were housed and tested in an Association for Assessment and Accreditation of Laboratory Animal Care-accredited facility in strict compliance with all applicable regulations. CDPPB was administered to male Sprague-Dawley rats (Taconic Farms, Germantown, NY) through a cannula implanted in the jugular vein (2 mg/kg i.v. in dimethyl sulfoxide) or by oral gavage (10 mg/kg in 1% methylcellulose). At several time points (range from 0.02–24 h), blood samples (400 μl) were withdrawn through a cannula implanted in the carotid artery and were then placed in EDTA tubes on ice. Plasma was separated from the red cells by centrifugation, removed from the red cells, and frozen at –70°C until assayed. An aliquot of each plasma sample was treated with acetonitrile containing an internal standard to precipitate the protein. Treated samples were centrifuged for 15 min at 13,500g. The supernatant was removed and dried, then reconstituted in 10% methanol/water containing 0.1% formic acid. Reconstituted samples were separated chromatographically using an Agilent 1100 series high-performance liquid chromatography (Agilent Technologies, Palo Alto, CA) Samples were injected onto a reverse-phase analytical column (Prodigy 5μ ODS3 100Å, 50 × 2.00 mm; Phenomenex, Torrance, CA). Mobile phase A consisted of 10% methanol/water containing 0.1% formic acid, and mobile phase B was 100% acetonitrile. Samples were eluted with a ballistic gradient ramping from 90% A/10% B to 10% A/90% B in 18 sec. Samples were detected by an in-line API 2000 triple quadrupole mass spectrometer with turbo ion spray used in the positive ionization mode (Applied Biosystems/MDS Sciex, Foster City, CA). Quantitation of CDPPB in the plasma samples was by comparison with a standard curve using ANALYST software (version 1.3.1; Applied Biosystems/MDS Sciex). Pharmacokinetic parameters were calculated using Watson software (Innaphase, Philadelphia, PA).
P-Glycoprotein (P-gp) Efflux Studies. The P-gp efflux studies were carried-out with LL-PK1 cells used as a control cell line and transfected with MDR1 (human) and mdr1a (mouse) P-gp. The in vivo brain penetration studies were carried out using mdr1a +/+ and –/– CF-1 mice. Methods for both the in vitro and in vivo approaches have been previously reported (Yamazaki et al., 2001). The quantitative analysis was performed using an API-4000 triple quadrupole mass spectrometer (Applied Biosystems/MDS Sciex) operated in the positive ion turbo-ion spray mode. The transition monitored for CDPPB was 365.2 to 129.9
Metabolite Profiling. CDPPB (100 μM) was incubated (37°C, 120 min) with rat, dog, monkey, and human hepatic microsomes (2 mg/ml microsomal protein) in 100 mM KPO4 buffer fortified with 1 mM of both NADPH and UDPGA. The incubates were quenched with 1.5 ml of acetonitrile, centrifuged (10 min at 4000 rpm), and the supernatant was collected and evaporated to dryness, then reconstituted in 200 μl of 25% acetonitrile. The liquid chromatography-mass spectrometry used a 3- × 50-mm 5-μm BetaBasic C18 column (Analytical Sales and Service, Pompton Plains, NJ) with a flow rate of 1.5 ml/min split 5:1 such that 300 μl entered the mass spectrometer ion source. The solvents used were A (5% acetonitrile and 0.1% formic acid) and B (95% acetonitrile and 0.1% formic acid), and the chromatographic conditions were to hold for the 1st min at 95% A, followed by a linear gradient to 5% A over 5 min that was held for 1 min followed by a return to 95% A in 0.5 min held for 1.5 min. A TSQ Quantum triple quadrupole mass spectrometer (Thermo Finnigan, San Jose, CA) operated in the positive ion electrospray mode and using full-scan mass spectrometry and full-scan tandem mass spectrometry analyses was used for the metabolite profiling experiments.
Behavioral Testing.Subjects. For all behavioral studies, male Sprague-Dawley rats (Taconic Farms) were used. Rats were group housed on a 12-h light/dark cycle (lights on at 6:00 AM) and allowed food and water ad libitum. Rats were housed and tested in an Association for Assessment and Accreditation of Laboratory Animal Care-accredited facility in strict compliance with all applicable regulations.
Locomotor activity. Activity was assessed as mean distance traveled (in centimeters) using standard 16 × 16 photocell open field testing chambers (MED Associates, St. Albans, VT). Rats were habituated to the testing chambers for 60 min prior to the s.c. administration of d-amphetamine sulfate (1 mg/kg base). Rats were administered vehicle (60:40, dimethyl sulfoxide:polyethylene glycol 400) or CDPPB (1–10 mg/kg s.c.) 20 min prior to amphetamine treatment. Activity was recorded throughout the habituation period and for an additional 2 h postamphetamine administration. Data are expressed as the mean ± S.E.M. distance traveled recorded in 5-min intervals over the test period. The data were analyzed using repeated measures analysis of variance followed by post hoc testing using the Dunnett procedure. A difference was considered significant when p ≤ 0.05.
Prepulse inhibition (PPI). Sensorimotor gating was assessed by evaluation of prepulse inhibition of the acoustic startle response as previously described (Kinney et al., 2003). Briefly, all rats received amphetamine (2 mg/kg base s.c.) followed 20 min later by either vehicle or CDPPB (3–30 mg/kg s.c.). Testing began 10 min following vehicle/CDPPB treatment. In each test session, rats were given a 5-min acclimation period during which a 65-db background noise was continuously present. This background noise remained present throughout the entire testing session. Following the acclimation period, the test session began, which consisted of 10 repetitions of trials. Six trial types were presented during the session. These consisted of a 10-ms prepulse at 70, 75, 80, or 85 db (i.e., 5–20 db above background noise) followed 100 ms later by a 118- to 120-db 40-ms startle pulse (prepulse pulse conditions), the startle pulse alone (pulse alone), and a period where no stimulus was presented. Previous studies in our laboratory had determined that these prepulse intensities were insufficient to induce any change in startle amplitude alone. The stimuli were presented in random order with interstimulus intervals averaging 15 sec. Startle amplitude was measured as the mean value during a 100-ms period beginning at the onset of the startle-eliciting stimulus. Levels of prepulse inhibition were determined by the formula 100 – [(prepulse pulse/pulse alone) × 100] and expressed as the percentage of prepulse inhibition ± S.E.M. Data were analyzed using repeated measures analysis of variance with the prepulse intensity used as the within factor, followed by analysis of simple main effects and post hoc analysis using the Hochberg procedure for multiple pair-wise comparisons (Hochberg, 1988). An effect was considered statistically significant when p ≤ 0.05.
Results
FLIPR Assays. In FLIPR384 assays, CHO cells expressing the cloned human mGluR5 show concentration-dependent increases in Fluo-4 fluorescence in response to glutamate, quisqualate, and (R,S)-3,5-dihydroxyphenylglycine (3,5-DHPG). These compounds seemed to act as full agonists, with potencies consistent with previously reported EC50 values (O'Brien et al., 2003, 2004). We have used this assay to screen for compounds that increase the response of mGluR5 to small amounts of glutamate (300 nM) but that do not elicit a response by themselves. Selective allosteric modulators of mGluR5 from two structural classes (3,3′-difluorobenzaldazine and CPPHA) have previously been identified using this method (O'Brien et al., 2003, 2004), and we now report that CDPPB, a member of a third structural class (Fig. 1), was also found to be active as an mGluR5 potentiator.
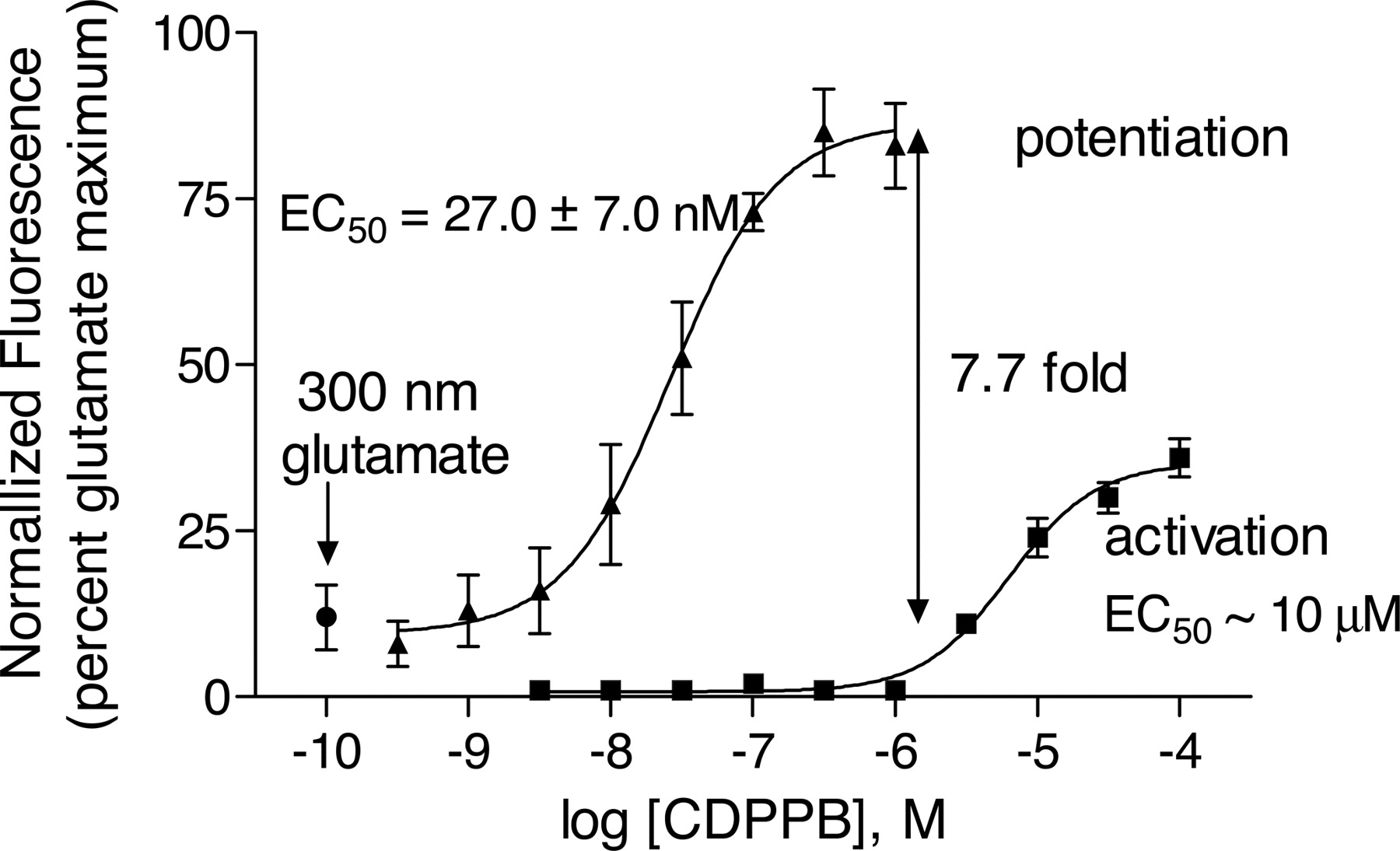
CDPPB caused a concentration-dependent increase in the response of both human and rat mGluR5 CHO cells to agonist in this assay. For example, the maximal potentiation of the response of human mGluR5 CHO cells to 300 nM glutamate by CDPPB was approximately 7.7-fold with an EC50 value for potentiation of 27.0 ± 7.0 nM (n = 3) (Fig. 2). Potencies of potentiation were similar for all agonists (glutamate, quisqualate, and 3,5-DHPG) as well as for both species (Table 1). In the absence of added agonist, CDPPB also caused an increase in the Fluo-4 fluorescence of mGluR5 CHO cells with an EC50 value of approximately 10 μM (Fig. 2). This agonist-like activity was not observed in other cells or in the parent cell lines at CDPPB concentrations up to 10 μM, well above the highest achievable brain concentrations estimated from the measured rat plasma and brain levels and pharmacokinetic parameters reported below. Further testing of CDPPB against a panel of 175 receptors, transporters, ion channels, and enzymes (MDS Panlabs, Bothell, WA) indicated no submicromolar activities.

CDPPB potentiates activation of mGluR5 by glutamate. CDPPB has partial agonist-like activity at concentrations above 1 μM. Human and rat mGluR5 CHO cells were plated in clear-bottomed 384-well plates in glutamate/glutamine-free medium, loaded the next day with the calcium-sensitive fluorescent dye Fluo-4, and placed in FLIPR384. A range of concentrations of CDPPB was added to cells (human mGluR5 CHO cells in this figure) after 10 s of baseline determination. Five minutes later, a fixed concentration (∼EC10 concentration) of agonist (glutamate in this figure) was added, and the Ca2+ response was measured by FLIPR384. For measurement of agonist-like activity, the FLIPR384 data were collected during the 5 min after the addition of CDPPB, before agonist was added to the plate. Concentration-response curves were generated from mean data of three experiments. Error bars are S.E.M. -Fold potentiation was calculated from the maxima and minima determined by nonlinear curve fitting of the meaned data. Results for glutamate, 3,5-DHPG, and quisqualate on both human and rat mGluR5 CHO cells are summarized in Table 1.
CDPPB potentiation of human and rat mGluR5 activation by glutamate, 3,5-DHPG, and quisqualate
Reversibility of CDPPB was determined by comparing the FLIPR response of mGluR5 CHO cells with 300 nM glutamate in the presence of a range of concentrations of CDPPB, with and without a wash-out step. The response to CDPPB was virtually abolished after washing, indicating CDPPB was reversible (data not shown).
Increasing concentrations of CDPPB caused a parallel, leftward shift of mGluR5 CHO cell glutamate concentration-response curves with no increase in maximal response. In the presence of 1 μM CDPPB, the EC50 for glutamate decreased 9.1-fold (Fig. 3). At this concentration of CDPPB, the basal fluorescence response of the mGluR5 CHO cells was increased compared with vehicle-treated cells. Similar shifts in agonist concentration responses were observed for quisqualate and 3,5-DHPG in both human and rat mGluR5 CHO cells, again with no increase in the maximal response to the agonists (Table 2). Note that the basal (no agonist) response at 1 μM CDPPB shown in Fig. 3 was greater than that observed in Fig. 2. The responses in Fig. 2 were measured immediately after addition of CDPPB, whereas the responses in Fig. 3 were measured 5 min later, upon addition of vehicle or agonist. Additional studies were performed to study this interesting, time-dependent property of the CDPPB interaction with mGluR5 CHO cells further. The basal response was completely inhibited by the negative allosteric modulator MPEP (100 nM; Fig. 4A), whereas the presence of the competitive (orthosteric) antagonist LY341495 [(2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid] (30 μM) had no effect on this response (Fig. 4B), suggesting the effect was not related to glutamate. Both LY341495 and MPEP exerted their expected respective effects at higher levels of added glutamate (Fig. 4, A and B).

Potentiation of mGluR5 activity by CDPPB is manifested as an increased sensitivity to agonist. Apparent agonist-like activity is observed at high concentrations, raising basal response. Human and rat mGluR5 CHO cells were plated in clear-bottomed 384-well plates in glutamate/glutamine-free medium, loaded the next day with the calcium-sensitive fluorescent dye Fluo-4, and placed in FLIPR384. Several fixed concentrations of CDPPB were added to cells (human mGluR5 CHO cells in this figure) after 10 s of baseline determination. Five minutes later, a range of concentrations of agonist (glutamate in this figure) was added. The basal activity of the glutamate concentration-response curves seems to increase at the highest concentration of CDPPB. Concentration-response curves were generated from mean data of three experiments. Error bars are S.E.M. Results for glutamate, 3,5-DHPG, and quisqualate on both human and rat mGluR5 CHO cells are summarized in Table 2, calculated from the EC50 values determined by nonlinear curve fitting of the meaned data.
Effect of CDPPB on FLIPR agonist concentration-response curves: human and rat mGluR5

The increased basal response caused by 1 μM CDPPB is inhibited by the negative allosteric modulator MPEP but not by the competitive antagonist LY341495. Conditions were the same as described for Fig. 3. A, vehicle, 1 μM CD-PPB, 100 nM MPEP, or 1 μM CDPPB plus 100 nM MPEP were added to the wells and mixed. Five minutes later, a range of concentrations of glutamate was added. B, vehicle, 1 μM CDPPB, 30 μM LY341495, or 1 μM CDPPB plus 30 μM LY341495 were added to the wells and mixed. Five minutes later, a range of concentrations of glutamate was added. Representative experiments are shown.
Radioligand Binding. CDPPB (up to 100 μM) had no effect on the binding of [3H]quisqualate (25 nM) to membranes prepared from CHO mGluR5 cells (Fig. 5), suggesting that this compound did not bind at the agonist binding site and did not increase the affinity of agonist binding. Nondisplaceable [3H]quisqualate binding was estimated in the presence of 1 mM glutamate. CDPPB did inhibit binding of the MPEP analog, 2 nM methoxy-PEPy (Cosford et al., 2003), to these membranes (Fig. 6). The binding of [3H]methoxyPEPy to these membranes was not affected by the presence or absence of glutamate [Kd values: 3.4 ± 0.5 nM (n = 3) in absence of glutamate, 3.3 ± 0.6 nM (n = 3) in presence of 600 nM glutamate]. The inhibition of [3H]methoxyPEPy binding by CDPPB was not affected by the presence or absence of glutamate [Ki values: 2.6 ± 0.5 μM (n = 3) in absence of glutamate, 1.7 ± 0.5 μM (n = 3) in presence of 600 nM glutamate], suggesting CDPPB binding affinity is not affected by agonist.

CDPPB has no effect on [3H]quisqualate binding to human mGluR5 CHO cell membranes. The assay conditions were the following. Membranes prepared from human mGluR5 CHO cells were incubated with [3H]quisqualate (25 nM final concentration in 20 mM HEPES, 2 mM CaCl2, and MgCl2, pH 7.2) in the presence of 100 μM CDPPB or vehicle for 60 min at room temperature. Samples were filtered onto glass fiber filters. Nondisplaceable binding (NSB) was estimated with the use of 1 mM glutamate. A representative experiment is shown. Error bars represent S.D.

CDPPB inhibits [3H]methoxyPEPy binding to human mGluR5 CHO cell membranes. Membranes prepared from human mGluR5 CHO cells were incubated with the radiolabeled MPEP analog [3H]3-methoxy-5-(2-pyridinylethynyl) pyridine (1 or 2 nM final concentration in 50 mM Tris and 0.9% NaCl, pH 7.4) for 60 min at room temperature in the presence of varying concentrations of CDPPB or MPEP. Samples were then filtered onto glass fiber filters. Nondisplaceable binding was estimated with 1 μM MPEP. A representative competition experiment is shown. Error bars are S.D.
Pharmacokinetic Studies. Pharmacokinetic studies in Sprague-Dawley rats indicated that after i.v. administration, CDPPB (2 mg/kg in dimethyl sulfoxide) had a 4.4-h plasma half-life with volume of distribution of 3.5 l/kg and clearance of 25.1 ml/min/kg. CDPPB had a low oral availability in rats (F = 2.9% after administration at 10 mg/kg in 1% methylcellulose). In vitro P-gp efflux studies indicated that CDPPB had high passive permeability (24.7 × 10–6 cm/s) and was not a P-gp substrate (B-A/A-B ratios were 0.6 for human and 0.8 for mouse). In vivo studies carried out with mdr1a +/+ and –/– CF-1 mice confirmed the lack of P-gp interaction in vivo and indicated that CDPPB was brain penetrant with a normalized brain to plasma AUC ratio of 1.7. Metabolite profiling in rat, dog, monkey, and human hepatic microsomes indicated that the major metabolic pathways for CDPPB were oxidation and glucuronidation of the diphenylpyrazole portion of the molecule.
Behavioral Studies. Further studies evaluated the efficacy of systemic CDPPB administration in two preclinical behavioral models sensitive to antipsychotic drug treatment. Specifically, we evaluated the ability of CDPPB to ameliorate amphetamine-induced locomotor activity and amphetamine-induced disruption of PPI in Sprague-Dawley rats. As shown in Fig. 7, pretreatment of rats with CDPPB 20 min prior to amphetamine administration significantly suppressed amphetamine-induced locomotor activity, without influencing spontaneous locomotor activity. Thus, when preamphetamine data were evaluated, no significant effects of treatment (p > 0.87) or treatment × time interaction (p > 0.93) were noted. Following amphetamine, however, a significant dose × time interaction was found [F(69,759) = 1.74, p < 0.001]. This significant interaction reflected a more efficacious effect of CDPPB at earlier time points (see results of post hoc analysis in Fig. 7). At the end of these studies, plasma samples and brains were analyzed for CDPPB levels. Plasma levels were 630 to 690 nM, and brain levels were 110 to 130 nM 120 min after 10 and 30 mg/kg s.c. administration. At the highest dose achievable (60 mg/kg s.c.), CDPPB levels were 980 nM in plasma and 270 nM in brain.

Effect of vehicle or CDPPB (3–30 mg/kg s.c.) on amphetamine (1 mg/kg s.c. base)-induced locomotor activity in rats. All rats were habituated to the testing chambers a minimum of 60 min before amphetamine administration. Vehicle (VEH) or CDPPB was administered 20 min prior to amphetamine injection, which occurred at time 0. Data are expressed, and the mean ± S.E.M. (error bars) distance traveled was recorded in 5-min intervals over the test period. Asterisks represent a significant difference from vehicle-treated animals; *, p < 0.05.
In a second assessment of behavioral efficacy, we examined the activity of CDPPB on amphetamine-induced disruption of PPI in rats. Consistent with previous studies, amphetamine (2 mg/kg s.c.) significantly disrupts PPI in these rats at all prepulse intensities tested (data not shown). As shown in Fig. 8A, CDPPB dose dependently reversed these amphetamine-induced deficits. This effect was confirmed by the finding of a significant treatment × prepulse intensity interaction [F(9,132) = 2.2, p < 0.027]. Post hoc analyses suggested that both 10 and 30 mg/kg had significant activity at prepulse intensities 10 to 20 dB above the background noise (see Fig. 8A for results of post hoc testing). It is notable that CDPPB had no effect on the response of these rats to the no-stimulus condition (p > 0.05) suggestive of a lack of effect on basal startle reactivity (Fig. 8B). A significant main effect of treatment was found in the pulse-alone condition [F(3,44) = 3.97, p < 0.02]; however, post hoc testing demonstrated that no active treatment group significantly differed from the vehicle-treated animals. In a companion paper, Lindsley et al. (2004) have replicated these results in a second study performed under the same conditions.

A, effect of CDPPB on amphetamine-induced deficits of prepulse inhibition in male Sprague-Dawley rats. All rats received amphetamine (2 mg/kg base s.c.) followed 20 min later by either vehicle or CDPPB (3–30 mg/kg s.c.). Testing began 10 min following vehicle/CDPPB treatment. Data are expressed as the mean ± S.E.M. (error bars) percentage of prepulse inhibition. Asterisks represent a significant difference from vehicle-treated animals; *, p < 0.05; **, p < 0.01. B, effect of CDPPB on startle amplitude during the no-stimulus and pulse-alone conditions. Animals were treated as described above. No significant differences were found relative to vehicle-treated animals; p > 0.05.
Discussion
We have been interested in developing compounds that selectively increase NMDAR function to test the hypothesis that NMDAR hypofunction plays a key role in the development and/or in the underlying symptoms of schizophrenia. Previous work has suggested that the metabotropic glutamate receptor mGluR5 is functionally linked to NMDAR activation (Alagarsamy et al., 1999; Awad et al., 2000; Salt and Binns, 2000). Mannaioni et al. (2001) used the group I-selective mGluR agonist DHPG to demonstrate that activation of mGluR5 increases NMDA receptor currents in rat hippocampal CA-1 pyramidal cells. Kinney et al. (2003) used the mGluR5-selective agonist (R,S)-2-chloro-5-hydroxyphenylglycine to show that mGluR5 plays a modulatory role in rodent PPI. Furthermore, several groups have shown that mGluR5 knock out mice demonstrate impairments in PPI (Kinney et al., 2003; Brody et al., 2004). These findings suggest that activation of mGluR5 may represent a rational approach to antipsychotic drug development. However, development of an agonist for chronic therapy must overcome several problems such as lack of subtype specificity, risk of toxicity from direct receptor activation and lack of receptor responses to normal increases and decreases of endogenous agonist levels, which may lead to receptor desensitization and down-regulation. Positive allosteric modulators, which act through allosteric sites on a receptor that are distinct from the normal agonist binding site (orthosteric site), may be expected to circumvent some of these problems (Christopoulos, 2002).
Positive allosteric modulators are expected to exhibit receptor subtype selectivity since they target sites that are distinct from highly conserved orthosteric agonist binding sites (Christopoulos, 2002). It is likely that such sites are under less evolutionary pressure to maintain high sequence homology. It is also possible to find compounds that may interact with similar binding sites on related receptors but exert different allosteric effects on different subtypes. An example of this is (–)-N-phenyl-7-(hydroxylimino)cycloprop-a[b]chromen-1a-carboxamide, an mGluR1-selective negative allosteric modulator that is also a positive allosteric modulator of mGluR4 (Maj et al., 2003; Marino et al., 2003). When the binding site for an allosteric modulator is saturated, no further allosteric effect for a given modulator is observed (Christopoulos, 2002). Thus, unlike agonists, allosteric modulators can be given in relatively high doses without concern that the receptor will be overstimulated. Positive allosteric modulators also exert their effects only in tissues in which the endogenous agonist exerts its effect. Since normal signaling in the CNS involves cycles of release and removal of neurotransmitter, an allosteric modulator is expected to have an effect when the neurotransmitter is released but would have a minimal effect when the neurotransmitter is removed. A drug targeting the orthosteric agonist site would continue to affect receptor activity without regard to the release or removal of the endogenous agonist, eventually leading to receptor desensitization or down-regulation (Christopoulos, 2002).
We have worked to identify positive allosteric modulators of mGluR5 with sufficient potency and solubility to test in behavioral models and have identified CDPPB as such a compound. CDPPB is similar to (but more potent than) mGluR5 allosteric modulators we have previously reported (O'Brien et al., 2003, 2004). It is reversible and selective. CDPPB binding does not affect binding of the mGluR5 agonist [3H]quisqualate but does compete with the binding of the mGluR5-selective negative allosteric modulator [3H]methoxyPEPy. Thus, similar to our other mGluR5 allosteric modulators, CDPPB is able to increase agonist functional potency without increasing agonist binding affinity (O'Brien et al., 2004). The presence of glutamate does not affect the binding of either [3H]methoxyPEPy or the inhibition of this binding by CDPPB. Unlike our previously described positive allosteric modulators, CDPPB possesses intrinsic agonist activity that activates mGluR5 to approximately 25 to 35% the levels observed for a full agonist. This agonist-like activity is not affected by the competitive (orthosteric site) antagonist LY341495 but is inhibited by the negative allosteric modulator MPEP. Mathematical modeling (using a modification of the Hall model of allosterism detailed in O'Brien et al., 2004) suggests that allosteric modulation by CDPPB is consistent with a mechanism involving an increase in activation cooperativity (similar to our other modulators), as well as some intrinsic activity of the modulator itself. The difference between the EC50 values reported for functional potentiation by CDPPB and the IC50 value for inhibition of [3H]methoxyPEPy binding is accounted for by nonlinearity in the activity-response relationship of the receptor. This difference is also observed in the difference between the binding affinity and the functional potency of the mGluR5 agonist quisqualate.
We had previously reported that the mGluR5 positive allosteric modulator CPPHA increased NMDAR currents in hippocampal and STN preparations (O'Brien et al., 2004). However, the potency and solubility profile of CPPHA was not adequate for in vivo studies. CDPPB has a more attractive profile, and in vivo studies in mice indicated that the compound was brain permeable with a brain/plasma ratio of 1.7. CDPPB was tested in two preclinical behavioral models that are sensitive to known antipsychotic drugs. In the first, an amphetamine-induced locomotion model, CDPPB (1, 3, and 10 mg/kg s.c.) was active in suppressing amphetamine-induced locomotor activity without affecting spontaneous locomotor activity, suggesting that the effect of CDPPB was not due to sedation. The second rodent model assesses sensorimotor gating through measurement of prepulse inhibition of the acoustic startle response (PPI), a model that is also sensitive to known antipsychotic drugs treatment (for review, see e.g. Geyer et al., 2001). Similar to results reported by Lindsley et al. (2004), CDPPB (3, 10, and 30 mg/kg s.c.) was found to reverse amphetamine-induced deficits in PPI at both 10 and 30 mg/kg. Furthermore, CDPPB did not differ from vehicle-treated animals on basal activity or on startle amplitude at any dose tested, suggesting a lack of overt sedative effects. Thus, just as activation of mGluR5 by the agonist CPHG reverses deficits in rodent models of PPI (Kinney et al., 2003), positive allosteric modulation of mGluR5 by CDPPB also reverses such deficits.
These results provide the first description of a potent, selective, and brain-penetrant positive allosteric modulator of mGluR5. In addition, we provide the first in vivo evidence that positive allosteric modulation of this receptor by small-molecule drug treatment results in efficacy in rodent behavioral models in a manner consistent with that of known antipsychotic drugs. These results are the first in vivo evidence that positive allosteric modulation of mGluR5 may be a valid approach to increase mGluR5 activity and should be evaluated as a potential novel strategy for treatment of schizophrenia.
Acknowledgments
We thank Masayo Yamazaki and Steven W. Louie for conducting the in vitro P-gp studies; Scott E. Fauty, Todd Killino, Tamara Pittman, and Anne H. Taylor for conducting the in vivo P-gp studies in mice; Blake A. Rowe for conducting the specificity studies for mGluR2 and 3; and Yvonne Leonard and Kenneth Anderson for conducting the pharmacokinetic studies in rats.
Footnotes
-
doi:10.1124/jpet.104.079244.
-
ABBREVIATIONS: CNS, central nervous system; NMDA, N-methyl-d-aspartate; NMDAR, NMDA receptor; mGluR, metabotropic glutamate receptor; CPPHA, N-{4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl] phenyl}-2-hydroxybenzamide; CDPPB, 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide; CHO, Chinese hamster ovary; FLIPR, fluorometric imaging plate reader; MPEP, 2-methyl-6-(phenylethynyl)-pyridine; P-gp, P-glycoprotein; PPI, prepulse inhibition; DHPG, (R,S)-3,5-dihydroxyphenylglycine; LY341495, (2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid.
- Received October 13, 2004.
- Accepted December 15, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}