


Abstract
β-Arrestin is an adaptor protein that has been shown to couple G protein-coupled receptors (GPCRs) to clathrin-coated pits and target them for subsequent internalization. More recently, β-arrestin 2 has also been shown to be involved in the activation of mitogen-activated protein kinase cascades by G protein-coupled receptors independently of G protein activation. Direct interactions between proteins can be monitored using enzyme complementation between two inactive deletion mutants of β-galactosidase (β-gal; Δα and Δω). In the present study, we have used fusion proteins of the human β2-adrenoceptor (C-terminal β-gal Δα) and β-arrestin 2 (β-gal Δω) to study directly the pharmacology of this interaction in C2C12 cells expressing the β2-adrenoceptor-β-gal Δα fusion protein at low physiological levels (38.2 ± 2.7 fmol · mg protein-1). Isoprenaline, noradrenaline, and adrenaline (-log EC50 = 5.9, 5.5, and 5.7, respectively) stimulated an association between the β2-adrenoceptor and β-arrestin 2 at much higher concentrations than required for activation of cAMP accumulation (-log EC50 = 7.6, 6.3, and 7.7, respectively). This was sensitive to inhibition by the β2-adrenoceptor antagonists propranolol, timolol, and ICI 118551. Both salbutamol and terbutaline behaved as partial agonists of β-gal complementation. Furthermore, the long-acting β2-agonist salmeterol (-log KD for inhibition of [3H]CGP12177 binding = 8.7) behaved as an antagonist of isoprenaline-stimulated β2-adrenoceptor-arrestin 2 interactions (-log KD = 8.0), whereas acting as a full agonist of cAMP accumulation in the same cells (-log EC50 = 9.2). These data suggest that salmeterol can discriminate between receptor-GS protein and receptor-arrestin 2 complexes (in terms of efficacy and affinity) in a way that is favorable for its long duration of action.
The β2-adrenoceptor is the most thoroughly investigated member of the G protein-coupled receptor (GPCR) family, which classically signals through activation of Gs heterotrimeric G proteins (Kobilka, 1992; Benovic, 2002). Agonist stimulation of the β2-adrenoceptor leads to activation of adenylyl cyclase, resulting in an increase in the intracellular levels of cAMP (Kobilka, 1992; Benovic, 2002). cAMP activates protein kinase A (PKA) and causes phosphorylation of multiple targets within the cell (Alto et el., 2002). Persistent stimulation of this receptor does not cause a continuous response from the cell, but instead, the cell adapts to the presence of the stimuli and becomes desensitized (Carman and Benovic, 1998).
Desensitization of the β2-adrenoceptor can be mediated either by a mechanism that is dependent upon agonist occupancy of the β2-adrenoceptor (homologous desensitization) or by a mechanism that involves the activation of PKA (heterologous desensitization; Carman and Benovic, 1998; Ferguson et al., 1998). However, phosphorylation of the β2-adrenoceptor by PKA only causes a partial uncoupling of the receptor from its G protein (Yuan et al., 1994). This phosphorylation can be triggered by low agonist occupancy of the receptor, since it only requires a small increase in cAMP to fully activate PKA (Clark et al., 1999). In contrast, homologous desensitization requires high agonist occupancy by a strong agonist and involves the recruitment of G protein-coupled receptor kinases (GRKs) to the receptor (Kahout and Lefkowitz, 2003; Marchese et al., 2003). This leads to phosphorylation of serine residues within the C-terminal tail of the β2-adrenoceptor (Seibold et al., 2000) and the subsequent binding of β-arrestins (Barak et al., 1997; Ferguson et al., 1998). Binding of arrestin causes a disruption of the receptor-Gs-protein interaction and prevents further signaling from the receptor via the Gs-protein (Barak et al., 1997).
Arrestin has also been found to act as an adaptor molecule coupling GPCRs to clathrin-coated pits and targeting the receptor for subsequent internalization (Luttrell and Lefkowitz, 2002; Marchese et al., 2003). More recently, β-arrestin 2 has also been shown to act as a scaffold for activation of MAP kinase cascades by GPCRs and can function to retain activated MAP kinase enzymes within the cytosol rather than allowing them to translocate to the nucleus (Luttrell et al., 2001; Seta et al., 2002; Tohgo et al., 2002; Wei et al., 2003). Furthermore, it is now clear that this interaction between GPCRs and β-arrestins can activate intracellular signaling via MAP kinase independently of the involvement of heterotrimeric G proteins (Seta et al., 2002; Azzi et al., 2003; Wei et al., 2003; Terrillon and Bouvier, 2004).
The translocation of arrestins to the plasma membrane and their physical interaction with GPCRs therefore represents an important early step in receptor internalization and G protein-independent receptor signaling. However, at the present time, only a few investigations have used technologies capable of detecting direct protein-protein interactions to probe specifically and quantitatively the characteristics of the interaction between GPCRs and β-arrestins. These have included the use of bioluminescence resonance energy transfer (Berglund et al., 2003) and the study of the translocation of fluorescently labeled arrestins to the plasma membrane (Oakley et al., 2002). In both of these examples, complex detection techniques (e.g., confocal microscopy or other ratiometric optical techniques) and/or algorithms are required to obtain quantitative information on the extent of these protein-protein interactions. In this study, we have used β-galactosidase (β-gal) complementation and a simple luminescence detection methodology to investigate the pharmacology of these interactions.
Direct interactions between different proteins can be monitored by use of enzyme complementation between two inactive deletion mutants of β-galactosidase (β-gal; Δα and Δω; Mohler and Blau, 1996). Both mutants of the bacterial enzyme contain inactivating mutations in different essential domains and have been expressed as a fusion protein to target proteins, which are known to associate via protein-protein interactions (Rossi et al., 1997, 2000). When the two target proteins interact the deletion mutants of the galactosidase enzyme are brought into proximity, allowing complementation and the recreation of an active enzyme (Rossi et al., 1997, 2000). The aim of the present study was to investigate the pharmacology of the β2-adrenoceptor-β-arrestin 2 interaction at low physiological levels of receptor expression using a cell line containing β-galactosidase mutants fused to either the C terminus of the β2-adrenoceptor (Δα) or the C terminus of β-arrestin 2 (Δω).
Materials and Methods
Materials. Cell culture reagents were from Sigma Chemical (Poole, Dorset, UK), except fetal calf serum (PAA Laboratories, Teddington, Middlesex, UK). [3H]Adenine, [3H]CGP 12177 and [14C]cAMP were obtained from GE Healthcare (Buckinghamshire, Little Chalfont, UK). Salmeterol, sotalol, CGP 12177, and propranolol were from Tocris Cookson Inc. (Avonmouth, Bristol, UK). The Gal-Screen reagent was from Applied Biosystems (Bedford, MA). All assay plates were obtained from Corning Glassworks (Corning, NY). Lipofectamine was obtained from Invitrogen (Paisley, UK), whereas Opti-MEM reduced serum medium was obtained from Invitrogen cell culture systems. The Hit hunter DiscoverX cAMP assay kit was purchased from DiscoverX (Cardiff, UK). All other reagents were supplied by Sigma Chemical.
Cell Culture. C2C12 (mouse myoblast) cells stably expressing the human β2-adrenoceptor-β-gal-Δα and β-arrestin 2-β-gal-Δω fusion proteins (Yan et al., 2002; Applied Biosystems) were grown at 37°C in Dulbecco's modified Eagle's medium (DMEM) supplemented with 4 mM l-glutamine and 20% FCS in a humidified 5% CO2: 95% air atmosphere. Expression of the β2-adrenoceptor-β-gal-Δα and β-arrestin 2-β-gal-Δω fusion proteins was maintained using resistance to geneticin (0.5 mg/ml) and hygromycin (0.2 mg/ml), respectively. In addition a cell line expressing only the β-arrestin 2-β-gal-Δω fusion protein (Yan et al., 2002; Applied Biosystems) was used to investigate the characteristics of the endogenous murine β2-adrenoceptor in C2C12 cells. Chinese hamster ovary (CHO) cells were grown at 37°C in Dulbecco's modified Eagle's medium/Nutrient mix F-12 (DMEM F12) supplemented with 2 mM l-glutamine and 10% FCS in a humidified 5% CO2, 95% air atmosphere.
Transient Transfection of β2AR-β-Gal-Δα in CHO Cells. CHO cells were grown to 60 to 70% confluence in T75 flasks and then transfected with 10 μg of β2AR-βgal-Δα (Applied Biosystems) in pcDNA3.1 using Lipofectamine and Opti-MEM according to the manufacturer's instructions. Cells were incubated overnight at 37°C, 5% CO2; after this time the transfection mix was removed and replaced with 20 ml of DMEM F12 media (containing 2 mM glutamine and 10% FCS). Cells were incubated for a further 24 h at 37°C, 5% CO2, after which time they were plated into 96-half-well white view plates.
[3H]cAMP Accumulation. Cells were grown to 80% confluence in 24-well plates then prelabeled with [3H]adenine (2 μCi ml-1) for 2 h at 37°C in 500 μl well-1 Hanks' balanced salt solution containing 20 nM HEPES, pH 7.4 (HBH). The [3H]adenine was removed, each well washed with 1 ml of HBH, and then incubated for 30 min with 1 ml of medium containing 3-isobutyl-1-methyl xanthine (100 μM). Agonists were added at 10 μl per well, and cells were incubated for a further 10 min before the reaction was terminated by the addition of 50 μl of concentrated HCl. [3H]Cyclic AMP was separated from other [3H]adenine nucleotides by acid alumina chromatography. Each column was corrected for efficiency by comparison with [14C]cAMP recovery as described previously (Alvarez and Daniels, 1992).
cAMP Accumulation Using the DiscoverX Assay. Cells were grown to 80% confluence in 96-half-well white view plates. The media was then aspirated and replaced with 20 μl well-1 of assay media (C2C12 cells, DMEM media containing 4 mM glutamine; CHO cells, DMEM F12 media containing 2 mM glutamine) containing agonist where appropriate. Cells were incubated for 30 min at 37°C. Cells were then lysed and monitored for cAMP content according to the manufacturer's instructions (DiscoverX).
β2-Adrenoceptor-β-Gal-Δα and β-Arrestin 2-β-Gal-Δω Fusion Protein Complementation. Cells were grown to 80% confluence in 96-well white view plates; media were aspirated and replaced with 180 μl well-1 assay media (DMEM media containing 4 mM glutamine), plus or minus antagonist. Before agonist addition, cells were incubated for 30 min at 37°C with antagonist. Agonists were added at 20 μl well-1, and cells were incubated for a further 1 h. Reactions were terminated via aspiration of all media. Cells were washed with phosphate-buffered saline (PBS) at 200 μl well-1, and lysed with the addition of Gal-Screen buffer/substrate (with the substrate diluted 1:25) at 100 μl well-1. Cells were then incubated at room temperature in Gal-Screen buffer/substrate (between 21 and 26°C) for 1 h. β-Galactosidase activity was determined using a TopCount microplate scintillation and luminescence counter.
[3H]CGP 12177 Binding. Cells were grown to confluence in 96-well white view plates. The media were removed, and 200 μl of serum-free media DMEM containing [3H]CGP 12177 (0.05–8 nM) and β2-ligands was added to each well. The plates were incubated for 1.5 h at 37°C in a 5% CO2 atmosphere. Nonspecific binding was determined using 1 μM ICI 118551. The media and drugs were then removed, and the cells were washed twice with 200 μl of PBS. Then, 200 μl of Microscint 20 was then added to each well, and the plates were counted on a TopCount.
Data Analysis. A maximal isoprenaline concentration was included in each separate experiment for both β-galactosidase complementation, cAMP, and [3H]cAMP accumulation to allow agonist responses to be expressed as a percentage of the isoprenaline maximum. cAMP data were corrected for recovery of tracer [14C]cyclic AMP, from acid alumina chromatography.
Agonist concentration response curves were fitted to a four-parameter logistic equation through the computer assisted nonlinear regression using the program Prism 2 as described previously (Baker et al., 2003a). Antagonist dissociation curves were assessed at fixed antagonist concentrations (assuming competitive antagonism) by observing the shift in the agonist concentration-response curve using the equation DR = 1 + [A]/KD, where DR (dose ratio) is the ratio of the concentrations of agonist required to produce an identical response in the presence and absence of antagonist, [A] is the concentration of antagonist, and KD is the antagonist dissociation constant.
Where antagonists produced a decrease in the maximum isoprenaline response, KD values were obtained as described by Christopoulos et al. (1999). Data were normalized against the isoprenaline standard included in each experiment, and the EC25 was then determined and used to calculate KD values using the above-mentioned equation. Where appropriate, partial agonist dissociation constants were estimated according to the method of Stephenson (1956). In the case of salmeterol inhibiting the β-galactosidase response to a fixed concentration of isoprenaline (10 μM), the apparent KD value was determined from the relationship C/C′ = (IC50/KD) + 1, where C is the concentration of isoprenaline (10 μM) and C′ is the concentration of isoprenaline (alone) that produces response equivalent to 50% of that produced by 10 μM isoprenaline in the presence of an IC50 concentration of salmeterol.
KD values were also obtained from inhibition of the specific binding of [3H]CGP 12177 using the relationship KD = IC50/(1 + [L]/KL), where [L] is the concentration of [3H]CGP 12177 and KL is its dissociation constant. Saturation binding curves were fitted to the following expression: specific binding (SB) = BMAX × [L]/(KL + [L]), where BMAX is the maximal specific binding capacity. The curve in Fig. 2 was also fitted to the expression SB = BMAX × [L]/(KL + [L]) + m × [L], where m is the slope of the line for specific binding in native cells only expressing the β-arrestin 2-β-gal-Δω fusion protein.
All data are represented as mean ± S.E.M. The “n” in the text refers to the number of separate experiments.
Results
Agonist-Stimulated Interactions between the β2-Adrenoceptor and β-Arrestin 2. Isoprenaline produced a marked stimulation of β-galactosidase activity (β2-adrenoceptor/β-arrestin 2 complementation) in C2C12 cells stably expressing the human β2-adrenoceptor-β-gal-Δα and β-arrestin 2-β-gal-Δω fusion proteins but not in native C2C12 cells (Fig. 1a). Similarly, no stimulation of β-galactosidase activity was detected in C2C12 cells expressing only the β-arrestin 2-β-gal-Δω fusion protein (n = 4; data not shown). However, it is interesting to note that in the native C2C12 cells, a considerable degree of endogenous β-galactosidase activity was detectable (Fig. 1a). The presence of endogenous β-galactosidase activity in mammalian cells has been detected previously (Fisher et al., 1967; Hendrikx et al., 1994; Weiss et al., 1997). Similar levels of endogenous β-galactosidase activity were detected in CHO-K1 cells expressing the human β2-adrenoceptor; however, stimulation of β2-adrenoceptors with isoprenaline did not enhance this β-galactosidase activity (data not shown; n = 4), despite a considerable stimulation by isoprenaline of [3H]cAMP accumulation in these cells (Baker et al., 2003a). Furthermore, direct stimulation of cAMP accumulation with forskolin (3 μM) did not alter β-galactosidase activity in either CHO-K1 cells (data not shown; n = 4) or in C2C12 cells (expressing both β-gal fusion proteins; Fig. 1b). These data confirm that the endogenous β-galactosidase activity is not regulated by β2-adrenoceptor stimulation or elevation in cAMP levels.
Isoprenaline stimulated cAMP accumulation in both C2C12 cells expressing the human β2-adrenoceptor-β-gal-Δα and β-arrestin 2-β-gal-Δω fusion proteins and in native C2C12 cells (Fig. 1c). This latter effect indicates that C2C12 cells express endogenous β2-adrenoceptors, but they are also not able to regulate endogenous β-galactosidase activity (Fig. 1a). The log EC50 for isoprenaline for this cAMP response in native C2C12 cells was -5.6 ± 0.0 and the maximum level of cyclic AMP generated was 1240 ± 297 fmol cAMP/well (n = 3). In the β2-adrenoceptor-β-gal-Δα-expressing cells, the log EC50 was an order of magnitude more sensitive (-6.7 ± 0.3) and the maximal response was larger (2387 ± 373 fmol/well; n = 4). Transient expression of the β2-adrenoceptor-β-gal-Δα fusion protein in CHO-K1 cells that do not endogenously express a β2-adrenoceptor (Baker et al., 2003a) confirmed that this receptor fusion protein was able to respond normally to β-agonists and to stimulate cAMP accumulation (Fig. 1d).
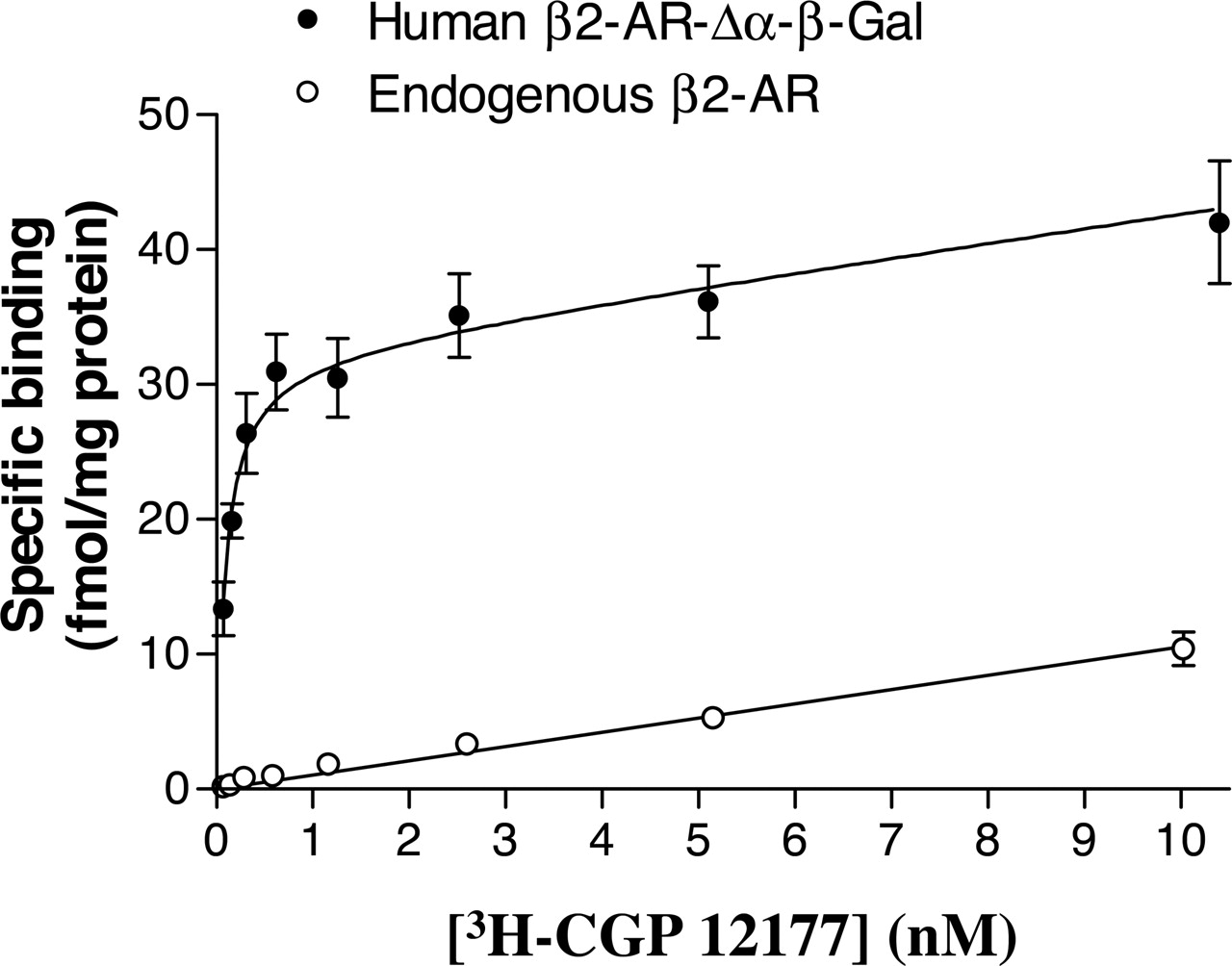
In the host C2C12 cells expressing the β-arrestin 2-β-gal-Δω fusion protein, but not the human β2-adrenoceptor-β-gal-Δα fusion protein, no high-affinity specific binding of [3H]CGP 12177 was detectable (Fig. 2), indicating a very low level of endogenous β2-adrenoceptor expression in C2C12 cells. Indeed, the binding that was sensitive to inhibition by 1 μM ICI 118551 in C2C12 cells expressing the endogenous β2-adrenoceptor was best fit to a straight line (Fig. 2). Furthermore, no displacement of this binding of [3H]CGP 12177 (1 nM) by β2-agonists was detectable in these cells, suggesting that it is nonspecific (n = 4). In C2C12 cells also expressing the β2-adrenoceptor-β-gal-Δα fusion protein, however, saturation binding analysis confirmed the expression of detectable levels of β2-adrenoceptors (38.2 ± 2.7 fmol · mg protein-1; n = 4; KD for [3H]CGP 12177 was 0.2 ± 0.1 nM; n = 4; Fig. 2). The β2-adrenoceptor-selective antagonist ICI 118551 was able to potently inhibit this specific binding of [3H]CGP 12177 with a -log KD of 9.17 ± 0.04 (n = 4). Interestingly, the linear low-affinity nonspecific component of ICI 118551-displaceable binding was also detected in the binding curve obtained in cells transfected with the human β2-adrenoceptor-β-gal-Δα fusion protein (Fig. 2).
β-Galactosidase complementation in C2C12 cells expressing the human β2-adrenoceptor-β-gal-Δα and β-arrestin 2-β-gal-Δω fusion proteins was also stimulated by adrenaline and noradrenaline, yielding very similar log EC50 values and maximal responses to that obtained with isoprenaline (Table 1). Isoprenaline responses were detectable as early as 5 min (Fig. 3) after agonist stimulation (1.86 ± 0.09-fold over basal, -log EC50 = 5.5 ± 0.2; n = 7) and maintained for up to an hour after addition of isoprenaline (2.1 ± 0.1-fold over basal, -log EC50 = 5.7 ± 0.2; n = 7). All three agonists were also able to elicit comparable stimulations of [3H]cAMP accumulation in the same cell line, although the EC50 values were substantially lower than those obtained for β-galactosidase complementation (Table 2).
Concentration-response parameters for agonist-stimulated complementation between β2-adrenoceptor-β-gal-Δα and β-arrestin 2-β-gal-Δω fusion proteins in C2C12 cells Values are mean ± S.E.M. Emax is the maximum response expressed as a percentage of the response to 0.1 mM isoprenaline, which was measured in each experiment. The response to salmeterol was only sufficiently large to obtain an EC50 value in seven of the eight experiments.
Concentration-response parameters for agonist-stimulated [3H]cAMP accumulation in C2C12 cells Values are mean ± S.E.M. Emax is the maximum response expressed as a percentage of the response to 0.1 mM isoprenaline, which was measured in each experiment.

β2-Adrenoceptor/β-arrestin 2 complementation (β-galactosidase activity) and cAMP accumulation stimulated by isoprenaline in C2C12 cells expressing the human β2-adrenoceptor-β-gal-Δα and β-arrestin 2-β-gal-Δω fusion proteins. a, β-galactosidase activity [in relative light units (RLU)] obtained in a single experiment after 1-h incubation with agonist in native C2C12 cells or C2C12 cells expressing the β2-adrenoceptor and β-arrestin 2 fusion proteins. Similar data were obtained in five (native C2C12 cells) or nine (fusion protein-containing cells) separate experiments. Data points are mean ± S.E.M. of triplicate determinations. b, β-galactosidase activity in C2C12 cells expressing the human β2-adrenoceptor-β-gal-Δα and β-arrestin 2-β-gal-Δω fusion proteins in response to isoprenaline and forskolin. Data are the mean values ± S.E.M. of four separate experiments. In each experiment, determinations were made in triplicate. c, cyclic AMP accumulation in response to isoprenaline in native C2C12 cells or in C2C12 cells expressing the human β2-adrenoceptor-β-gal-Δα and β-arrestin 2-β-gal-Δω fusion proteins. Values are mean ± S.E.M. of six replicates in a single experiment. Similar data were obtained in three (C2C12 cells) or four (fusion protein-containing cells) separate experiments. cAMP was measured using the DiscoverX assay as described under Materials and Methods. In the cells expressing the β2-adrenoceptor and β-arrestin 2 fusion proteins, parallel studies were made of the effect of isoprenaline on the cAMP activity in the absence of the DiscovereX cAMP antibody and cAMP ED reagents. This background galactosidase activity contributed less than 6000 RLU to the overall signal and was not influenced by agonist concentration. d, effect of isoprenaline and salmeterol on cAMP accumulation (DiscoverX) in CHO-K1 cells transiently transfected with the human β2-adrenoceptor-β-gal-Δα fusion protein. Data are expressed as a percentage of the response to 3 μM forskolin (measured in each individual experiment). Values are mean ± S.E.M. of 16 (isoprenaline) and 18 (salmeterol) separate experiments.
To determine the extent to which the agonist-stimulated β-galactosidase complementation was maintained after removal of agonist, cells were stimulated for either 10 or 30 min (Fig. 4) with isoprenaline. After this period of time, the agonist was washed out, the media were replaced, and the assay continued for a further 50 or 30 min in the absence of agonist. Compared with a standard isoprenaline response measured over 1 h, responses were markedly reduced after washout of isoprenaline (Fig. 4). The decrease in the maximal response to isoprenaline after this treatment was 52 ± 2% (10 min, -log EC50 = 5.4 ± 0.4; n = 5) and 60 ± 1% (30 min, -log EC50 = 5.3 ± 0.3; n = 6) respectively. The residual, but reduced, maximal responses may well reflect internalization of receptor-arrestin complexes, but this requires further investigation.

Specific binding of [3H]CGP 12177 to C2C12 cells expressing β-arrestin 2-β-gal Δω-galactosidase and either the endogenous murine β2-AR or this endogenous receptor and the human β2-adrenoceptor-Δα-β-galactosidase fusion protein (β2-AR-β-gal Δα). Specific binding was taken as that displaceable by 1 μM ICI 118551. Values represent mean ± S.E.M. from eight (β2-AR) or four (β2-AR-β-gal Δα) separate experiments. In each individual experiment, quadruplicate determinations were made of total and nonspecific binding at each [3H]CGP 12177 concentration. The combined endogenous β2-AR data were fitted by linear regression (slope, 1.06 ± 0.04). The mean data from the β2-AR-β-gal Δα cells were fitted to an equation based on a single binding site with an additional linear component (slope, 1.06). The BMAX and KD values from this analysis were 38.2 ± 2.7 fmol · mg protein-1 and 0.09 ± 0.01 nM, respectively.
The β2-adrenoceptor antagonists timolol and ICI 118551 (Fig. 5a) shifted the isoprenaline concentration-response curve for galactosidase complementation to the right, but also reduced the maximum response. This was consistent with a hemi-equilibrium between slowly dissociating antagonists and an agonist response with a low receptor reserve (Christopoulos et al., 1999; Baker et al., 2003a). Estimation of -log KD values according to the method of Christopoulos et al. (1999) produced -log KD values of 9.7 ± 0.2 (n = 6) and 8.7 ± 0.2 (n = 5) for timolol and ICI 118551, respectively. In contrast, low concentrations of the β-adrenoceptor antagonist propranolol (Fig. 5b) produced shifts in the concentration-response curve to isoprenaline that were more consistent with competitive antagonism (-log KD = 9.0 ± 0.1; n = 6) and its more rapid dissociation from the receptor (Motulsky and Mahan,1984). The lower affinity β-adrenoceptor antagonist sotalol (-log KD = -6.0 ± 0.1; n = 9; Fig. 5c) also produced a shift that was more compatible with competitive antagonism as would be predicted by its more rapid dissociation. CGP 12177 was without any agonist effects, but it proved to be a potent antagonist of the isoprenaline response (-log KD = -9.3 ± 0.2; n = 6; Fig. 5d).

Comparison of isoprenaline-induced β2-adrenoceptor/β-arrestin 2 complementation obtained after different times of agonist stimulation in C2C12 cells. Data are expressed as a percentage of the response to 0.1 mM isoprenaline in each experiment and represent mean ± S.E.M. from seven separate experiments.
Partial Agonist-Induced β-Galactosidase Complementation. Partial agonists of the β2-adrenoceptor have previously been reported to induce only a small desensitization and internalization of the β2-adrenoceptor via GRK phosphorylation and β-arrestin recruitment (January et al., 1997; Clark et al., 1999; Baker et al., 2003b). In these C2C12 cells expressing low levels of the human β2-adrenoceptor-β-gal-Δα fusion protein, terbutaline and salbutamol were able to elicit [3H]cAMP responses of a similar size to those obtained with isoprenaline (Table 2). However, in the case of β2-adrenoceptor-β-arrestin 2 galactosidase complementation responses, the maximum responses to these low-efficacy agonists were markedly lower than that obtained with isoprenaline (Table 1; Fig. 6a). Salmeterol is a long-acting β2-agonist that has been proposed to interact additionally with an exocite on the receptor via which its action is prolonged (Green et al., 1996). This agonist was very weak at producing an effective interaction between the β2-adrenoceptor and β-arrestin 2. The maximum response obtained accounted for only 14% of the maximal response to isoprenaline (0.1 mM) measured in the same experiment (Fig. 6b; Table 1). In those experiments in which it could be determined, the mean -log EC50 value for salmeterol was 7.6 ± 0.2 (n = 7). However, salmeterol was able to elicit a full cAMP response after activation of the β2-adrenoceptor-β-gal-Δα conjugate in the same cells (Table 2). In keeping with its very low efficacy for activation of β2-adrenoceptor/β-arrestin 2 complexes, salmeterol was able to antagonize galactosidase responses to the more efficacious agonist isoprenaline (Fig. 7). Thus, salmeterol was able to shift isoprenaline concentration-response curves to higher agonist concentrations if added simultaneously with isoprenaline (Fig. 7b) or if applied 30 min before isoprenaline (Fig. 7a). Estimation of -log KD values according to the method of Stephenson (1956) produced values of 8.3 ± 0.2 (n = 7) and 8.2 ± 0.3 (n = 9) for simultaneous addition or preincubation with salmeterol, respectively. The effect of pretreatment (30 min) with different concentrations of salmeterol on the β-galactosidase complementation response to a fixed concentration (10 μM) of isoprenaline was also tested (Fig. 8). The -log IC50 value for this inhibitory effect was 6.9 ± 0.2 (n = 21) and yielded an estimated -log KD value of 7.8 ± 0.1.

β2-Adrenoceptor/β-arrestin 2 complementation (β-galactosidase activity) after washout (w/o) of isoprenaline in C2C12 cells expressing the human β2-adrenoceptor-β-gal-Δα and β-arrestin 2-β-gal-Δω fusion proteins. Cells were initially stimulated for 10 (a) or 30 (b) min with isoprenaline, after which time all media were removed, the cells were washed in fresh media, and then incubations were continued for a further 50 or 30 min in the absence of agonist. Control cells were maintained in the presence of isoprenaline for 1 h. Data points are expressed as a percentage of the response to 0.1 mM isoprenaline measured after 1-h continuous stimulation in each experiment. Data points represent mean ± S.E.M. (triplicate determinations in each individual experiment) from six separate experiments.
[3H]CGP 12177 Whole Cell Binding. To establish the binding affinities of the various agonists studied for the β2-adrenoceptor-β-gal-Δα fusion protein in these cells, whole cell binding experiments were performed using [3H]CGP 12177 (which did not detect any endogenous β2-adrenoceptors in the C2C12 cell line). The β2-adrenoceptor selective antagonist ICI 118551 was able to potently inhibit the specific binding of [3H]CGP 12177 with a log -KD of 9.2 ± 0.0 (n = 4). As expected, the majority of the agonists used (isoprenaline, salbutamol, and terbutaline) had low binding affinities (Table 3). However, the exception was salmeterol with a -log KD of 8.65 ± 0.03 (n = 4).
-Log KD values determined from inhibition of [3H]CGP 12177 binding to intact C2C12 cells Values represent mean ± S.E.M. of -log KD values obtained from inhibition of the specific binding of [3H]CGP12177 (1 nM). Nonspecific binding was determined with 1 μM ICI 118551. n gives the number of separate experiments.
[3H]cyclic AMP Accumulation in Cells Expressing only the β-Arrestin 2-β-Gal-Δω Fusion Protein. Despite the very low expression level of endogenous murine β2-adrenoceptors, these receptors were able to stimulate [3H]cAMP accumulation in cells expressing only the β-arrestin 2-β-gal-Δω fusion protein (Table 2). A notable feature of these data was that cAMP responses were only elicited by high agonist concentrations and that noradrenaline, salbutamol, terbutaline, and salmeterol were all partial agonists (Table 2). These data were in marked contrast to higher potencies and apparent efficacies obtained with these ligands in cells where the human β2-adrenoceptor-β-gal-Δα fusion protein was also expressed (Table 2). In the cells expressing only the β-arrestin 2-β-gal-Δω fusion protein, the maximum response to isoprenaline was 164.6 ± 14.8% (n = 4) of the response to 3 μM forskolin. In the C2C12 cells also expressing the human β2-adrenoceptor-β-gal-Δα fusion protein, the maximum response to isoprenaline was 210.9 ± 7.5% (n = 4) of the 3 μM forskolin response. The endogenous receptor was, however, of the β2-adrenoceptor subtype since the cAMP responses to isoprenaline was potently antagonized by ICI 118551 (-log KD = 9.6 ± 0.1; n = 4). In marked contrast, no significant antagonism was obtained with the selective β1-adrenoceptor antagonist CGP 20712A (100 nM; n = 6).
Discussion
Recruitment of β-arrestin to cell surface β2-adrenoceptors is associated with the desensitization and internalization of this receptor (Barak et al., 1997; Ferguson et al., 1998; Kahout and Lefkowitz, 2003). However, only a few investigations have used technologies capable of detecting direct protein-protein interactions to probe specifically and quantitatively the characteristics of the interaction between GPCRs and β-arrestin 2. These have included the use of bioluminescence resonance energy transfer (Berglund et al., 2003) and study of the translocation of fluorescently labeled arrestins to the plasma membrane (Oakley et al., 2002). However, at the present time very little information is available concerning the pharmacological characteristics of receptor-arrestin interactions in terms of both agonist (efficacy and potency) and antagonist (affinity) properties. In this study we have used β-galactosidase complementation and a simple luminescence detection method to investigate the pharmacology of these interactions.

β-Galactosidase complementation induced by isoprenaline in the presence and absence of 10 nM, 100 nM, or 1 μM ICI 118551 (a); 1, 10, or 100 nM propranolol (b); 100 μM sotalol (c); or 100 nM CGP 12177 (d). Antagonists were added 30 min prior to isoprenaline. Values are expressed as a percentage of the control response to 1 mM isoprenaline (a–c) or 0.1 mM isoprenaline (d) measured in each experiment. Data points are mean ± S.E.M. from five (a), six (b), three (c), and six (d) separate experiments.
Isoprenaline was able to stimulate a rapid association between the human β2-adrenoceptor-β-gal-Δα and β-arrestin 2-β-gal-Δω fusion proteins expressed in C2C12 cells. However, an unexpected feature of these studies was the high basal galactosidase activity detected in mammalian cells. This was due to endogenous mammalian galactosidase activity since it could be detected in cells (CHO-K1 and C2C12) that had not been transfected with galactosidase-containing constructs. Endogenous galactosidase activity has been detected previously in mammalian cells and tissues (Fisher et al., 1967; Hendrikx et al., 1994; Weiss et al., 1997). However, in CHO cells expressing high levels of the human β2-adrenoceptor (Baker et al., 2003a, 2004), no isoprenaline-stimulated increase in this endogenous galactosidase activity was detected. This was also true of the native C2C12 cells that express endogenous murine β2-adrenoceptors at low levels, but can nevertheless stimulate a cAMP response. A similar observation was made with C2C12 cells expressing the β-arrestin 2-β-gal-Δω fusion protein. It was only when the human β2-adrenoceptor-β-gal-Δα fusion protein was also present that agonist-stimulated β-galactosidase activity was detected.
A similar sized β-galactosidase response to isoprenaline was observed with adrenaline and noradrenaline as agonists. It was notable, however, that the EC50 values for β2-adrenoceptor-stimulated cAMP accumulation for all three agonists were 2 orders of magnitude lower than those required for β-galactosidase complementation. This reflects greater signal amplification for cAMP accumulation and the need for higher agonist occupancy for receptor-arrestin interactions (January et al., 1997). These data also confirm that the C-terminal modification to the β2-adrenceptor to create the β-galactosidase-Δα fusion protein did not interfere with its ability to signal to Gs and adenylyl cyclase. This was also demonstrated by expressing the human β2-adrenoceptor-β-gal-Δα fusion protein in CHO-K1 cells and generating potent isoprenaline and salmeterol-stimulated cAMP accumulation. It should be noted that the cell surface expression level of the β2-adrenoceptor-β-galactosidase fusion protein was low in the transfected C2C12 cells at ≈38.2 fmol/mg protein and was therefore very similar to the endogenous levels found in native airway smooth muscle cells (70 fmol/mg protein; Green et al., 1995).

β2-Adrenoceptor/β-arrestin 2 complementation (β-galactosidase activity) stimulated by salbutamol (a) or salmeterol (b) in C2C12 cells expressing the human β2-adrenoceptor-β-gal-Δα and β-arrestin 2-β-gal-Δω fusion proteins. Data points are mean ± S.E.M. (triplicate determinations) from a single experiment and represent nine (a) or eight (b) separate experiments. Bars represent the β-galactosidase complementation from unstimulated cells and from cells stimulated with 0.1 mM isoprenaline (Iso), respectively.

β2-Adrenoceptor/β-arrestin 2 complementation induced by isoprenaline in the presence and absence of 1 μM salmeterol. In a, salmeterol was preincubated for 30 min before addition isoprenaline, whereas in b, the two agents were added simultaneously. Data points are mean ± S.E.M. (triplicate determinations) from a single experiment and represent seven (a) and nine (b) separate experiments, respectively. The bar represents the β-galactosidase complementation from unstimulated and cells stimulated with 1 mM salmeterol, respectively.
In C2C12 cells transfected with the β-arrestin 2-β-gal-Δω fusion protein, but not the human β2-adrenoceptor-β-gal-Δα fusion protein, the levels of endogenous murine β2-adrenoceptors were not detectable by [3H]CGP 12177 binding. However, a poorly coupled [3H]cAMP response was measurable that was characterized by submaximal (relative to isoprenaline) agonist responses to noradrenaline, salbutamol, terbutaline, and salmeterol. The expression in these cells of the human β2-adrenoceptor-β-gal-Δα fusion protein, however, produced a leftward shift in the cAMP concentration-response curves to all agonists and increased their maximal responses to the same level as that obtained with isoprenaline.

β2-Adrenoceptor/β-arrestin 2 complementation induced by isoprenaline in the presence of increasing concentrations of salmeterol. The two ligands were added simultaneously. Data points are mean ± S.E.M. (triplicate determinations) from a single experiment representative of 21 separate experiments.
The isoprenaline-stimulated β2-adrenoceptor/β-arrestin 2 galactosidase complementation was antagonized effectively by a number of different β2-adrenoceptor antagonists. However, although low concentrations of propranolol seemed to produce a parallel rightward shift of the isoprenaline concentration-response curves consistent with competitive antagonism, timolol, ICI 118551, and higher concentrations of propranolol also substantially decreased the maximal agonist responses. It is known that both ICI 118551 and timolol dissociate slowly from the receptor due to their lipophilic nature (Becker and Porzig, 1984; Baker et al., 2003a) and it is likely that this, combined with the low β2-adrenoceptor expression and the lack of signal amplification of the receptor-arrestin interactions in these cells, is the reason for the antagonist behavior. ICI 118551 and timolol are thus effectively removing receptor from the agonist accessible pool needed for activation of the arrestin recruitment. Similar behavior of ICI 118551 has been reported for cAMP accumulation (Baker et al., 2003a). In contrast, it is well documented that propranolol dissociates more quickly from the β2-adrenoceptor (Motulsky and Mahan, 1984), which explains why this antagonist effect was surmountable at low concentrations. In keeping with this proposal, the low-affinity β-antagonist sotalol (which should also dissociate more rapidly) produced shifts with a better maintained maximal response. Calculation of the antagonist KD values for ICI 118551 and timolol by the method of Christopoulos et al. (1999), however, provided values in keeping with values obtained previously for β2-adrenoceptor-mediated responses (Baker et al., 2003a).
Low-efficacy agonists of the β2-adrenoceptor such as terbutaline, salbutamol, and salmeterol have previously been described as having little effect on both GRK phosphorylation and internalization of the β2-adrenoceptor (January et al., 1997). In the present study, the use of a galactosidase complementation assay has enabled the pharmacological characteristics of β2-adrenoceptor-β-arrestin 2 interactions induced by low efficacy agonists to be directly compared. At low physiological levels of receptor expression, both salbutamol and terbutaline were able to act as partial agonists of β2-adrenoceptor-mediated arrestin complementation. However, the most striking finding was that, at these levels of receptor expression, the long-acting β2-agonist salmeterol had very little effect on arrestin complementation but nevertheless was able to act as a potent and full agonist of cAMP-mediated responses. This is despite the similarity of its efficacy to that of both salbutamol and terbutaline for the stimulation of cAMP accumulation via the endogenous β2-adrenoceptor in C2C12 cells expressing only the β-arrestin 2-β-gal-Δω fusion protein (Table 2). Furthermore, in the C2C12 cells also expressing the human β2-adrenoceptor, salmeterol was able to act as an antagonist of the arrestin complementation response to more efficacious β2-agonists (e.g., isoprenaline). The log dissociation constant for salmeterol obtained from this interaction was ca. -8.0 (average of the mean values obtained in the three different assay formats). This is an order of magnitude higher than the log EC50 value for salmeterol-stimulated cAMP accumulation (-9.2) and higher than the log KD value determined from inhibition of [3H]CGP 12177 binding (-8.7). These data suggest that salmeterol may be able to distinguish between the receptor conformations required for Gs-protein activation and that required for β-arrestin 2 binding both in terms of efficacy and affinity. The data obtained with [3H]CGP 12177 binding confirm that salmeterol has a higher affinity (compared with the other agonists measured) for the β2-adrenoceptor in intact C2C12 cells. The values obtained from [3H]CGP 12177 binding probably represent those for the low-affinity agonist resting state (compared with the high-affinity GS-coupled active state) of the receptor, since the intracellular surfaces of the receptor in intact cells will be exposed to normal physiological levels of GTP.
As mentioned above, low-efficacy agonists such as salmeterol, salbutamol, and terbutaline do not readily phosphorylate the β2-adrenoceptor via GRK (January et al., 1997). This may be because partial agonists stabilize receptor conformations that differ in their ability to act as substrates for GRKs (January et al., 1997). It is known that arrestins possess recognition domains that discriminate between active and inactive receptors as well as between phosphorylated and unphosphorylated forms of the receptor (Gurevich and Gurevich, 2004). As a consequence, the reduced receptor phosphorylation by GRK obtained with low-efficacy agonists may significantly contribute to the low potency of these ligands to activate receptor-arrestin association. Alternatively, the nature of the receptor-arrestin interaction may not be entirely dependent upon receptor phosphorylation and the pharmacology of the β2-adrenoceptor-arrestin interaction may differ from that of the β2-adrenoceptor-Gs protein interaction in a manner similar to that described for agonist trafficking between different G proteins (Kenakin, 1995). However, the phosphorylation status of the β2-adrenoceptor-β-gal-Δα fusion is unknown in this study, and it remains to be established whether the extent of receptor phosphorylation by each agonist correlates with their efficacy in stimulating receptor-arrestin interactions. The study of receptor-arrestin interactions using enzyme complementation does, however, provide a means by which some of these important questions can now be directly addressed.
In the present study, the use of a galactosidase complementation assay has also provided clear evidence that the long-acting β2-adrenoceptor agonist salmeterol (Green et al., 1996) has a very low efficacy for stimulating β2-adrenoceptor-β-arrestin 2 complementation at physiological levels of receptor expression. These properties are clearly beneficial for an agonist that is designed to provide long-acting agonism of Gs-mediated responses but without substantial receptor internalization. Furthermore, these data suggest that salmeterol may be able to discriminate between receptor-GS protein and receptor-arrestin 2 complexes (in terms of efficacy and affinity) in a way that is favorable for its long duration of action.
Acknowledgments
We thank Applied Biosystems for provision of the C2C12 (mouse myoblast) cells stably expressing the human β2-adrenoceptor-β-gal-Δα and β-arrestin 2-β-gal-Δω fusion proteins (InteraX system) and Dr. Melissa Gee (Applied Biosystems) for technical discussion. We also thank GlaxoSmithKline for financial support.
Footnotes
-
A.A.C. holds a Biotechnology and Biological Sciences Research Council research studentship.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.105.088914.
-
ABBREVIATIONS: GPCR, G protein-coupled receptor; PKA, protein kinase A; GRK, G protein-coupled receptor kinase; MAP, mitogen-activated protein; β-gal, β-galactosidase; CGP 12177, (-)-4-(3-tert-utylamino-2-hydroxypropoxy)-benzimidazol-2-one; DMEM, Dulbecco's modified Eagle's medium; DMEM F12, Dulbecco's modified Eagle's medium Ham's F-12; β-AR, β2-adrenoceptor; FCS, fetal calf serum; CHO, Chinese hamster ovary; HBH, Hanks' balanced salt solution; PBS, phosphate-buffered saline; ICI 118551, (-)-1-(2,3-[dihydro-7-methyl-1H-inden-4-yl]oxy)-3-([1-methylethyl]-amino)-2-butanol; CGP 20712A, 2-hydroxy-5-(2-[{hydroxyl-3-(4-[1-methyl-4-trifluoromethyl-2-imidazolyl]phenoxy)-propyl}amino]ethoxy)benamide.
- Received May 2, 2005.
- Accepted July 26, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}