


Abstract
Group II metabotropic glutamate receptors (mGluRs), mGluR2 and mGluR3, play a number of important roles in mammalian brain and represent exciting new targets for certain central nervous system disorders. We now report synthesis and characterization of a novel family of derivatives of dihydrobenzo[1,4]diazepin-2-one that are selective negative allosteric modulators for group II mGluRs. These compounds inhibit both mGluR2 and mGluR3 but have no activity at group I and III mGluRs. The novel mGluR2/3 antagonists also potently block mGluR2/3-mediated inhibition of the field excitatory postsynaptic potentials at the perforant path synapse in hippocampal slices. These compounds induce a rightward shift and decrease the maximal response in the glutamate concentration-response relationship, consistent with a noncompetitive antagonist mechanism of action. Furthermore, radioligand binding studies revealed no effect on binding of the orthosteric antagonist [3H]LY341495 [2S-2-amino-2-(1S,2S-2-carboxycyclopropan-1-yl)-3-(xanth-9-yl)propionic acid]. Site-directed mutagenesis revealed that a single point mutation in transmembrane V (N735D), previously shown to be an important residue for potentiation activity of the mGluR2 allosteric potentiator LY487379 [N-(4-(2-methoxyphenoxy)phenyl)-N-(2,2,2-trifluoroethylsulfonyl)pyrid-3-ylmethylamine], is not critical for the inhibitory activity of negative allosteric modulators of group II mGluRs. However, this single mutation in human GluR2 almost completely blocked the enhancing activity of biphenyl-indanone A, a novel allosteric potentiator of mGluR2. Our data suggest that these two positive allosteric modulators of mGluR2 may share a common binding site and that this site may be distinct from the binding site for the new negative allosteric modulators of group II mGluRs.
The eight known subtypes of metabotropic glutamate receptors (mGluRs) have been classified based on sequence homology, pharmacology, and signal transduction. These include group I (mGluR1 and 5), group II (mGluR2 and 3), and group III receptors (mGluR4, 6, 7, and 8). The group I receptors couple to Gαq and phospholipase C, whereas group II and group III mGluRs couple to Gαi (Conn and Pin, 1997; Schoepp et al., 1999). A large body of in vitro and in vivo preclinical studies suggest that specific mGluR subtypes play a broad range of neuromodulatory roles in different central nervous system circuits and that specific subtypes may provide viable targets for novel treatment strategies for a range of neurological and psychiatric disorders, including anxiety (Linden et al., 2005; Swanson et al., 2005), pain (Fisher et al., 2002; Adwanikar et al., 2004), Parkinson's disease (Conn et al., 2005), schizophrenia (Moghaddam, 2004; Kinney et al., 2005; Shipe et al., 2005), and cognitive disorders (Campbell et al., 2004; Homayoun et al., 2004; Moghaddam, 2004). Thus, compounds that are selective for a specific subtype of mGluR could provide a valuable tool to further investigate the involvement of these receptors in various diseases.
Group II mGluRs (mGluR2/3) are abundantly expressed in forebrain regions, such as cortex, hippocampus, striatum, and amygdala (Ohishi et al., 1998). Activation of group II mGluRs by group-selective agonists, including (-)-2-oxa-4-aminobicyclo[3.1.0]hexane-4,6-dicarboxylate (LY379268) and methyl substitution of 2-aminobicyclo[3.1.0]hexane 2,6-dicarboxylate (LY354740), leads to robust anxiolytic-like effects and antipsychotic-like activity in rodents (Carter et al., 2004; Linden et al., 2005) and has also shown anxiolytic activity in humans (Grillon et al., 2003). Whereas the therapeutic significance of group II mGluR antagonists has not been widely investigated, recent studies of selective competitive group II mGluR antagonists, including 2S-2-amino-2-(1S,2S-2-carboxycyclopropan-1-yl)-3-(xanth-9-yl)propionic acid (LY341495) and (1R,2R,3R,5R,6R)-2-amino-3-(3, 4-dichlorobenzyloxy)-6-fluorobicyclo[3.1.0] hexane-2,6-dicarboxylic acid (MGS0039), have suggested that these compounds exhibit antidepressant-like activity and antiobsessive-compulsive disorder-like effects in animal models (Chaki et al., 2004; Shimazaki et al., 2004; Palucha and Pilc, 2005).
Due to the high level of conservation of the orthosteric binding site of mGluRs, it has proven difficult to develop subtype-specific ligands for these receptors. As such, the most widely used orthosteric antagonists for group II mGluRs show some level of activity at all mGluR subtypes (Kingston et al., 1998). However, major advances have been made in developing highly selective antagonists of group I mGluRs by targeting allosteric sites on the receptor to non-competitively block receptor function (Gasparini et al., 1999; Lavreysen et al., 2003). The ability to achieve higher selectivity with these compounds is probably due to the fact that they bind within the seven-transmembrane (TM)-spanning domain of the mGluR, which is less highly conserved than the glutamate binding pocket. Moreover, allosteric antagonists may provide other advantages in that their activity is not altered by the presence of competing orthosteric agonists.
Recently, the discovery of two highly selective allosteric potentiators of mGluR2, N-(4-(2-methoxyphenoxy)phenyl)-N-(2,2,2-trifluoroethylsulfonyl)pyrid-3-ylmethylamine (LY487379) (Johnson et al., 2003) and biphenyl-indanone A (BINA) (Galici et al., 2006), has provided excellent tools to selectively activate this group II mGluR subtype. Less progress has been achieved with the discovery of negative allosteric modulators (allosteric antagonists) of group II mGluRs. However, preliminary report presented in abstract form suggested that a series of dihydro-benzo[1,4]diazepin-2-one derivatives may have allosteric activity at the group II mGluRs (Gatti et al., 2001). Based on this finding, we synthesized a series of compounds based on the scaffold described by Adam et al. (2003) and determined the activity of these molecules at group II mGluRs. Here we report that these compounds provide a novel family of potent and negative allosteric modulators that are active at both group II mGluR subtypes but are without effects on group I and group III mGluRs. These compounds are useful in both cell lines and in blocking electrophysiological effects of group II mGluRs in brain slices. Interestingly, these compounds do not alter binding to the orthosteric site and may act at an allosteric site that is distinct from that of recently described positive allosteric modulators that are selective for mGluR2.
Materials and Methods
Materials. All tissue culture reagents were obtained from Invitrogen (Carlsbad, CA). G418 sulfate was obtained from Mediatech, Inc., (Herndon, VA). [3H]LY341495 (28.28 Ci/mmol) was obtained from American Radiolabeled Chemicals, Inc. (St Louis, MO). l-Glutamate, DCG-IV, l-AP4 [l-(+)-2-amino-4-phosphonobutyric acid], and LY341495 were obtained from Tocris (Ellisville, MO). Methotrexate was purchased from Calbiochem (La Jolla, CA). BINA was synthesized as described previously (Galici et al., 2006). The Calcium 3 Assay Kit was obtained from Molecular Devices (Sunnyvale, CA). The indicator dye fluo-4 was obtained from Invitrogen. Probenecid, dimethyl sulfoxide (DMSO), puromycin dihydrochloride, GDP, and guanosine 5′-3-O-(thio)triphosphate were purchased from Sigma-Aldrich, Inc., (St. Louis, MO). Unifilter-96 GF/B plates and MicroScint-20 were obtained from PerkinElmer Life and Analytical Sciences (Boston, MA). BioCoat poly-d-lysine 96-well culture plates were obtained from BD Biosciences Discovery Labware (Bedford, MA). QuikChange site-directed mutagenesis kit and Pfu Ultra high fidelity DNA polymerase were obtained from Stratagene (La Jolla, CA). Complimentary oligonucleotides were obtained from Operon Biotechnologies (Huntsville, AL). All test compounds were synthesized as described previously (Adam et al., 2003); the synthesis of three representative compounds is described here.
MNI-135.[3-(7-iodo-4-oxo-4,5-dihydro-3H-benzo[1,4]diazepin-2-yl)-benzonitrile]. Under N2, trifluoroacetic acid (30 mmol) was slowly added at 0°C to a solution of [4-iodo-2-[3-(2-cyano-pyridin-4-yl)-3-oxo-propionylamino]-phenyl]-carbamic acid tert-butyl ester (1.9 mmol) in dichloromethane (20 ml). The reaction was stirred 7 h at room temperature. The mixture was washed with 10% aqueous NaHCO3, the organic layer was dried over Na2SO4, and the solvent was removed on the rotary evaporator. The residue was purified by flash chromatography on silica gel using 60:40 hexane-ethyl acetate to yield 610 mg (80%) of MNI-135 as a white solid. Mp: 226°C; 1H NMR (DMSO), δ ppm: 3.60 (2H, s), 7.21 (1H,d, J = 9.2 Hz), 7.55–7.57 (2H, m), 7.74 (1H,t, J = 7.6 Hz), 8.03 (1H,d, J = 7.6 Hz), 8.35 (1H, d, J = 8 Hz), 8.46 (1H, s), 10.62 (1H, s); 13C NMR (DMSO) δ ppm: 40.0, 91.4, 112.1, 118.3, 129.8, 130.0, 130.2, 131.2, 131.6, 132.1, 132.7, 134.5, 138.0, 138.6, 157.2, 165.9; Anal. (C16H10IN3O) C, H, N, MS: 388.3 (M + 1).
MNI-136.[7-bromo-4-(3-pyridin-3-yl-phenyl)-1,3-dihydro-benzo [1,4] diazepin-2-one]. Under N2, N-(4-bromo-2-nitro-phenyl)-3-oxo-3-(3-pyridin-3-yl-phenyl)-propionamide (1.9 mmol) was dissolved in ethanol, and tin chloride (5.7 mmol) was added. The mixture was stirred 7 h at reflux. The solvent was evaporated, and the residue was basified with 1 N NaOH (pH 7–8) and extracted with ethyl acetate. The organic layer was dried over Na2SO4, and the solvent was removed on the rotary evaporator. The residue was purified by chromatography on silica gel using 70:30 hexane-ethyl acetate to yield 650 mg (86%) of MNI-136 as a white solid. Mp: 218°C; 1H NMR (DMSO) δ ppm 3.66 (2H, s), 7.16 (1H,d, J = 8.8 Hz), 7.46 (1H, dd, J = 6 Hz, J = 9.2 Hz), 7.53–7.56 (1H, m), 7.64 (1H,d, J = 2.3 Hz), 7.69 (1H,t, J = 7.6 Hz), 7.95 (1H, d, 8.8 Hz), 8.12 (1H,d, J = 7.6 Hz), 8.18 (1H,d, J = 8 Hz), 8.36 (1H, s), 8.62–8.64 (1H, m), 8.97–8.99 (1H, m), 10.71 (1H, s); 13C NMR (DMSO) δ ppm: 40.1, 115.8, 123.9, 124.0, 126.2, 127.3, 128.9, 129.6, 129.7, 129.8, 129.9, 134.3, 135.0, 137.6, 137.8, 140.7, 147.8, 148.9, 159.5, 166.0; Anal. (C20H14BrN3O) C, H, N, MS: 393.6 (M + 1).
MNI-137.[4-(7-bromo-4-oxo-4,5-dihydro-3H-benzo[1,4]diazepin-2-yl)-pyridine-2-carbonitrile]. Under N2, trifluoroacetic acid (30 mmol) was slowly added at 0°C to a solution of [4-bromo-2-[3-(2-cyanopyridin-4-yl)-3-oxo-propionylamino]-phenyl]-carbamic acid tert-butyl ester (3 mmol) in dichloromethane (20 ml). The reaction was stirred 7 h at room temperature. The mixture was washed with 10% NaHCO3, the organic layer was dried over Na2SO4, and the solvent was removed on the rotary evaporator. The residue was purified by chromatography on silica gel using 60:40 hexane-ethyl acetate to yield 610 mg (80%) of MNI-137 as a white solid. Mp: 238°C; 1H NMR (DMSO) δ ppm: 3.65 (2H, s), 7.42 (3H, m), 8.25 (1H, dd, J = 1 Hz, J = 5 Hz), 8.54 (1H, s), 8.92 (1H,d, J = 5 Hz), 10.79 (1H, s); 13C NMR (DMSO), δ ppm 40.2, 117.3, 119.5, 124.2, 125.1, 126.6, 127.1, 130.1, 131.7, 133.6, 137.8, 145.2, 152.3, 155.7, 165.7; Anal. (C15H9BrN4O) C, H, N, MS: 342.3 (M + 1).
Cell Culture and Transfections. Baby hamster kidney cells stably expressing the rat mGluR1a (rmGluR1a) were generously provided by Dr. Betty Haldeman (Zymogenetics, Seattle, WA). Cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 5% heat-inactivated fetal bovine serum (FBS), 2 mM l-glutamine (GlutaMAX I; Invitrogen), antibiotic-antimycotic (100 U of penicillin, 100 μg of streptomycin, and 0.25 μg of amphotericin B), 1 mM sodium pyruvate, 20 mM HEPES, and 250 nM methotrexate. Rat mGluR2 and the promiscuous G protein, Gqi5, were cotransfected into HEK293A cells using Lipofectamine 2000 (Invitrogen) and were grown in DMEM containing 10% heat-inactivated FBS, 2 mM l-glutamine, antibiotic-antimycotic, 0.1 mM nonessential amino acids, and 20 mM HEPES. Chinese hamster ovary (CHO) cells stably expressing human mGluR2 (hmGluR2), which were transiently transfected with Gqi5, and CHO cells stably expressing both the hmGluR4 and Gqi5 were grown in DMEM containing 10% heat-inactivated dialyzed FBS, 2 mM l-glutamine, antibiotic-antimycotic, 1 mM sodium pyruvate, 20 mM HEPES, 5 nM methotrexate, and 20 μg/ml l-proline. CHO cells stably expressing the rmGluR3 were grown in DMEM containing 10% heat-inactivated FBS, 2 mM l-glutamine, 100 U of penicillin, 100 μg of streptomycin, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, and 20 mM HEPES. The rat mGluR8/Gqi9/CHO cell line was a generous gift of Jarda Wroblewska (Georgetown University, Washington, DC) and was maintained in 90% DMEM, 10% FBS, 100 U/ml penicillin/streptomycin, 20 mM HEPES (pH 7.3), 1 mM sodium pyruvate, and 2 mM glutamine supplemented with 800 μg/ml G418. HEK cells stably expressing rat mGluR7a were grown in 45% DMEM, 45% Ham's F12, 10% FBS, 100 U/ml penicillin/streptomycin, 20 mM HEPES (pH 7.3), 1 mM sodium pyruvate, 2 mM glutamine, 600 ng/ml puromycin dihydrochloride, and 700 μg/ml G418 at 37°C in the presence of 5% CO2.
Functional Calcium Mobilization Assay for mGluRs 1, 2, 4, and 5. Recombinant cell lines, including mGluR1, 2, 4, and 5, were plated at a seeding density of 7 to 8 × 105 cells/well in clear-bottomed, poly-d-lysine-coated 96-well plates. Cells were then incubated in glutamate/glutamine-free medium overnight at 37°C in an atmosphere of 95% O2/5%CO2, with the exception of baby hamster kidney cells stably expressing rmGluR1a, which were maintained in regular medium. Cells were loaded with calcium indicator dye (Calcium 3 Assay Kit) at 37°C for 1 h. Dye was removed and replaced with the appropriate volume of assay buffer containing 1× Hanks' balanced salt solution (HBSS), 20 mM HEPES, and 2.5 mM probenecid, pH 7.4. At this stage, cells were used for the calcium mobilization assay.
All test compounds were dissolved in 100% DMSO and then serially diluted into assay buffer containing 0.1% bovine serum albumin fora5× stock. The stock solution was added to the assay plate to a final DMSO concentration of 0.1%. Glutamate and l-AP4 were prepared as a 10× stock solution in assay buffer before addition to assay plates. Calcium mobilization was measured using the FLEXstation II (Molecular Devices). The signal amplitude was normalized as a percentage of the response to the nearly maximal concentration of glutamate (EC80) or as a percentage of the maximal response to glutamate (10 μM).
Functional Calcium Mobilization Assay for mGluR8. The rat mGluR8/Gqi9/CHO cell lines were plated at a seeding density of 4 × 105 cells/well in clear-bottomed, poly-d-lysine-coated 384-well plates (black-walled) in DMEM containing 10% FBS, 100 U/ml penicillin/streptomycin and 20 mM HEPES. Cells were plated in the morning and incubated at 42°C for 2 h in the afternoon before analysis (“heat shock”) and then incubated overnight at 37°C in the presence of 5% CO2. On the day of the assay, the medium was removed, and 0.9 μM indicator dye fluo-4 was added in HBSS containing 20 mM HEPES, pH 7.3 (20 μl/well). Cells were incubated for 1 h at room temperature, and the dye was replaced with 20 μl/well HBSS. Test compounds were dissolved as a 10 mM stock in 100% DMSO and serially diluted in DMSO. Test compound plates were prepared in HBSS at 2.5× their final concentration in 0.25% DMSO; l-AP4 was at 5× the final concentration to be assayed in HBSS. Cell plates and compound plates were loaded onto a Hamamatsu FDSS 6000 kinetic imaging plate reader (Hamamatsu Corporation, Bridgewater, NJ). The assay was initiated by collecting 10 images at 1 Hz before the addition of the test compound. Test compound (20 μl/well) was then added, and data were collected for 5 min. After this 5-min period, 10 μl of vehicle or agonist was added, and data were collected for an additional 2 min. Functional response was quantified using Microsoft Excel by calculating the maximal fluorescence generated during the time window after agonist addition and subtracting the fluorescence obtained in the vehicle control wells. The signal amplitude was then normalized as the percentage of the response to the EC80 concentration of l-AP4.

All test compounds produced a concentration-dependent inhibition of glutamate-induced responses in HEK293A cells expressing rm-GluR2, as assessed by calcium mobilization assay (A) or in membranes expressing rmGluR3, as assessed by [35S]GTPγS binding assay (B). Cells were preincubated for 5 min with various concentrations of test compounds before the addition of a nearly maximal concentration (EC80) of glutamate. Changes in calcium mobilization were measured using the FLEXstation II. For the [35S]GTPγS assay, membranes were prepared from CHO cells stably expressing rmGluR3. The assay mixtures contained 10 μg of membrane protein, test compound, glutamate, 0.1 nM [35S]GTPγS, and assay buffer. The mixtures were incubated at 30°C for 60 min, and the binding equilibrium was terminated by rapid filtration through Unifilter-96 GF/B filter plates. The radioactivity was determined by TopCount NXT. Data were normalized as a percentage of the maximal response to 31.6 or 10 μM glutamate for [35S]GTPγS binding and calcium mobilization assays, respectively. Data are presented as the mean of three individual experiments performed in triplicate, with error bars representing mean ± S.E.M.
Functional Cyclase Inhibition Assay-mGluR7. HEK cells stably expressing rat mGluR7a were plated in clear-bottomed, poly-d-lysine-coated 96-well plates 1 day before assay. After serum starving for 2 h, cells were equilibrated to assay buffer (DMEM, 20 mM HEPES, and 0.025% ascorbic acid) for 10 min at room temperature. Compounds were serially diluted in 100% DMSO. Cells were then incubated with 500 μM 3-isobutyl-1-methylxanthine and test compounds or appropriate vehicle for 30 min at 37°C. Forskolin (15 μM) and an EC80 concentration, l-AP4, were then added to cells and incubated at 37°C for an additional 20 min. Drug solutions were decanted from cells, and 3% trichloroacetic acid was added for >2 h at 4°C to extract intracellular cAMP. Samples were then incubated with bovine protein kinase A and [3H]cAMP (1 nM) for 2 h on ice. After harvesting samples onto a GF/B filter plate (PerkinElmer Life and Analytical Sciences), samples were counted in a Packard TopCount (PerkinElmer Life and Analytical Sciences, Waltham, MA), and [cAMP] in each sample was determined based on a standard curve of unlabeled cAMP. The effect of the test compounds was then normalized as a percentage of the response to the EC80 concentration of l-AP4.

A, similar to results observed with rmGluR2, test compounds exhibited inhibitory effects in a calcium mobilization assay in cells expressing hmGluR2. B, a statistically significant (p < 0.0001) correlation was observed between the IC50 values obtained from cells expressing rmGluR2 and hmGluR2 for all test compounds evaluated in a glutamate-induced calcium mobilization assay. Data were analyzed using linear regression analysis and are the mean of three independent experiments performed in triplicate. Error bars represent mean ± S.E.M.
Membrane Preparation. Membranes from cell lines expressing hmGluR2 or rmGluR3 were prepared as described previously (Johnson et al., 1999; Schaffhauser et al., 2003). In brief, confluent cells of both cell lines were washed once with ice-cold phosphate-buffered saline. hmGluR2-expressing cell lines were harvested and resuspended in ice-cold binding buffer containing 10 mM potassium phosphate monobasic and 100 mM potassium bromide, pH 7.6, whereas rmGluR3-expressing cell lines were harvested and resuspended in ice-cold binding buffer 1 (20 mM HEPES and 10 mM EDTA, pH 7.4). Once harvested, cells were homogenized on ice using a Polytron homogenizer (Tekmar, Inc., Cincinnati, OH) for 2 s. The homogenate was centrifuged at 40,000g for 20 min at 4°C. This last step was repeated two additional times, homogenizing between centrifugations for 10 and 5 s, respectively, with the exception of rmGluR3 pellets, which were resuspended in ice-cold binding buffer 2 (20 mM HEPES and 0.1 mM EDTA, pH 7.4) in the final centrifugation. The final pellets were resuspended and homogenized on ice using a Dounce glass homogenizer (Fisher Scientific Co., Pittsburgh, PA) and were stored in aliquots at -80°C until use. Protein concentrations were measured using the Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules, CA) using serum albumin as the standard (Pierce, Rockford, IL).
Radioligand Binding Studies. After thawing, the hmGluR2 membranes were resuspended and homogenized in ice-cold binding buffer as described above using a Dounce glass homogenizer. Binding experiments were prepared as described previously (Johnson et al., 1999). Test compounds were dissolved in 100% DMSO and then serially diluted in binding buffer to yield a 5× stock. The stock solution was added to the assay plate to a final DMSO concentration of 0.1%. All binding reactions were performed in 96-well deep plates in a final volume of 100 μl. [3H]LY341495 binding assay mixtures contained 15 μg of membrane protein, 1 nM [3H]LY341495, and various concentrations of test compounds. The assay mixtures were incubated on ice for 30 min. Nonspecific binding was determined in the presence of 1 mM glutamate.

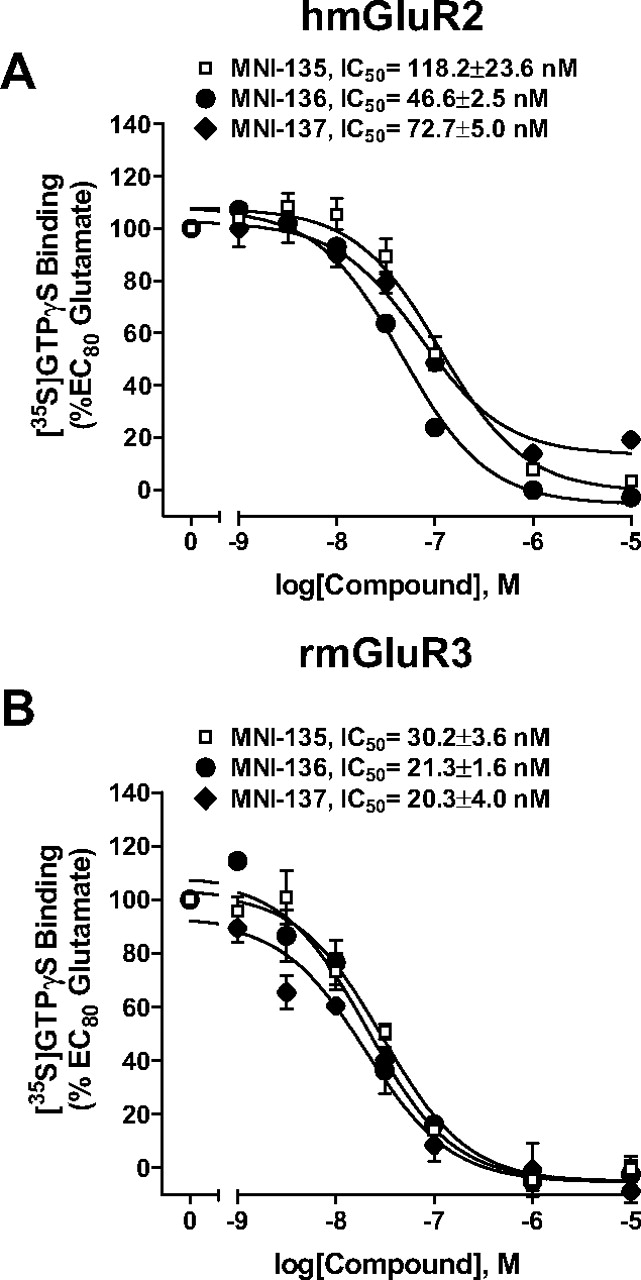
Three representative antagonists inhibited an EC80 concentration of glutamate-induced [35S]GTPγS binding in membranes from cell lines stably expressing hmGluR2 or rmGluR3 in a concentration-dependent manner. Results are expressed as the percentage of the maximal stimulation induced by 1 mM or 31.6 μM glutamate for hmGluR2 and rm-GluR3, respectively. Data are presented as the mean of three individual experiments performed in triplicate with error bars representing mean ± S.E.M.
The binding equilibrium was terminated by rapid filtration through Unifilter-96 GF/B filter plates (presoaked with ice-cold binding buffer), and the filter plates were washed three times with ice-cold binding buffer using a 96-well Brandel harvester (Brandel Inc., Gaithersburg, MD). Filter plates were dried and filled with 30 μl of MicroScint-20, and the radioactivity was determined by TopCount NXT microplate scintillation and luminescence counter (PerkinElmer Life and Analytical Sciences).
[35S]GTPγS Binding Studies. rmGluR3 membranes were thawed and homogenized using a glass homogenizer in ice-cold binding buffer containing 50 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 150 mM NaCl, 1 mM EDTA, 10 μg/ml saponin, and 1 μM GDP. Assay mixtures contained 10 μg of membrane protein, test compound, glutamate, 0.1 nM [35S]GTPγS, and assay buffer to yield a total volume of 100 μl. Nonspecific binding was determined in the presence of 10 μM unlabeled guanosine 5′-3-O-(thio)triphosphate. The assay mixtures were incubated at 30°C for 60 min. The reaction was terminated in a manner similar to that described above in radioligand binding studies.
Site-Directed Mutagenesis. The cDNA encoding human mGluR2 in the pGTh backbone (Grinnell et al., 1991) was generously provided by Dr. M. Baez (Eli Lilly & Co., Indianapolis, IN). Point mutations were generated using the QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. Complimentary oligonucleotides were designed to contain the desired mutations and a novel restriction site, which does not alter the amino acid sequence, to be used for screening purposes. Sense and antisense oligonucleotides were based on the following sequences and were used to introduce three point mutations into the human mGluR2 sequence. 5′-CTGCCTGGCACTTATACTAGTCCAGCTGCTCATCGTG-3′ was used for generation of two consecutive point mutations (S688L, G689V) along with a novel SpeI site, and 5′-GGCTCGCTGGCCTACGACGTCCTCCTCATCGCGCTC-3′ was used to introduce a third single mutation (N735D) in combination with a novel AatII site. PCR amplification was performed using Pfu Ultra high-fidelity DNA polymerase. The mutated hmGluR2 sequence was dropped out of the pGTh backbone using flanking SalI sites and subcloned into the pcDNA 3.1+ vector from Invitrogen at the complimentary XhoI site. Final constructs were verified by sequencing using an Applied Biosciences DNA analysis system at the Vanderbilt University DNA sequencing facility.
Field Potential Recording in Hippocampus Slice. Hippocampal brain slices were prepared from 3 to 4-week-old male Sprague-Dawley rats as described previously with minor modifications (Macek et al., 1996, 1998). Brains were rapidly removed and submerged in an ice-cold sucrose replacement solution containing 200 mM sucrose, 2.5 mM KCl, 1.2 mM NaH2PO4, 1.3 mM MgCl2, 8 mM MgSO4, 20 mM glucose, and 46 mM NaHCO3, equilibrated with 95% O2/5% CO2. Coronal hippocampal slices (400-μm thick) were made using a microtome (Leica Microsystems Inc., Bannockburn, IL). The slices were placed in a holding chamber containing artificial cerebrospinal fluid (ACSF) comprising 124 mM NaCl, 2.5 mM KCl, 1.3 mM MgSO4, 1.0 mM NaH2PO4, 2 mM CaCl2, 20 mM glucose, and 26 mM NaHCO3, equilibrated with 95% O2/5% CO2, which was maintained at room temperature. To increase slice viability, 5 μM glutathione, 500 μM pyruvate, and 250 μM kynurenic acid were added to the sucrose replacement buffer, maintaining ACSF buffer in all experiments.

Three representative antagonists exhibited no effect toward rmGluR1, rmGluR5, hmGluR4, rmGluR7, or rmGluR8 in functional assays. Increasing concentrations of antagonists were unable to inhibit an EC80 concentration of glutamate- or l-AP4-induced function in cells lines expressing rmGluR1 (A), rmGluR5 (B), and hmGluR4 (C), or rmGluR8 (D) using calcium mobilization assay or rmGluR7 (E) using a cyclase inhibition assay. Data were analyzed using nonlinear regression analysis and are the mean of three separate experiments performed with a number of three (A-C), four (E), or eight (D) independent replicates per experiment. Error bars represent mean ± S.E.M.
After a recovery period of at least 1 h, a slice was transferred to a submerged brain slice recording chamber, where it was perfused continuously with oxygenated ACSF at 2 to 3 ml/min at room temperature. Recording electrodes were pulled on a Narishige (East Meadow, NY) vertical patch pipette puller from a borosilicate glass and filled with 2.5 mM NaCl, in which the electrode resistance ranged from 0.3 to 1.0 MΩ. A bipolar tungsten electrode (FHC, Bowdoinham, ME) and recording electrode were placed in the middle third layer of the molecular layer at the dentate gyrus for stimulation of the medial perforant path (MPP) fibers. Stimuli (0.1 ms in duration) were delivered at 0.05 Hz using Grass S48 stimulator and Grass isolator (Grass Technologies, West Warrick, RI). In each experiment, an input-output curve was performed, and the stimulus intensity was adjusted to produce a field excitatory postsynaptic potential (fEPSP) approximately 50 to 70% of the maximal response.
The fEPSPs were recorded by an Axon MultiClamp 700B amplifier (Molecular Devices) in current clamp mode. Data were filtered at 2 kHz, digitized with DigiData 1322A (Molecular Devices), and acquired by the pClampex 9.2 program (Molecular Devices). The fEPSP slope was measured by pClampfit 9.2 program (Molecular Devices). The starting six consecutive slopes were averaged to define the baseline fEPSP slope value for a given experiment. The subsequent fEPSP slopes in the experiment were normalized to the baseline fEPSP slope value and expressed as a percentage. Six consecutive fEPSP slopes after 12 min of application of DCG-IV were averaged and defined as the value of applying DCG-IV. All normalized fEPSP slopes for each concentration of DCG-IV were then expressed as the mean ± S.E.M. All compounds were dissolved in double deionized water with 1 N sodium hydroxide, with the exception of MNI-137, which was dissolved in DMSO, and was then diluted with ACSF to the desired concentration. All compounds were added to the recording chamber via addition to the perfusion solution.

Concentration-response curves of glutamate-induced calcium mobilization were inhibited in a noncompetitive manner by three representative antagonists (31.6 nM) in HEK293A cells expressing rmGluR2. Data are expressed as a percentage of the maximal response to glutamate (10 μM) and are the mean of three individual experiments performed in triplicate. Error bars represent mean ± S.E.M.
Data Analysis. For calcium mobilization, radioligand binding and [35S]GTPγS binding studies, data analysis was performed using GraphPad Prism 3.0 (GraphPad Software Inc., San Diego, CA). Concentration-response curves were analyzed using a nonlinear regression analysis and were generated from the means of three separate experiments. Inhibition curves were fitted by nonlinear regression analysis using the one-site competition equation. Error bars represent the mean ± S.E.M. For the electrophysiology experiments, data were analyzed using pClampfit 9.2. All results are expressed as mean ±S.E.M., and statistical significance was determined using Student's t test.
Results
Negative Allosteric Modulators Have Inhibitory Activity for Group II mGluRs. We synthesized a range of novel molecules based on the structure reported by Adam et al. (2003). We first examined their inhibitory activity on cells expressing rmGluR2 by employing a calcium mobilization assay. Consistent with action as antagonists of group II mGluRs, all test compounds blocked a glutamate-induced increase in calcium (Fig. 1A). A similar effect was observed using [35S]GTPγS binding studies to measure activation of rmGluR3 overexpressed in CHO cells, as [35S]GTPγS binding was attenuated in these cells following an application of test compounds (Fig. 1B). Inhibitory effects of these compounds on calcium mobilization were also observed in CHO cells stably expressing hmGluR2 (Fig. 2A). For the calcium mobilization assay, a highly significant positive correlation (r2 = 0.958, p < 0.0001) was observed between the IC50 values obtained from cells expressing rmGluR2 and hmGluR2 (Fig. 2B), suggesting that there is no species specificity exhibited within this series.

Three representative antagonists had no effect on [3H]LY341495 binding. Membranes were prepared from CHO cells stably expressing hm-GluR2 and were incubated with 1 nM [3H]LY341495 in a final volume of 100 μl for 30 min at 4°C in the presence of varying concentrations of LY341495 or three representative antagonists. Bound and free radioligand were separated by vacuum filtration through Unifilter-96 GF/B filter plates. Nonspecific binding was determined in the presence of 1 mM glutamate. The radioactivity was determined by TopCount NXT. A, binding of 1 nM [3H]LY341495 was displaced by cold LY341495 but not by the three representative compounds tested. B, the presence of three representative compounds (1 μM) did not affect the displacement of [3H]LY341495 by glutamate. The graphs summarize data from three independent experiments performed in triplicate. Error bars represent mean ± S.E.M.
Negative Allosteric Modulators Are Selective for Group II Relative to Group I and III mGluRs. Three compounds (MNI-135, MNI-136, and MNI-137) that exhibited the most potent inhibitory activity in the calcium mobilization assay at rmGluR2 were selected for further studies aimed at determining their selectivity for specific mGluR subtypes. Similar to their activity observed in the calcium mobilization assay, these three representative compounds inhibited glutamate-induced [35S]GTPγS activation in cells expressing hmGluR2 (Fig. 3A). Comparison of these data with the effects of these compounds on rmGluR3 (Fig. 3B), as assessed by [35S]GTPγS binding, revealed that these compounds were slightly more potent at inhibiting glutamate-induced activation of mGluR3 relative to mGluR2. However, none of the compounds provided sufficient selectivity between the group II mGluRs to be useful for differentiating between the group II mGluR subtypes. To further characterize the selectivity of these compounds, we used the calcium mobilization assay with cells expressing mGluR1, 4, 5, or 8, as well as a cyclase inhibition assay with cells expressing mGluR7. None of these compounds exhibited inhibitory activity induced by activation of any of these receptor subtypes at concentrations up to 10 μM (Fig. 4, A–E).
MNI-135, MNI-136, and MNI-137 Are Noncompetitive Group II mGluR Antagonists. To characterize the mechanism of inhibition of the novel negative allosteric modulators, the effect of one single concentration (31.6 nM) of each of these compounds on the glutamate concentration-response curves was assessed. In cells expressing rmGluR2, all three compounds shifted glutamate concentration-response curves in a calcium mobilization assay to the right with a concomitant decrease in the maximal response (Fig. 5). These results are consistent with a noncompetitive mode of action. To confirm that these compounds act as noncompetitive antagonists, we determined their ability to bind to the orthosteric site by measuring their ability to displace the binding of [3H]LY341495, a potent orthosteric antagonist of group II mGluRs. [3H]LY341495 binding was measured in CHO cell membranes stably expressing hmGluR2. As shown in Fig. 6A, the three compounds tested had no effect on binding of 1 nM [3H]LY341495 at concentrations up to 10 μM, whereas radioactive LY341495 potently displaced binding of the radioligand. The ability of glutamate to displace the binding of 1 nM [3H]LY341495 was not altered in the presence of these compounds (Fig. 6B). For comparison, the potency of inhibition of all test compounds on glutamate-induced responses for both calcium mobilization and [35S]GTPγS binding assay are summarized in Table 1.
IC50 values for the inhibition of an EC80 concentration of glutamate-induced response in both calcium mobilization and [35S]GTPγS binding assay on cells expressing rmGluR2, hmGluR2 or rmGluR3. Error expressed as S.E.M.
MNI-137 Inhibits Group II mGluR Agonist-Mediated Responses at Medial Perforant Path-Dentate Gyrus Synapses in Rat Hippocampal Slices. Previous findings revealed that group II mGluRs, the predominant subtype at the medial perforant path-dentate granule cell synapse (MPP-DG) inhibits excitatory synaptic transmission by reducing glutamate release from presynaptic terminals (Macek et al., 1996, 1998). Thus, the MPP-DG synapses of the rat hippocampus were employed as a native system for determining the effects of MNI-137, a representative novel negative allosteric modulator identified in this study, on group II mGluR-mediated responses.
To assess the effects of activation of group II mGluRs on synaptic transmission at the MPP-DG synapse, extracellular field potentials were recorded from the middle third of the molecular layer in the dentate gyrus. Consistent with our previous results, bath application of the group II agonist DCG-IV induced a concentration-dependent decrease in fEP-SPs at this synapse (data not shown) (Galici et al., 2006). 1 μM DCG-IV inhibited the fEPSP slope by 60.31 ± 2.58% (n = 12) (Fig. 7, A and C). In addition, the effect of 1 μM DCG-IV was blocked by the orthosteric mGlu2/3 receptor antagonist LY341495 (1 μM), (data not shown). Consistent with the finding that MNI-137 inhibited glutamate-induced increases in [35S]GTPγS binding in cells expressing hmGluR2, with a maximal effect in the low micromolar range, 3 μM MNI-137 significantly blocked the effect of 1 μM DCG-IV on fEPSP slope (16.84 ± 3.01%; n = 8; *, p < 0.01) (Fig. 7, B and C).
Point Mutation of Residue Asn735 Located in Transmembrane V Abolishes Potentiation Activities of Positive Allosteric Modulators of mGluR2 but Not Negative Allosteric Modulators of Group II mGluRs. The present functional and radioligand binding studies suggest that these negative allosteric modulators do not bind to the orthosteric binding site (i.e., glutamate binding site). A previous study reported that the amino acid residues forming the binding pocket for a novel positive allosteric modulator for mGluR2, LY487379, are Ser688, Gly689, and Asn735 (Schaffhauser et al., 2003). We next sought to investigate whether these residues are also critical for the inhibitory activity of our novel negative allosteric modulators.

MNI-137 blocked DCG-IV-induced inhibition of the fEPSP slope at MPP-DG synapses in rat hippocampal slices. A, representative fEPSP traces depicting the effect of a 12-min application of 1 μM DCG-IV alone on the synaptic transmission at MPP-DG synapses. B, representative fEPSP traces depicting the effect of 1 μM DCG-IV following a 10-min preincubation with 3 μM MNI-137. Each representative fEPSP trace is an average of six consecutive evoked responses at this synapse. C, the bar graph illustrates the effects of application of 1 μM DCG-IV (n = 12) in the absence or presence of 3 μM MNI-137 (n = 8) on the fEPSP slope at MPP-DG synapses. The average of the fEPSP slope obtained from six traces immediately before and after compound application was compared to determine the effects of test compounds. All values are expressed as mean ± S.E.M, Calibration bar: 0.2 mV/5 ms. *, p < 0.01; Student's t test.
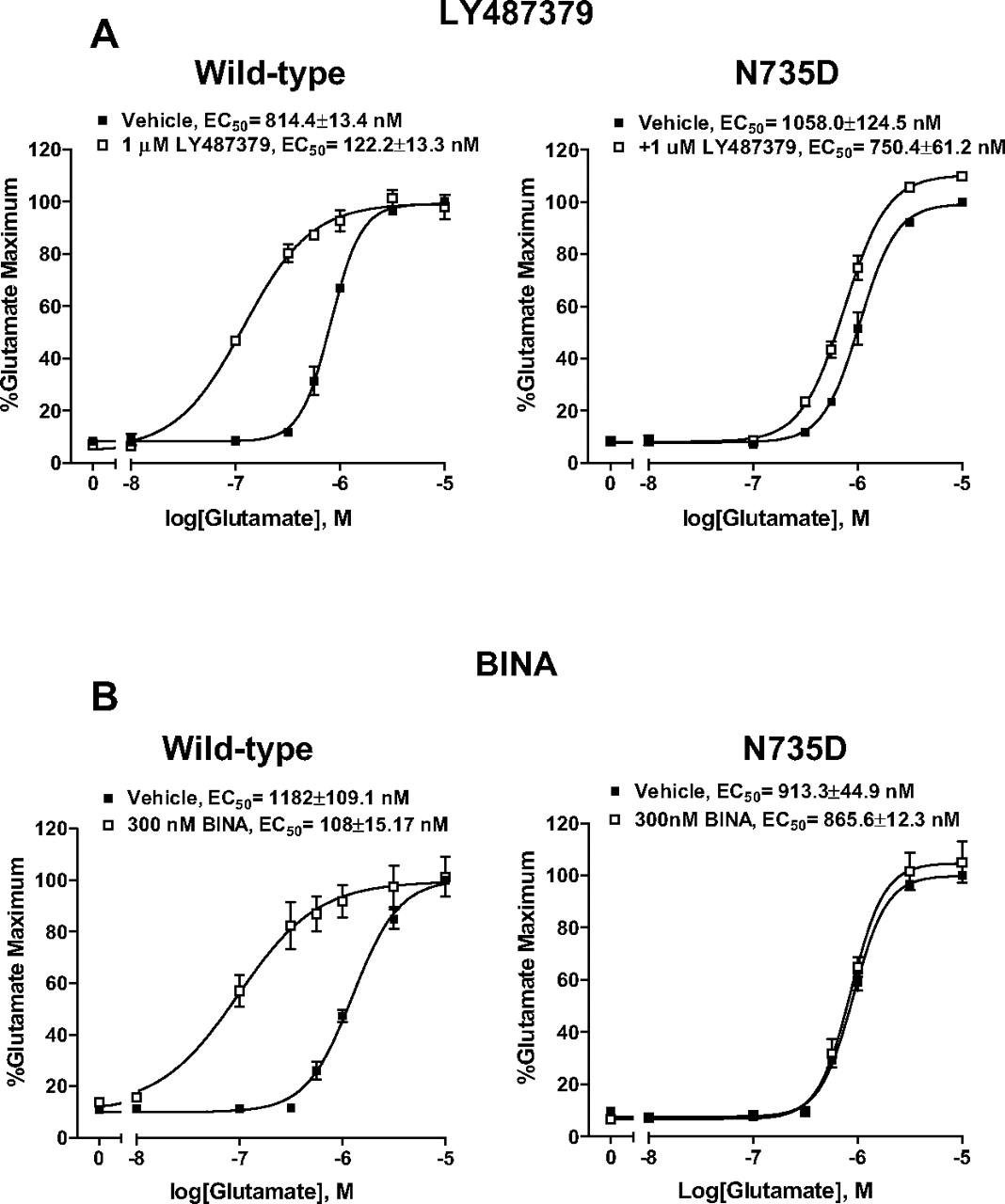
We first examined the enhancing property of LY487379 in HEK293A cells transiently expressing wild-type hmGluR2 or mutant hmGluR2 containing a single (N735D in TM V), double (S688L/G689V in TM IV), or triple (S688L/G689V/N735D) point mutation in the presence of single concentration of LY487379 (1 μM) as a control experiment. Consistent with previous studies, LY487379 induced parallel shifts of the glutamate concentration-response curve approximately 6 to 7-fold to the left in HEK293A cells transiently expressing wild-type hmGluR2 (Fig. 8A, left). The single point mutation, N735D, in TM V of hmGluR2 markedly attenuated the potentiation activity of LY487379 compared with wild type (Fig. 8A, right). In addition, approximately 30 and 85% reduction of the potentiation by LY487379 of glutamate-induced calcium mobilization was observed in mutant hm-GluR2 containing the double mutation (S688L/G689V) or triple mutation (S688L/G689V/N735D), respectively (data not shown). These results suggest that residue Asn735 plays a critical role for the enhancing activity of this modulator. Based on this finding, we sought to examine whether this residue was also critical for potentiation activity of a structurally distinct newly reported positive allosteric modulator of mGluR2, BINA. Similar to LY487379, BINA induced parallel shifts of the glutamate concentration-response curve approximately 11-fold to the left in HEK293A cells transiently expressing wild-type hmGluR2 (Fig. 8B, left) (data were from Galici et al. (2006). Potentiation activity of BINA was almost completely abolished in mutant hmGluR2 containing N735D (Fig. 8B, right).
To determine whether residues Ser688, Gly689, and Asn735 contribute to the inhibitory activity of our novel negative allosteric modulators of group II mGluRs, we used calcium mobilization and mutagenesis approaches similar to those performed with LY487379 and BINA. We found that the ability of MNI-135, MNI-136, and MNI-137 to block an EC80 glutamate-induced response was not altered in HEK293A cells transiently expressing mutant hmGluR2 containing N735D (Fig. 9, right) compared with wild-type (Fig. 9, left). Similar results were also observed with the double mutant (S688L/G689V) or triple mutant (S688L/G689V/N735D) (data not shown), suggesting that these residues are not critical for the inhibitory activity of our novel negative allosteric modulators.
Discussion
In the present study, we synthesized and rigorously characterized a novel family of noncompetitive antagonists (negative allosteric modulators) of group II mGluRs based on the general structure reported by Adam et al. (2003). These studies confirm a preliminary report suggesting that compounds in this structural class have allosteric antagonist activity at group II mGluRs (Gatti et al., 2001). These compounds are chemically and structurally unrelated to the competitive group II antagonist, LY341495 or MGS0039, or to the allosteric mGluR2 potentiator, LY487379. These novel compounds exhibited nanomolar potency for both rat and human mGluR2 and rat mGluR3 expressed in cell lines and also blocked activation of group II mGluRs at the MPP-DG synapse in rat hippocampal slices in a manner similar to that observed in the recombinant systems.

Potentiator activity of both LY487379 (A) and BINA (B), novel selective positive allosteric modulators of mGluR2, on glutamate-induced calcium mobilization in HEK293A cells is dependent on residue Asn735 in TM V of mGluR2. HEK293A cells were transiently transfected with the wild-type mGluR2 or a mutant mGluR2 containing a single point mutation N735D. LY487379 and BINA caused a parallel leftward shift of the glutamate concentration-response curve in cells expressing wild-type mGluR2 (A and B, left). However, the parallel leftward shift of the glutamate concentration curve was markedly reduced with the N735D mutant mGluR2, indicating a loss of potentiation activity at this mutant (A and B, right). The fluorescence responses were normalized as a percentage of the maximal response to glutamate (10 μM) and are the mean of four independent experiments performed in triplicate. Error bars are mean ± S.E.M.
These compounds were found to be noncompetitive antagonists of glutamate action on group II mGluRs, and they did not displace [3H]LY341495 from its binding site. Interestingly, the affinity of glutamate to displace the binding of [3H]LY341495 was not affected in the presence of these compounds, suggesting that they do not act by allosterically regulating binding of agonists to the orthosteric site. This is similar to the effects of previously identified allosteric antagonists of group I mGluRs, which do not alter glutamate binding but may act by altering conformational changes of the seven-transmembrane domain that in turn can inhibit coupling to G proteins (Kuhn et al., 2002).

The effect of novel negative allosteric group II mGluRs (i.e., MNI-135, MNI-136, and MNI-137) on glutamate-induced calcium mobilization in HEK293A cells is independent of N735D in TM V of mGluR2. HEK293A cells were transiently transfected with the wild-type hmGluR2 or a mutant mGluR2 containing a single point mutation, N735D. These compounds produced a concentration-dependent inhibition of glutamate-induced responses in cells expressing wild-type mGluR2 (left). Single point mutation of residue Asn735 in hmGluR2 did not alter the inhibitory activity of these novel compounds (right). The fluorescence responses were normalized as a percentage of the maximal response to glutamate (10 μM) and are the mean of four independent experiments performed in triplicate. Error bars are mean ± S.E.M.
Importantly, these novel compounds did not inhibit glutamate- or l-AP4-induced function in cell lines expressing mGluR1, 4, 5, or 8 by using calcium mobilization assay or mGluR7 by using a cyclase inhibition assay, suggesting that they are selective group II mGluR antagonists. However, it is important to note that the calcium fluorescence assay used for measuring mGluR4 and mGluR8 activity relies on coupling to a promiscuous G protein that is not the native G protein to which these receptors normally couple. Although unlikely, it is conceivable that this could affect the response of these receptors to antagonists
These novel compounds identified in the present study the most selective noncompetitive antagonists of group II mGluRs available to date. The most commonly used antagonist of these receptors is LY341495, which acts as a competitive antagonist at the orthosteric glutamate binding site (see Schoepp et al., 1999 for review). Although selective for group II mGluRs relative to other mGluR subtypes, this compound has antagonist activity at all known mGluRs and blocks mGluR8 with a potency similar to its potency at mGluR2 and mGluR3 (Kingston et al., 1998). The present findings are consistent with a growing body of literature suggesting that allosteric antagonists provide a novel approach for developing both antagonists and potentiators for mGluRs that have higher subtype selectivity than traditional orthosteric ligands. With the group I and group III mGluRs, this has extended to the discovery of ligands that differentiate between members within a group (Kuhn et al., 2002; Marino et al., 2003; Mitsukawa et al., 2005). Unfortunately, the present compounds did not exhibit selectivity that would distinguish between mGluR2 and mGluR3. Furthermore, there was no indication from the present studies that the structure-activity relationship within this series could lead to subtype-specific compounds that differentiate among these closely related subtypes.
An especially interesting finding in the current studies was preliminary evidence suggesting that two structurally distinct allosteric potentiators of mGluR2 may share a common binding site and that mutations that disrupt activity of these potentiators are without effect on the response to the new allosteric antagonists. Recent findings by Schaffhauser et al. (2003) suggest that residues Ser688, Gly689, and Asn735 are part of the binding pocket for a novel positive allosteric modulator for mGluR2, LY487379, and are important for its potentiation activity. Therefore, we next sought to investigate whether these residues are also critical for the inhibitory activity of our novel negative allosteric modulators. Consistent with this previous report, the enhancing effect of LY487379 on glutamate-induced calcium mobilization was markedly abolished by the single point mutation N735D, whereas the double mutation S688L/G689V induced a slight reduction. Moreover, the triple mutation, S688L/G689V/N735D, further abolished LY487379-induced responses compared with the single N735D mutant, suggesting that residue Asn735 plays a critical role for the enhancing activity of this allosteric modulator. Interestingly, we also found that residue Asn735 is also critical for potentiation activity of a newly reported selective allosteric potentiator of mGluR2, BINA. These data suggest that these two chemically unrelated allosteric potentiators of mGluR2, LY487379, and BINA may share a common binding site. In contrast, the single mutation N735D failed to block the inhibitory effect of our novel negative allosteric modulators of group II mGluRs. These studies are consistent with growing evidence that other mGluR subtypes can contain multiple allosteric regulatory sites. For instance, we recently reported that two families of allosteric potentiators of mGluR1 share a common binding site and that this site is distinct from the binding site that is common to multiple allosteric antagonists of mGluR1 (Hemstapat et al., 2006). Furthermore, we have provided evidence that N-{4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl}-2-hydroxybenzamide, an allosteric potentiator of mGluR5, acts at a site that is distinct from that of the site for the allosteric antagonist 2-methyl-6-(phenylethynyl)-pyridine (O'Brien et al., 2004). However, it is also clear that other classes of compounds can bind to the 2-methyl-6-(phenylethynyl)-pyridine site with a range of activities, including positive and negative allosteric modulators as well as neutral ligands (O'Brien et al., 2003; Kinney et al., 2005; Rodriguez et al., 2005).
In summary, we have thoroughly characterized a novel family of negative allosteric modulators of group II mGluRs. Although few reports have described the therapeutic significance of group II mGluR antagonists, recent studies have demonstrated that they may exhibit antidepressant-like activity and antiobsessive-compulsive disorder-like effects (Chaki et al., 2004; Shimazaki et al., 2004). Our current finding of negative allosteric modulators of group II mGluRs suggests that targeting of allosteric sites provides a viable approach for developing highly selective antagonists of these receptors.
Acknowledgments
We thank Kari Myers for the construction of the mGluR7/HEK cell lines and Douglas Sheffler for help with the cyclase assays.
Footnotes
-
This work was supported by grants from National Institute of Mental Health; NINDS, National Institutes of Health; The National Alliance for Research on Schizophrenia and Depression; and the Stanley Foundation. Vanderbilt University is a site in the National Institutes of Health-supported Molecular Libraries Screening Center Network.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.106.117093.
-
ABBREVIATIONS: mGluR, metabotropic glutamate receptor; CHO, Chinese hamster ovary; HEK, human embryonic kidney; TM, transmembrane; DCG-IV, (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine; LY341495, 2S-2-amino-2-(1S,2S-2-carboxycyclopropan-1-yl)-3-(xanth-9-yl)propionic acid; LY487379, N-(4-(2-methoxyphenoxy)-phenyl-N-(2,2,2-trifluoroetylsulfonyl)-pyrid-3-ylmethylamine; BINA, biphenyl-indanone A or 3′-(((2-cyclopropyl-6,7-dimethl-1-oxo2,3-dihydro-1H-inden-5-yl)oxy)methyl)biphenyl-4-carboxylic acid; DMSO, dimethyl sulfoxide; [35S]GTPγS, guanosine 5′-O-(3-[35S]thio)triphosphate; ACSF, artificial cerebrospinal fluid; l-AP4, L(+)-2-amino-4-phosphonobutyric acid; FBS, fetal bovine serum; DMEM, Dulbecco's modified Eagle's medium; MPP, medial perforant path; fEPSP, field excitatory postsynaptic potential; LY379268, (-)-2-oxa-4-aminobicyclo[3.1.0]hexane-4,6-dicarboxylate; LY354740, (1S,2S,5R,6S)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid; MGS0039, (1R,2R,3R,5R,6R)-2-amino-3-(3,4-dichlorobenzyloxy)-6-fluorobicyclo[3.1.0]hexane-2,6-dicarboxylic acid; MNI-135, 3-(7-iodo-4-oxo-4,5-dihydro-3H-benzo[b][1,4]diazepin-2-yl)-benzonitrile; MNI-136, 7-bromo-4-(3-pyridin-3-yl-phenyl)-1,3-dihydro-benzo[b][1,4]diazepin-2-one; MNI-137, 4-(7-bromo-4-oxo-4,5-dihydro-3H-benzo[b][1,4]diazepin-2-yl)-pyridine-2-carbonitrile; HBSS, Hanks' balanced salt solution; DG, dentate granule; hmGluR, human GluR; rmGluR, rat GluR.
- Received November 14, 2006.
- Accepted April 5, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}