


Abstract
The glucagon-like peptide (GLP)-1 receptor is a promising target for the treatment of type 2 diabetes and obesity, and there is great interest in characterizing the pharmacology of the GLP-1 receptor and its ligands. In the present report, we have applied bioluminescence resonance energy transfer2 assays to measure agonist-induced recruitment of βarrestins and G-protein-coupled receptor kinase (GRK) 2 to the GLP-1 receptor in addition to traditional measurements of second messenger generation. The peptide hormone oxyntomodulin is described in the literature as a full agonist on the glucagon and GLP-1 receptors. Surprisingly, despite being full agonists in GLP-1 receptor-mediated cAMP accumulation, oxyntomodulin and glucagon were observed to be partial agonists in recruiting βarrestins and GRK2 to the GLP-1 receptor. We suggest that oxyntomodulin and glucagon are biased ligands on the GLP-1 receptor.
A great deal of the signaling molecules implicated in appetite regulation and energy homeostasis exert their effect through seven-transmembrane (7TM) G-protein-coupled receptors. One of the most interesting new targets in the management of diabetes and obesity is the 7TM glucagon-like peptide (GLP)-1 receptor (Holst, 2004; Davidson et al., 2005). GLP-1 is created through posttranslational processing of the proglucagon peptide, a processing that also creates the peptides GLP-2, glucagon, oxyntomodulin, and glicentin (Mayo et al., 2003; Sinclair and Drucker, 2005). The 37-amino acid residue oxyntomodulin peptide is an eight-residue C-terminally extended glucagon peptide that is cosecreted with GLP-1 from intestinal L-cells as a response to food intake (Mayo et al., 2003). In vitro data demonstrates that oxyntomodulin binds to and activates both the glucagon and GLP-1 receptor although with severely reduced affinity compared with the cognate agonists (Bataille et al., 1982; Baldissera et al., 1988; Gros et al., 1993; Schepp et al., 1996; Baggio et al., 2004). Oxyntomodulin is receiving an increasing amount of attention after in vivo data have demonstrated that it reduces food intake in mice, rats, and human subjects (Dakin et al., 2001, 2002, 2004; Cohen et al., 2003; Baggio et al., 2004; Wynne et al., 2005). We describe that oxyntomodulin and glucagon are full agonists in GLP-1 receptor-mediated cAMP accumulation but partial agonists in recruiting G-protein-coupled receptor kinase (GRK) 2, βarr1, and βarr2 to the receptor, suggesting that oxyntomodulin and glucagon are biased ligands on the GLP-1 receptor.
Materials and Methods
Molecular Biology and Peptides. Human βarr2 N-terminally tagged with green fluorescent protein (GFP)2 and the Renilla luciferase (RLuc) cDNA were purchased from PerkinElmer Life and Analytical Sciences (Wellesley, MA). Human βarr1 cDNA was purchased from Origene (Rockville, MD) and subcloned in a 3′ position of GFP2 lacking the stop codon. Human GRK2 and GRK5 were cloned from a hypothalamic cDNA library. GRK2 lacking the stop codon was made by standard molecular biology techniques and inserted in a 5′ position of GFP2. Human GLP-1 receptor and rat glucagon receptor constructs lacking the stop codon were made by standard molecular biology techniques and subcloned in a 5′ position of Renilla luciferase cDNA. Mutations (R393E and R395E) in βarr2 were introduced in human GFP2-βarr2 using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). All cDNA clones were verified by sequencing. All peptides were obtained from Bachem (Bubendorf, Switzerland).
Tissue Culture and cAMP Measurements. HEK293 and COS-7 cells were obtained from the European Collection of Animal Cell Cultures and maintained according to protocol. Agonist-induced cAMP accumulation was measured as described previously (Elling et al., 1999).
Bioluminescence Resonance Energy Transfer2 Assays. We measured bioluminescence resonance energy transfer (BRET)2 by using the GFP2 blue-shifted variant of GFP. BRET2 measurement was done using a Mithras LB 940 plate reader (Berthold Technologies, Bad Wildbad, Germany). To enhance the BRET2 signal in the βarr2 recruitment assay, a GFP2-βarr2(R393E;R395E) mutant was used (Vrecl et al., 2004) (except in Fig. 3). Receptor-RLuc cDNA was transiently transfected into HEK293 cells stably expressing GFP2-βarr2(R393E;R395E) (hereafter referred to as βarr2) for measurements of βarr2 recruitment or transiently cotransfected with GFP2-βarr1 or GRK2-GFP2 into HEK293 cells for measurements of βarr1 or GRK2 recruitment, respectively. The commercial use of the βarrestin2(R393E;R395E) mutant in BRET assays requires a license from 7TM Pharma. To potentiate the BRET2 signal as described previously (Jorgensen et al., 2005), human GRK5 cDNA was cotransfected in a receptor-RLuc/GRK5 cDNA ratio of 1:1 in βarr recruitment assays (except in Fig. 3). The reading time was 10 min after agonist addition for βarr recruitment and 5 min for GRK2 recruitment. For measurements of the antagonistic effect of oxyntomodulin or exendin(9–39), the antagonist was added 2 min before the agonist (10 nM GLP-1). For the kinetic study, variable reading times were used, and agonist was added with an injector. The background signal from RLuc was determined by coexpressing the RLuc construct with empty vector, and the BRET2 ratio generated from this transfection was subtracted from all other BRET2 ratios.
Receptor Internalization Assay. GLP-1 receptor internalization assay was based on a protocol described previously (Vrecl et al., 1998). In brief, HEK293 cells transiently transfected with GLP-1 receptor cDNA were first incubated in serum-free Hepes-modified Dulbecco's modified Eagle's medium containing 0.1% bovine serum albumin, pH 7.4, before GLP-1 receptor internalization was induced by 0.1 μM GLP-1 or 10 μM oxyntomodulin at 37°C for time intervals ranging from 2 min to 1 h. Stimulation was stopped by washing the cells with ice-cold phosphate-buffered saline followed by a 6-min acid wash (50 mM acetic acid and 150 mM NaCl, pH 2.8) to remove surface-bound ligand. Cells were then subjected to 125I-exendin(9–39) (PerkinElmer Life and Analytical Sciences) binding at 4°C for 3 h, and the radioactivity was measured. The time-dependent loss of the surface GLP-1 receptor was determined relative to unstimulated cells. Nonspecific binding for each time point was determined in the presence of 1 μM GLP-1.
Data Analysis. Data were analyzed using Prism (GraphPad Software Inc., San Diego, CA).
Results
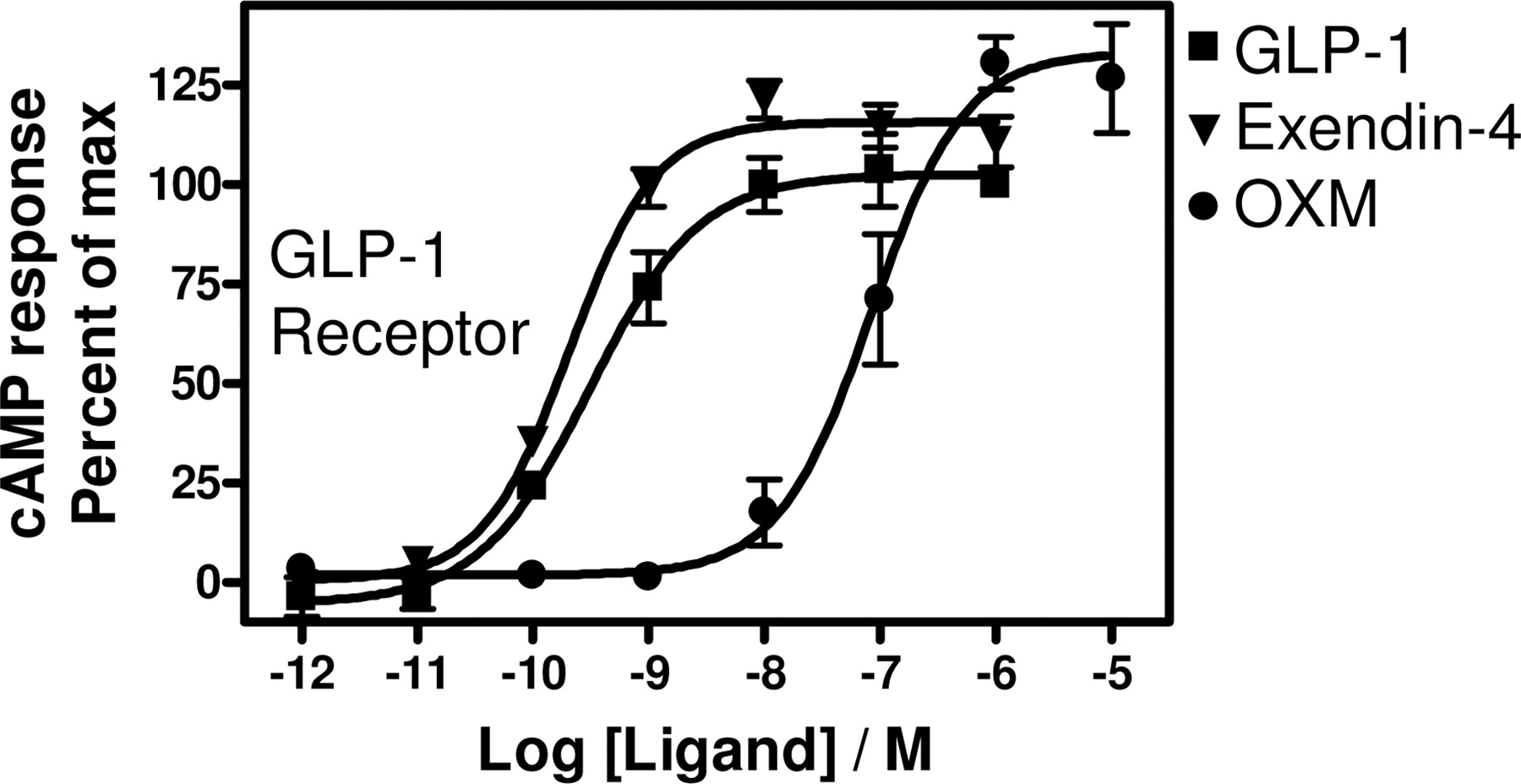
To expand the in vitro pharmacological analysis of oxyntomodulin, we studied in parallel the pharmacological profile of oxyntomodulin, glucagon, and GLP-1(7–36)amide (hereafter GLP-1) on the glucagon and GLP-1 receptors in cAMP accumulation and βarr recruitment-based BRET assays. Both the glucagon and GLP-1 receptors are coupled to Gαs, and the activation of these receptors therefore up-regulates intracellular cAMP levels. Oxyntomodulin was observed to be a full glucagon receptor agonist in a cAMP assay with a potency of 4.1 ± 2.0 nM relative to 1.2 ± 0.14 nM for glucagon (Fig. 1A). As has been reported elsewhere (Runge et al., 2003), GLP-1 did not stimulate glucagon receptor-mediated cAMP accumulation (Fig. 1A). GLP-1, glucagon, and oxyntomodulin have been reported to be full agonists on the GLP-1 receptor in stimulating cAMP production (Gros et al., 1993, 1995; Schepp et al., 1996; Runge et al., 2003; Baggio et al., 2004). This was confirmed in the present study, where all three ligands were full agonists on the GLP-1 receptor with a relative potency distribution of GLP-1 > oxyntomodulin > glucagon (EC50 of 0.029 ± 0.0028 nM for GLP-1, 9.2 ± 0.34 nM for oxyntomodulin, and 66 ± 1.9 nM for glucagon; Fig. 1B).

Ligand-induced cAMP accumulation. Dose-response curves for ligand-induced cAMP accumulation in COS-7 cells transiently transfected with cDNA encoding either the wild-type glucagon receptor (A) or GLP-1 receptor (B). Response is expressed as percentage of maximal glucagon-induced response (A) or maximal GLP-1-induced response (B). OXM, oxyntomodulin. Results are the means ± S.E.M. of duplicate determinations in at least three independent experiments.
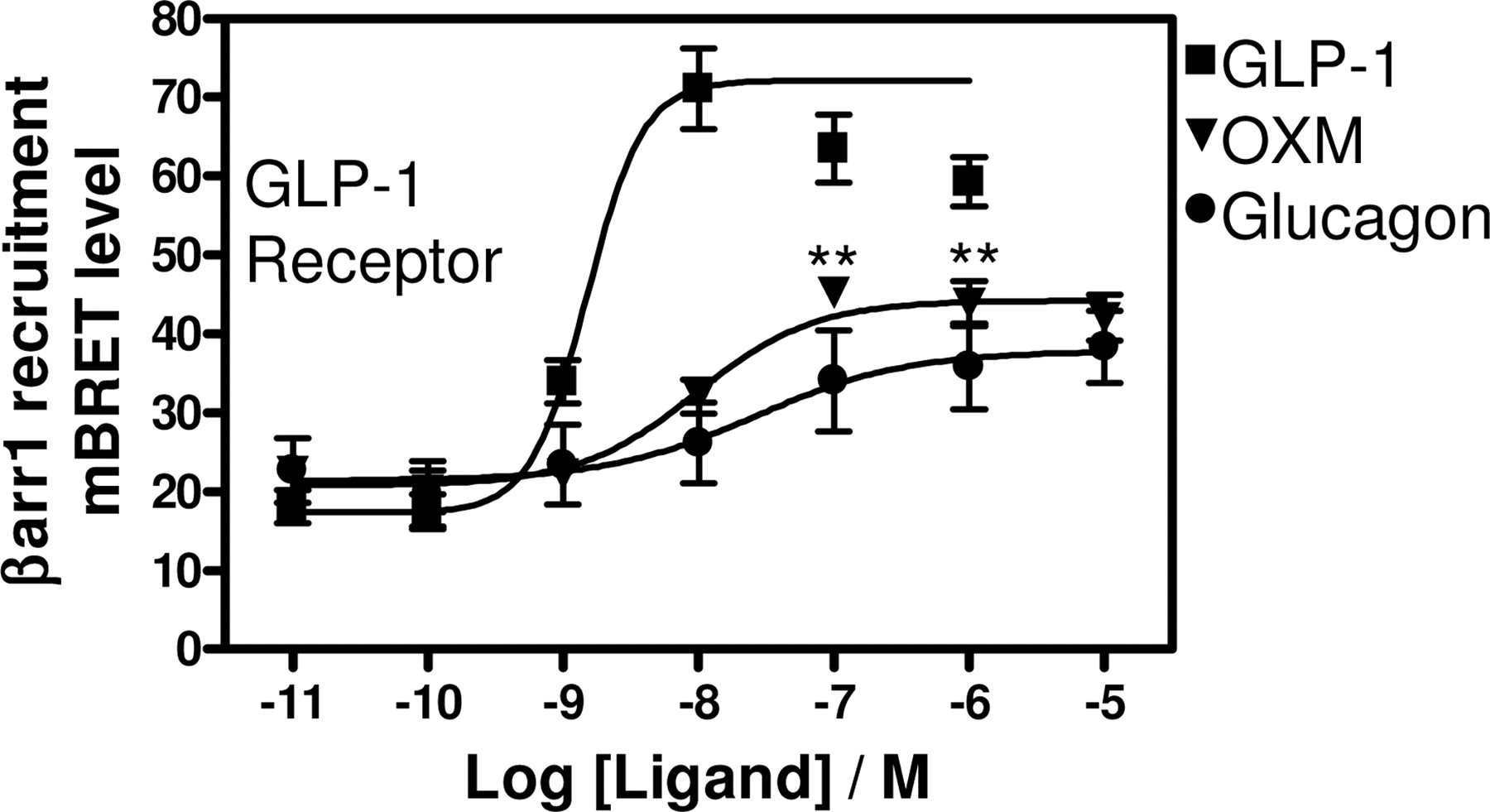
To analyze the ability of oxyntomodulin, glucagon, and GLP-1 to recruit βarr2 to the glucagon and GLP-1 receptors, we used a proximity-based BRET2 assay that measures energy transfer from a RLuc-tagged protein to a GFP-tagged protein upon addition of the RLuc substrate Deep Blue C (Mercier et al., 2002; Heding, 2004; Vrecl et al., 2004). The BRET2 signal is highly distance dependent and is obtained if the RLuc energy donor and the GFP acceptor are within approximately 100 Å or less of each other. Glucagon- and oxyntomodulin-induced recruitment of βarr2 to the glucagon receptor with identical efficacies and potencies of 2.1 ± 0.38 and 76 ± 21 nM, respectively (Fig. 2A). Consistent with the cAMP results, GLP-1 did not induce βarr2 recruitment to the glucagon receptor. On the GLP-1 receptor, GLP-1, oxyntomodulin, and glucagon peptides all induced βarr2 recruitment to the receptor. Surprisingly, we observed that oxyntomodulin and glucagon, compared with GLP-1, were partial agonists in recruiting βarr2 to the GLP-1 receptor (BRET-max of 190 ± 9.5 mBRET for GLP-1, 129 ± 8.4 mBRET for oxyntomodulin, and 127 ± 5.3 mBRET for glucagon; Fig. 2B). The relative potency profile (GLP-1 > oxyntomodulin > glucagon) was conserved in βarr2 recruitment (EC50 of 1.7 ± 0.17 nM for GLP-1, 10 ± 1.1 nM for oxyntomodulin, and 66 ± 21 nM for glucagon; Fig. 2B) compared with cAMP accumulation. Oxyntomodulin was found to be a functional antagonist of the GLP-1-induced agonist response on the GLP-1 receptor antagonizing the full GLP-1-induced response down to the maximal oxyntomodulin-induced signal with an IC50 of 53 ± 1.6 nM (Fig. 2B). We have used here a mutant of βarr2 as well as cotransfection with GRK5 to enhance the BRET2 signal window in βarr2 recruitment (see Materials and Methods). When the GLP1R-RLuc construct was coexpressed with GFP2-βarr2(wt) without GRK5 cotransfection, we still observed oxyntomodulin and glucagon to be significantly less efficacious than GLP-1 in recruiting βarr2 to the GLP-1 receptor (64 and 55% efficacy, respectively, compared with GLP-1, Fig. 3). The EC50 under these conditions was 2.2 ± 0.46 nM for GLP-1, 133 ± 10 nM for oxyntomodulin, and 371 ± 48 nM for glucagon (Fig. 3). Hence, a reduction in potency of oxyntomodulin and glucagon compared with the measurements with GRK5 coexpression (approximately 13- and 6-fold reduction in potency for oxyntomodulin and glucagon, respectively) without a concomitant change in the potency of GLP-1. Because two nonvisual βarrs (βarr1 and βarr2) are ubiquitously expressed in mammalian cells, we also investigated the ligand-induced recruitment of GFP2-βarr1 to the GLP1R-RLuc construct in a BRET2 assay. GLP-1, oxyntomodulin, and glucagon all induced recruitment of βarr1 to the GLP-1 receptor with potencies similar to the βarr2 recruitment (EC50 of 1.5 ± 0.039 nM for GLP-1, 9.6 ± 4.5 nM for oxyntomodulin, and 27 ± 147 nM for glucagon; Fig. 4). As observed in βarr2 recruitment, oxyntomodulin and glucagon were partial agonists in inducing βarr1 recruitment to the GLP-1 receptor (Fig. 4).

βarr2 recruitment-based BRET2 assay. Dose-response curves for ligand-induced GFP2-βarr2(R393E;R393E) recruitment to the glucagonR-RLuc (A) and GLP1R-RLuc (B). OXM, oxyntomodulin. Results are the means ± S.E.M. of duplicate determinations in at least three independent experiments. **, p < 0.01; ***, p < 0.001 for GLP-1-versus oxyntomodulin-treated cells (unpaired Student's t test).

βarr2 recruitment-based BRET2 assay. Dose-response curves for ligand-induced GFP2-βarr2 recruitment to GLP1R-RLuc. GFP2-βarr2(wt) was transiently coexpressed with GLP1R-RLuc in HEK293 cells. OXM, oxyntomodulin. Results are the means ± S.E.M. of duplicate determinations in five independent experiments. *, p < 0.05; **, p < 0.01 for GLP-1 versus oxyntomodulin- or glucagon-treated cells (unpaired Student's t test).

βarr1 recruitment-based BRET2 assay. Dose-response curves for ligand-induced GFP2-βarr1 recruitment to GLP1R-RLuc. The BRET2 value obtained at 10 nM GLP-1 was taken as the maximal effect and fixed as a constant. OXM, oxyntomodulin. Results are the means ± S.E.M. of duplicate determinations in at least three independent experiments. **, p < 0.01 for GLP-1-versus oxyntomodulin-treated cells (unpaired Student's t test).
GRK-mediated receptor phosphorylation is known to precede βarr recruitment to activated 7TM receptors (Penn et al., 2000), and the BRET technique has formerly been employed to demonstrate agonist-induced 7TM receptor recruitment of GRK2 (Hasbi et al., 2004). To analyze a potential interaction of GRK2 with the GLP-1 receptor, we measured BRET2 between GLP1R-RLuc and GRK2-GFP2. Similar to the observations with βarr recruitment, oxyntomodulin and glucagon were observed to be partial agonists in inducing recruitment of GRK2 to the GLP-1 receptor (BRETmax of 67 ± 2.6 mBRET for GLP-1, 52 ± 1.7 mBRET for oxyntomodulin, and 52 ± 1.9 mBRET for glucagon) (Fig. 5). The peptides were observed to induce GRK2 recruitment to the GLP-1 receptor with an EC50 of 3.8 ± 0.42 nM for GLP-1, 60 ± 6.3 nM for oxyntomodulin, and 94 ± 12 nM for glucagon (Fig. 5).
We next expanded the pharmacological analysis of ligand-induced βarr recruitment to the GLP-1 receptor to include the agonist exendin-4 and the antagonist exendin(9–39) (an N-terminally truncated version of exendin-4). Exendin-4 is a 39-amino acid peptide found in the venom of the Gila monster, Heloderma suspectum. It shares approximately 50% sequence identity with GLP-1 and is reported to be a full agonist on the GLP-1 receptor (Göke et al., 1993). As seen in Fig. 6, GLP-1 and exendin-4 were equipotent agonists in inducing recruitment of βarr1 and βarr2 to the GLP-1 receptor (EC50 of 1.5 ± 0.039 versus 1.7 ± 0.17 nM for GLP-1 in recruiting βarr1 and βarr2, respectively, and 1.4 ± 0.067 versus 1.4 ± 0.24 nM for exendin-4 in recruiting βarr1 and βarr2, respectively). Interestingly, exendin-4 was observed to be marginally but significantly less efficacious than GLP-1 in βarr recruitment assays (BRETmax of 172 ± 1.8 mBRET for exendin-4 and 190 ± 9.5 for GLP-1 in βarr2 recruitment; Fig. 6). The BRET signal for βarr1 recruitment was observed to decrease at the highest concentrations of GLP-1 and exendin-4, resulting in bell-shaped curves (Fig. 6B). The signal decrease at high agonist concentrations may be caused by receptor internalization. Consistent with its described role as a GLP-1 receptor antagonist, exendin(9–39) was not observed to have efficacy in inducing βarr1 or βarr2 recruitment to the GLP-1 receptor (Fig. 6) but was observed to be an antagonist of GLP-1-induced βarr1 and βarr2 recruitment to the GLP-1 receptor with an IC50 of 30 ± 1.7 and 27 ± 6.3 nM for βarr1 and βarr2 recruitment, respectively (Fig. 6).

GRK2 recruitment-based BRET2 assay. Dose-response curve for ligand-induced GRK2-GFP2 recruitment to GLP1R-RLuc. OXM, oxyntomodulin. Results are the means ± S.E.M. of duplicate determinations in at least four independent experiments. **, p < 0.01 for GLP-1-versus oxyntomodulin- or glucagon-treated cells (unpaired Student's t test).
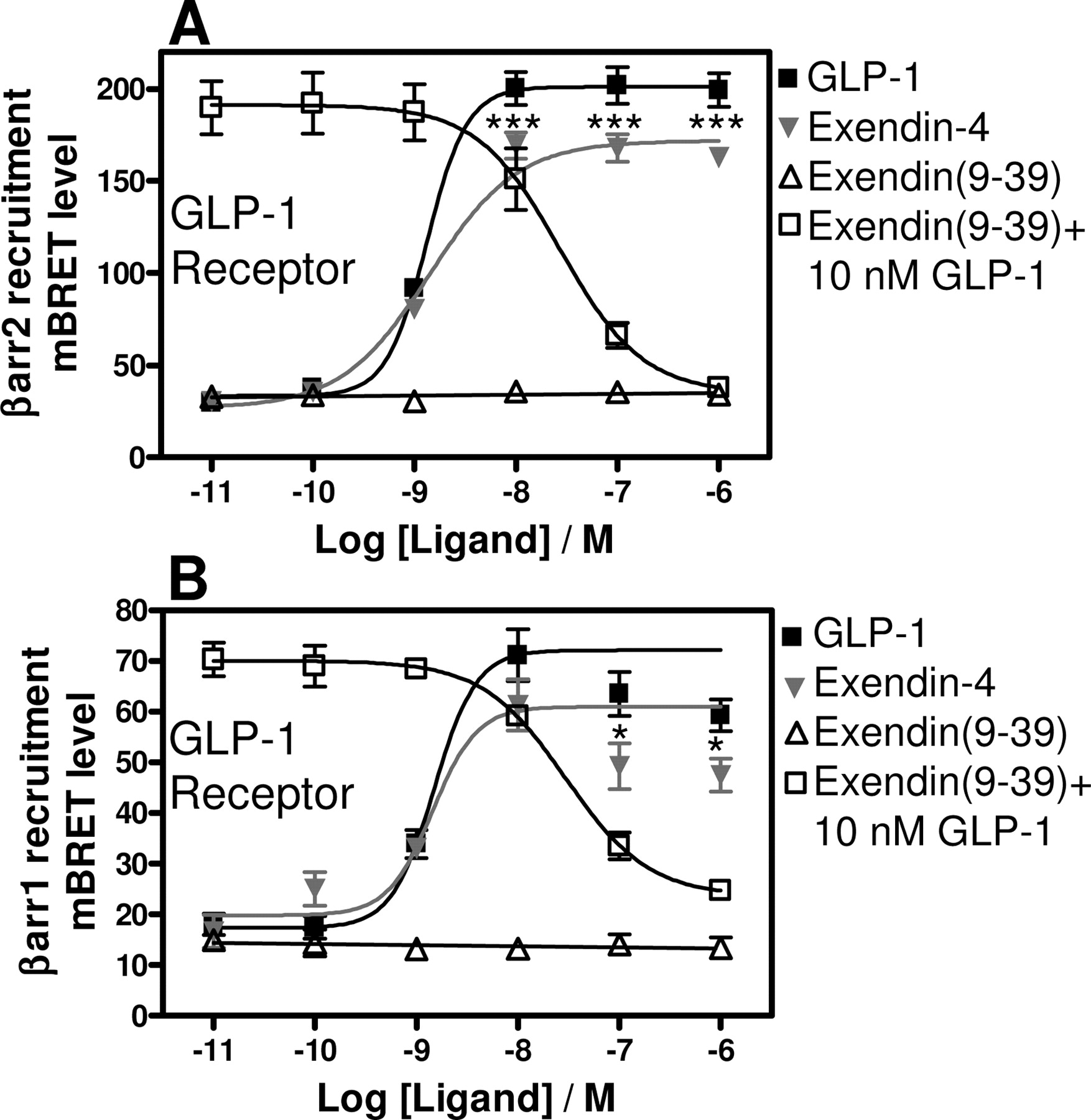
βarr1 and βarr2 recruitment-based BRET2 assays. Dose-response curves for ligand-induced GFP2-βarr2(R393E;R395E) (A) or GFP2-βarr1 (B) recruitment to GLP1R-RLuc. The GLP-1 dosis response curve in B is identical to the curve shown in Fig. 4. The BRET2 value obtained at 10 nM GLP-1 or exendin-4 was taken as the maximal effect and fixed as a constant in B. OXM, oxyntomodulin. Results are the means ± S.E.M. of duplicate determinations in at least three independent experiments. *, p < 0.05; ***, p < 0.001 for GLP-1-versus exendin-4-treated cells (unpaired Student's t test).
To determine whether the chosen time point for measuring BRET2 dose-response curves was representative for observing the partial efficacy of oxyntomodulin, we measured βarr2 recruitment to the GLP-1 receptor at consecutive time points after the addition of saturating concentrations of GLP-1 or oxyntomodulin (Supplemental Fig. 1). We observed that GLP-1 and oxyntomodulin-induced recruitment of βarr2 to the GLP-1 receptor with similar kinetics (Supplemental Fig. 1). Furthermore, oxyntomodulin was observed to be a partial agonist at all of the time points after ∼2 min, where the maximal signal was reached (Supplemental Fig. 1). We next assessed the ability of GLP-1 and oxyntomodulin to induce internalization of the GLP-1 receptor (measured by loss of surface receptor expression). GLP-1 and oxyntomodulin used at saturating concentrations were observed to induce GLP-1 receptor internalization with similar kinetics and efficacy (t1/2 of 2.6 ± 0.9 min and 54 ± 8% maximal internalization for GLP-1 versus t1/2 of 3.0 ± 1.1 min and 55 ± 3% maximal internalization for oxyntomodulin; Fig. 7).
Because the BRET2 measurements of βarr recruitment were done with GRK5 cotransfection favoring βarr interaction and not G-protein signaling, we assessed whether exendin-4 and oxyntomodulin were full agonists in inducing cAMP accumulation also under these conditions (same conditions as in Figs. 2B and 6A). Oxyntomodulin and exendin-4 were full agonists in inducing GLP1R-RLuc-mediated cAMP accumulation when GFP2-βarr2 and GRK5 were coexpressed (Fig. 8). The EC50 was 0.39 ± 0.16 nM for GLP-1, 0.20 ± 0.022 nM for exendin-4, and 78 ± 22 nM for oxyntomodulin, hence, an approximately 10-fold reduction in potency of both GLP-1 and oxyntomodulin compared with measurements with the GLP-1 receptor (wt) without GRK5 and βarr2 coexpression, which is consistent with the reported effect of GRK5 and βarr2 on GLP-1 receptor-mediated G-protein signaling (Jorgensen et al., 2005).

GLP1- and oxyntomodulin-induced GLP-1 receptor internalization. Loss of cell surface receptor binding in HEK 293 cells transiently transfected with GLP-1 receptor cDNA. Cells were incubated with either 0.1 μM GLP1 or 10 μM oxyntomodulin at 37°C for varying periods of time (2–60 min) and then subjected to antagonist 125I-exendin(9–39) binding at 4°C for 3 h. OXM, oxyntomodulin. Results are the means ± S.E.M. of triplicate determinations in three independent experiments.
Ligand-induced cAMP accumulation. Dose-response curves for ligand-induced cAMP accumulation in HEK293 cells stably expressing GFP2-βarr2(R393E;R395E) and transiently transfected with cDNA encoding GLP1R-RLuc and GRK5(wt). Response is expressed as percentage of maximal GLP-1-induced response. OXM, oxyntomodulin. Results are the means ± S.E.M. of duplicate determinations in four independent experiments.
Discussion
We have performed here a concurrent analysis of the ability of oxyntomodulin to mediate cAMP accumulation and induce βarr recruitment to the glucagon and GLP-1 receptors in comparison with their cognate ligands. Oxyntomodulin was found to be a full agonist in inducing glucagon receptor-mediated cAMP accumulation with a potency only 3-fold less than glucagon itself. This is similar to the published potency of oxyntomodulin in inducing cAMP accumulation in rat liver membranes (Bataille et al., 1982). Oxyntomodulin was found to be a full agonist in recruiting βarr2 to the glucagon receptor with a slightly decreased potency compared with glucagon.
Consistent with previous reports, we confirmed that oxyntomodulin and glucagon compared with GLP-1 are full but relatively low-potency agonists in GLP-1 receptor-mediated cAMP accumulation. Surprisingly, both oxyntomodulin and glucagon were partial agonists in recruiting βarr2 to the GLP-1 receptor (62 and 59% efficacy, respectively, compared with GLP-1 based on signal window). Consistent with the properties of a partial agonist, oxyntomodulin was observed to be a functional antagonist of the full GLP-1-induced agonist response in βarr2 recruitment. Oxyntomodulin and glucagon were also observed to be partial agonists in recruiting βarr1 to the GLP-1 receptor. Furthermore, using a BRET2 assay, we describe that GRK2 interacts with the GLP-1 receptor in an agonist-dependent manner. Similar to our observations with βarr recruitment, oxyntomodulin and glucagon were observed to be partial agonists in recruiting GRK2 to the GLP-1 receptor. The observation that oxyntomodulin and glucagon display partial efficacy in both GRK2 and βarr recruitment assays is consistent with the known role of GRK2 in promoting βarr recruitment to activated 7TM receptors. The observed partial efficacy of oxyntomodulin and glucagon in BRET2 assays could potentially be caused by different properties of these peptides, compared with GLP-1, in inducing GLP-1 receptor internalization. However, when measuring the time course of GLP-1 or oxyntomodulin-induced βarr2 recruitment to the GLP-1 receptor, we observed oxyntomodulin to be a partial agonist at all time points after the maximal signals had been reached. Both the GLP-1 and oxyntomodulin-induced BRET2 signals decreased slightly over time after agonist addition (∼20% decrease at 10 min relative to maximum), which may be caused by receptor internalization (Supplemental Fig. 1). In the βarr2 recruitment experiments (except in Fig. 3), we used a GFP2-βarr2(R393E;R395E) mutant that does not bind to the adaptor protein-2 component of clathrin-coated pits and thus does not mediate internalization of an interacting receptor (Vrecl et al., 2004). The lack of interaction with components of the internalization machinery reduces the effect of βarr2-mediated receptor internalization on the BRET2 signal (Vrecl et al., 2004). Furthermore, when measuring loss of cell surface GLP-1 receptors, GLP-1 and oxyntomodulin were observed to induce GLP-1 receptor internalization with similar kinetics and efficacy. Thus, the observed partial efficacy of oxyntomodulin in BRET2 assays is not secondary to different abilities of GLP-1 and oxyntomodulin to induce GLP-1 receptor internalization. The observation that oxyntomodulin is a partial agonist in recruiting βarrs to the GLP-1 receptor but a full agonist in inducing receptor internalization is supportive of a recent report suggesting that the GLP-1 receptor can use βarr-independent internalization pathways (Syme et al., 2006).
The exendin-4 peptide, which has been reported to be a full agonist on the GLP-1 receptor in functional assays, was observed here to be less efficacious than GLP-1 in the ability to recruit both βarr1 and βarr2 to the GLP-1 receptor (90% efficacy in βarr2 recruitment based on signal window, p < 0.001). A synthetic version of exendin-4 has recently been approved by the United States Food and Drug Administration for the treatment of noninsulin-dependent diabetes mellitus (Davidson et al., 2005). Under conditions with βarr2 and GRK5 overexpression, which favor βarr2 interaction, GLP-1 receptor-mediated cAMP accumulation is markedly reduced (Jorgensen et al., 2005). Exendin-4 and oxyntomodulin were observed to be full agonists, compared with GLP-1, also under these conditions. Thus, the observed differential efficacy of oxyntomodulin and exendin-4 in GLP-1 receptor-mediated cAMP signaling and βarr recruitment does not seem to be caused by a difference in experimental conditions of the two assays. Interestingly, examples of ligand-specific signaling where biased ligands selectively activate specific signaling pathways is now appearing in the literature (Kenakin, 2003). For the family B parathyroid hormone (PTH) 2 receptor, it has been demonstrated that, although the peptide agonists PTH and TIP39 both induced G-protein signaling, only TIP39 induced recruitment of βarrs to the receptor (Bisello et al., 2004). Likewise, PTH-1 receptor-biased ligands have been identified that activate G-protein signaling but not βarr recruitment (Bisello et al., 2002; Gesty-Palmer et al., 2006). By their differential efficacy profile in GLP-1 receptor-mediated cAMP accumulation versus GRK2 and βarr recruitment, oxyntomodulin, glucagon, and exendin-4 seem to be biased ligands on the GLP-1 receptor. Although partial agonism can be monitored when measuring on secondary messenger levels, it can potentially be disguised by spare receptors and amplification steps subsequent to receptor activation. In contrast, the here-applied BRET2 assays measure directly on GRK2 and βarr recruitment to the activated receptor. Thus, alternative to being true biased ligands on the GLP-1 receptor, it is possible that the partial efficacy observed here for oxyntomodulin, glucagon, and exendin-4 in βarr recruitment has remained unappreciated with other methods of measuring GLP-1 receptor activity such as cAMP accumulation. In either case, the different GLP-1 agonists would seem to preferentially stabilize distinct conformations of the GLP-1 receptor. This provides important information about structure activity relations of GLP-1 receptor ligands and could potentially improve the basis for design of GLP-1 receptor ligands, especially when targeting specific aspects of receptor function.
Interestingly, without GRK5 cotransfection, the potency of oxyntomodulin and glucagon in inducing βarr2 recruitment to the GLP-1 receptor was decreased approximately 10-fold, whereas the potency of GLP-1 remained unaffected. We have formerly described that GRK5 levels are crucial in obtaining a GLP-1 receptor/βarr2 complex that displays increased affinity for glucagon but not GLP-1 (Jorgensen et al., 2005). Thus, it seems that additional to factors inherent to the receptor, local concentrations of GRK5 might influence the pharmacological phenotype of the GLP-1 receptor and leave it more or less prone to oxyntomodulin- and glucagon-induced signaling. The apparent GRK5 effect on GLP-1 receptor pharmacology exemplifies how accessory proteins can function as receptor specificity-changing proteins and suggests an effect of GRK5 in regulating receptor signaling that extends the classic role in receptor desensitization. No cognate oxyntomodulin receptor has yet been identified, and experiments with transgenic mice lacking the GLP-1 receptor suggest that the inhibitory effect of oxyntomodulin on food intake is mediated by the cloned GLP-1 receptor (Baggio et al., 2004). However, the effect of oxyntomodulin was observed to be differential from the effect of exendin-4 (Baggio et al., 2004). Biased ligand properties of oxyntomodulin on the GLP-1 receptor could potentially constitute the basis for oxyntomodulin having physiological effects different from those of classic GLP-1 receptor agonists when administered in pharmacological doses. It remains possible that accessory proteins such as receptor activity-modifying protein or GRKs can change the pharmacological profile of the GLP-1 receptor toward an oxyntomodulin-preferring receptor (Sexton et al., 2006). Oxyntomodulin, especially when administered in pharmacological doses, could potentially exert effects through yet-unidentified cognate oxyntomodulin receptors or oxyntomodulin-preferring GLP-1 receptors in complex with accessory proteins. Although the GLP-1 receptor is a validated pharmaceutical target, it is still unknown whether the pharmaceutical use of oxyntomodulin will be affected by effects mediated by receptors other than the GLP-1 receptor.
Receptor interaction with βarr1 and βarr2 has been appreciated to not only exclude further G-protein interaction but in parallel mediate a number of signaling events by recruiting and scaffolding a multitude of proteins to the activated receptor (Lefkowitz and Shenoy, 2005). It has been demonstrated that βarrs can mediate G-protein independent activation of mitogen-activated protein (MAP) kinases upon activation through some 7TM receptors (Ren et al., 2005; Shenoy et al., 2006). GLP-1 and glucagon receptors have been reported to activate MAP kinase signaling through a G-protein-dependent mechanism, but the contribution of βarr-mediated signaling through these receptors has so far not been described (Jhala et al., 2003; Dalle et al., 2004). Recently, it has been demonstrated for the dopamine D2 7TM receptor that βarr2 interaction can mediate distinctive physiological effects independently of MAP kinases but dependent on a scaffolding function of βarr2 (Beaulieu et al., 2005). The finding described here that GLP-1 receptor agonists differentially affect GLP-1 receptor signaling pathways could aid future research on the contribution of specific signaling pathways to GLP-1 receptor-mediated cellular effects and physiology.
Acknowledgments
We thank Susanne Hummelgaard, Kate Hansen, and Helle Iversen for excellent technical assistance.
Footnotes
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.107.120006.
-
ABBREVIATIONS: 7TM, seven transmembrane; GLP, glucagon-like peptide; GRK, G-protein-coupled receptor kinase; GFP, green fluorescent protein; βarr, β-arrestin; RLuc, Renilla luciferase; BRET, bioluminescence resonance energy transfer; wt, wild type; PTH, parathyroid hormone; MAP, mitogen-activated protein.
-
↵
 The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material. - Received January 16, 2007.
- Accepted March 28, 2007.

- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}