


Abstract
The functional characteristics of human proton coupled folate transporter (hPCFT)/heme carrier protein (HCP) 1 were investigated. hPCFT/HCP1 expressed transiently in human embryonic kidney 293 cells mediated the transport of folate at an acidic extracellular pH of 5.5 in a manner independent of Na+ and insensitive to membrane potential, but its transport activity was absent at near-neutral pH. Folate transport mediated by hPCFT/hHCP1 at pH 5.5 was saturable with a Km of 1.67 μM and extensively inhibited by reduced folates, such as folinate, 5-methyltetrahydrofolate, and methotrexate (MTX). Sulfobro-mophthalein and 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid were also found to be potent inhibitors of hPCFT/hHCP1, but hemin was found to exhibit only minimal inhibitory effect. When expressed stably as a protein fused with green fluorescent protein (GFP-hPCFT/HCP1) in MDCKII cells, GFP-hPCFT/HCP1 was mainly localized at the apical membrane, and the cellular accumulation of MTX was higher from the apical side than from the basal side. These functional features of hPCFT/HCP1 are consistent with those of the well characterized carrier-mediated folate transport system in the small intestine, suggesting that hPCFT/HCP1 is responsible for the intestinal absorption of folate and also MTX. We also found that sulfasalazine is a potent inhibitor of hPCFT/HCP1, which would interfere with the intestinal absorption of MTX when coadministered in therapy for rheumatoid arthritis as well as folate.
Methotrexate (MTX), an analog of folate, has been widely used by oral administration in cancer chemotherapy. In addition, oral MTX has been proven to be effective at a lower dose for rheumatoid arthritis and, thus, also recognized as a disease-modifying anti-rheumatic drug (DMARD) (Cronstein, 1996). Such oral use of MTX is in practice because of its fairly good intestinal absorption, which has been suggested to be mediated by a carrier-mediated transport system specific to folate and its analogs (Sirotnak and Tolner, 1999). Therefore, it is important to clarify the carrier-mediated folate transport mechanism, including its molecular entity, to achieve effective therapy using MTX.
Earlier studies demonstrated that the transport of folate in the intact small intestine occurs in a saturable manner with an acidic luminal pH optimum (pH 5.0–6.0) and is competitively inhibited by MTX (Steinberg, 1984). Such a pH-sensitive and saturable transport has been also observed for MTX in studies using the brush-border membrane vesicles and everted sacs of the small intestine (Strum, 1977; Selhub and Rosenberg, 1981). It is notable that the folate transport system has been suggested to be also shared by reduced folates, such as 5-methyltetrahydrofolate and folinate, with affinities comparable with that of folate. Thus, the carrier-mediated intestinal folate transport system has been well characterized functionally. However, its molecular entity has not been fully clarified yet.
Reduced folate carrier (RFC) 1 is the first cloned transporter that was found to transport folates (Williams et al., 1994; Sirotnak and Tolner, 1999). This transporter is expressed ubiquitously in the body and, as an observation in the mouse small intestine, expressed more abundantly in the epithelial cells of the villus tip, localizing at the apical membrane, than in the immature cells of the crypt, suggesting its possible role in absorption process (Chiao et al., 1997). However, the functional features of RFC1 are different from those of the intestinal folate transport system in that it functions optimally at around neutral pH and favors reduced folates, as its name stands for, and MTX than folate as substrates. In addition, Goldman and co-workers (Zhao et al., 2004) demonstrated in HeLa cells that deletion of RFC1 gene from the genome does not result in any significant alteration of folate transport activity. Thus, the involvement of RFC1 in the intestinal folate transport is in question, and there has been an argument that some other transporter might be responsible for that.
Proton-coupled folate transporter (PCFT)/heme carrier protein (HCP) 1 is another folate transporter identified quite recently. This transporter was originally isolated from the mouse duodenum as a heme transporter (Shayeghi et al., 2005). Therefore, it was designated as HCP1 at that time. This transporter was shown to mediate the transport of heme and its analogs such as zinc protoporphyrin, although with a relatively low affinity (Michaelis constant of 125 μM for heme). It has also been reported that an enhanced expression of the transporter was accompanied with an increase in heme uptake in Caco-2, human colon cancer cell line, when treated with CdCl2 to induce heme oxygenase, supporting its suggested role as a heme transporter and implying its relevance to heme metabolism (Latunde-Dada et al., 2006).
However, in a very recent study (Qiu et al., 2006), PCFT/HCP1 was reidentified as a PCFT. They found that the human ortholog of PCFT/HCP1 (hPCFT/HCP1) is mainly expressed in the proximal part of the small intestine and can transport not only folate (oxidized form) but also reduced folates and MTX very efficiently in the presence of a proton gradient. It is noteworthy that a loss of the function of hPCFT/HCP1 due to genetic mutation leads to hereditary folate malabsorption, indicating that hPCFT/HCP1 plays a critical role in the absorption of dietary folate.
We also have been working on this controversial issue about the intestinal folate transport system and found that hPCFT/HCP1 heterologously expressed in mammalian cells has the characteristic of specific and efficient folate transporter with an acidic pH optimum. Our results in mammalian cells are quite consistent with those in Xenopus laevis oocytes reported by Qiu et al. (2006), strongly supporting the suggestion that the major role of PCFT/HCP1 is to mediate the transport of folate and analogs rather than heme. We further examined the effect of various drugs, including several nonsteroidal anti-inflammatory drugs (NSAIDs) and DMARDs that could be coadministered with MTX in therapy for rheumatoid arthritis, on hPCFT/HCP1-mediated MTX transport to gain information about possible interactions at PCFT/HCP1, which may be of clinical significance in oral MTX therapy.
Materials and Methods
Materials. [3H]Methotrexate (46.8 Ci/mmol) and [3H]folate (30 Ci/mmol) were obtained from Moravek Biochemicals (Brea, CA), [3H]cimetidine (25.0 Ci/mmol) was from GE Healthcare (Chalfont St. Giles, UK) and [3H]estrone sulfate (57.3 Ci/mmol) and [3H]taurocholate (1.2 Ci/mmol) were from PerkinElmer Life Sciences, Inc. (Boston, MA). Folate, methotrexate, 5-methyltetrahydrofolate, folinate, geneticin, and sulfasalazine were obtained from Sigma-Aldrich Co. (St. Louis, MO). Zinc protoporphyrin IX (Calbiochem, San Diego, CA) was obtained from Merck (Darmstadt, Germany). Restriction enzymes were obtained from TOYOBO (Tokyo, Japan). All other reagents were of analytical grade and commercially obtained.
Cell Culture. Human embryonic kidney (HEK)293 and MDCKII cells were maintained at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Isolation of hPCFT/HCP1. The cDNA of hPCFT/HCP1 was cloned from the human intestine total RNA (Clontech, Palo Alto, CA) by reverse transcription and subsequent PCR. In brief, a reverse transcription reaction was carried out using 3 μg of the total RNA, an oligo(dT) primer, and ReverTra Ace (TOYOBO) as a reverse transcriptase. The cDNA of hPCFT/HCP1 was isolated from the human intestine cDNA by PCR using KOD plus polymerase (TOYOBO) and the following primers: forward primer, 5′-GCT CCG CCG CGC ACG CAC AT-3′; and reverse primer, 5′-TCA GGG GCT CTG GGG AAA CTG C-3′. These primers were designed based on the sequence in GenBank (accession no., NP_542400). PCR was performed using the following conditions: 94°C for 2 min and 32 cycles of 1) 94°C for 20 s, 2) 58°C for 20 s, and 3) 72°C for 1.5 min. The amplified cDNA product was subcloned into pME18 vector and transferred into a mammalian expression vector, pCI-neo (Promega, Madison, WI). The sequence of the amplified cDNA product was determined with an automated sequencer (ABI PRISM 3100; Applied Biosystems, Foster City, CA) and confirmed to be identical to that in GenBank.
To generate an hPCFT/HCP1 fused with green fluorescent protein (GFP-hPCFT/HCP1), the coding sequence of hPCFT/HCP1 was amplified from the pCI-neo-based plasmid carrying the cDNA, using a forward primer containing an EcoRI restriction site (underlined), 5′-GGG AAT TCC ATG GAG GGG AGC GCG AGC-3′, and the T3 reverse primer for pCI-neo vector. Then the amplified product was introduced at the EcoRI and XbaI sites into pEGFP-C1 vector (Clontech). The sequences were determined with an automated sequencer.
Transport Study in HEK293 Cells Transiently Expressing hPCFT/HCP1. HEK 293 cells (1.5 × 105 cells/well initially) were grown on 24-well plates coated with poly-l-lysine for 24 h, transfected with the plasmid carrying hPCFT/HCP1 cDNA by the calcium phosphate coprecipitation method, and cultured for 24 to 36 h for transient expression. In regular transport assays, cells were preincubated in substrate-free uptake buffer [140 mM NaCl, 5 mM KCl, 0.4 mM KH2PO4, 0.8 mM MgSO4, 1.0 mM CaCl2, 25 mM glucose, and 10 mM 2-(N-morpholino) ethanesulfonic acid, pH 5.5] for 5 min, and then transport assays were started by replacing the substrate-free uptake buffer with one containing a 3H-labeled substrate (0.25 ml). When necessary, the uptake buffer was modified as indicated. When the effect of various compounds on substrate uptake was examined, test compounds were added only to the buffer for uptake period. All the procedures were conducted at 37°C. Assays were stopped by addition of ice-cold substrate-free uptake buffer (2 ml), and the cells were washed two times with 2 ml of the same buffer. The cells were solubilized in 0.5 ml of 0.2 N NaOH solution containing 0.5% sodium dodecyl sulfate, and associated radioactivity was measured by liquid scintillation counting. Cellular protein content was determined by the method of Lowry et al. (1951) using bovine serum albumin as the standard. HEK293 cells transfected with empty pCI-neo vector were used to determine the basal activity and are designated as mock cells.
The determination of zinc protoporphyrin was performed by high-performance liquid chromatography with fluorescence detection (Lim et al., 1988). In brief, the cells were lysed with 500 μl of mobile phase (described below) for 30 min at room temperature. The lysed sample was transferred into a sample tube and then centrifuged (16,000g, 10 min, 4°C). The supernatant was injected (50 μl) into the high-performance liquid chromatography system: column, YMC-pack ODS-A (150 × 4.6 mm; YMC Inc., Wilmington, NC); mobile phase, 88% methanol in 1 M ammonium acetate, pH 5.16; flow rate, 0.8 ml/min; and detection wavelength, 415 nm for excitation and 583 nm for emission (SPD-10A; Shimadzu Co., Kyoto, Japan).
Preparation of HEK293 Cells and MDCKII Cells Stably Expressing hPCFT/HCP1 or GFP-hPCFT/HCP1. HEK 293 cells and MDCKII cells were transfected with the plasmid carrying cDNA of hPCFT/HCP1 and GFP-hPCFT/HCP1, respectively, by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA), according to the manufacturer's instructions, and cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum and 800 μg/ml geneticin for 2 to 3 weeks. Antibiotic-resistant clones were selected and tested for transport of [3H]MTX. To confirm the expression of GFP-hPCFT/HCP1 in MDCKII cells, their emitting fluorescence intensity was examined by using a fluorescence microscope.
Transport Study in HEK293 Cells Stably Expressing hPCFT/HCP1. HEK293 cells stably expressing hPCFT/HCP1 (1.5 × 105 cells/well initially) were seeded on 24-well plates coated with poly l-lysine, cultured for 48 h, and used for transport assays in the same manner as described for those transiently expressing hPCFT/HCP1.
Transport Study in MDCKII Cells Stably Expressing GFP-hPCFT/HCP1. MDCKII cells stably expressing GFP-hPCFT/HCP1 were seeded at a density of 1 × 105 cells on each polycarbonate membrane insert of Transwell (12-mm i.d., 3.0-μm pore size; Costar, Cambridge, MA) and grown to confluence for 5 days. The medium was replaced with substrate-free uptake buffer (0.5 and 1.5 ml, respectively, for apical and basal compartments) for preincubation for 5 min. Transport assays were started by replacing the buffer in either apical or basal compartment with one containing [3H]MTX (10 nM). Assays were stopped by transferring the cells, with a membrane insert, into ice-cold substrate-free uptake buffer (20 ml). The cells on membrane insert were washed two times with the same buffer and solubilized in 0.5 ml of 0.2 N NaOH solution containing 0.5% sodium dodecyl sulfate. The radioactivity in the solubilized sample was measured by liquid scintillation counting.
Immunofluorescence Analysis. MDCKII cells stably expressing GFP-hPCFT/HCP1 were seeded at a density of 1 × 105 cells on each polycarbonate membrane insert of Transwell and grown to confluence for 5 days. The cells were washed twice with ice-cold phosphate-buffered saline (PBS), fixed in 3% paraformaldehyde, and permeabilized with 0.1% Triton X-100 in PBS for 30 min at room temperature. After washing twice with PBS, the cells were blocked with PBS containing 1 mg/ml bovine serum albumin (BSA) for 1 h at room temperature and then incubated with a 1:200 dilution of primary antibody/mouse anti-Na+/K+ ATPase (Santa Cruz Biotechnology, Santa Cruz, CA) for 1 h at room temperature. The cells were washed three times with PBS containing BSA and then stained with 1:500 dilution of Alexa 546-labeled goat anti-mouse IgG (Invitrogen) for 1 h at room temperature. The cells were washed three times with PBS containing BSA and then mounted on a glass slide in 9:1 glycerol/PBS. The localizations of immunofluorescent-labeled Na+/K+ ATPase and GFP-hPCFT/HCP1 were visualized by using a confocal laser scanning microscope (LSM510; Zeiss, Jena, Germany).
Data Analysis. To estimate the kinetic parameters of the Michaelis constant (Km, half-saturation concentration) and the maximal transport rate (Vmax) for the saturable transport, the following equation was fitted to the profile of uptake rate (v) versus substrate concentration (s) by means of nonlinear least-squares regression analysis using WinNonlin (Pharsight Co., Mountain View, CA): v = Vmax × s/(Km + s). Parameters are represented as computer-fitted parameters with S.E. Experimental data are represented as the means ± S.E., and statistical analysis was performed using two-tailed, unpaired Student's t test or, when multiple comparisons were needed, analysis of variance followed by Dunnett's test, with p < 0.05 considered significant.
Results
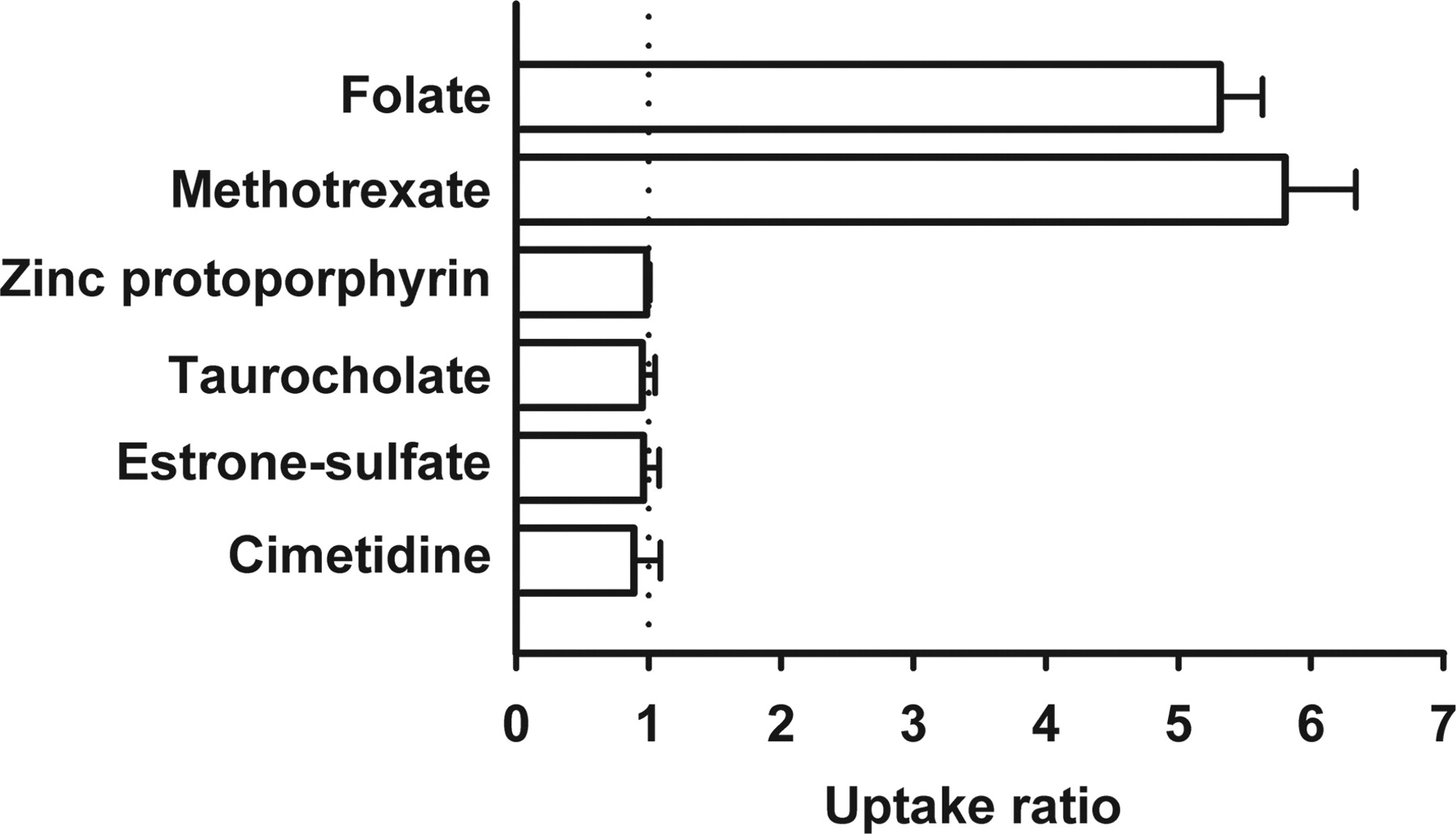
Functional Characteristics of hPCFT/HCP1. We examined the uptake of folate (oxidized form) and, for comparison, those of several compounds in HEK293 cells transiently expressing hPCFT/HCP1 (hPCFT/HCP1-HEK293 cells) under an acidic extracellular condition of pH 5.5 (Fig. 1). It was found that folate is very efficiently transported in hPCFT/HCP1-HEK293 cells, exhibiting 5-fold greater uptake compared with that in mock cells. A similar result was obtained for MTX. Zinc protoporphyrin was, unexpectedly, not transported by hPCFT/HCP1, even though this fluorescent heme analog is reportedly a substrate of mouse PCFT1/HCP1 (Shayeghi et al., 2005). Appreciable uptake mediated by hPCFT/HCP1 was not observed for other organic anions, such as taurocholate and estrone sulfate, and cimetidine as an organic cation. These results suggest that folate and MTX are specific and more preferential substrates of hPCFT/HCP1 than heme.

Uptake of various organic anions in HEK293 cells transiently expressing hPCFT/HCP1. hPCFT/HCP1-specific uptake was evaluated by dividing the uptake in hPCFT/HCP1-transfected cells by that in mock cells. Uptakes of [3H]folate (10 nM), [3H]methotrexate (10 nM), zinc protoporphyrin (5 μM), [14C]taurocholate (0.4 μM), [3H]estrone sulfate (10 nM), and [3H]cimetidine (20 nM) were evaluated at 37°C and pH 5.5 for 10 min in both cells. Data are represented as the means ± S.E. (n = 4).
The pH-sensitive characteristics of folate transport mediated by hPCFT/HCP1 were examined in a range of extracellular pHs between 4.0 and 8.0. As shown in Fig. 2A, the uptake of folate in hPCFT/HCP1-HEK293 cells was greatest at pH 4.5 and comparably high in a range from pH 4.0 to 5.5, at which regular transport assays were performed. Above pH 5.5, it decreased with an increase in pH and reached a negligibly small level at neutral pH and above. The uptake of folate in mock cells was minimal over the entire range of pH, although it also decreased with an increase in pH. Thus, folate transport mediated by hPCFT/HCP1 was found to be highly sensitive to extracellular pH. To examine the possible involvement of H+ as a driving force of hPCFT/HCP1-mediated transport, we examined folate uptake in hPCFT/HCP1-HEK293 cells in the presence of nigericin, a K+/H+ exchanger (ionophore), in the uptake buffer in which NaCl (Na+) was replaced by KCl (K+), named KCl buffer here, to dissipate H+ gradient across the plasma membrane (Fig. 2B). This treatment significantly reduced folate uptake, indicating that an inwardly directed H+ gradient is required. However, when the KCl buffer was used without nigericin only to depolarize the plasma membrane, folate uptake was not influenced. A similar result was obtained when 2 mM Ba2+ (BaCl2), a nonselective K+ channel inhibitor that can depolarize the plasma membrane, was added to the uptake buffer (data not shown). The hyperpolarization of the membrane induced by the use of valinomycin added in the regular up-take buffer containing NaCl did not alter folate uptake, either. Furthermore, when NaCl in the uptake buffer was iso-osmotically replaced with mannitol or K gluconate, folate uptake was not altered either, indicating that Na+ and Cl– are not involved in hPCFT/HCP1-mediated folate transport (Fig. 2B). Thus, hPCFT/HCP1-mediated folate transport was suggested to be of cotransport with H+ but insensitive to membrane potential.
Kinetic Features of pH-Sensitive Folate Transport by hPCFT/HCP1. The time courses of folate uptake were examined in hPCFT/HCP1-HEK293 cells and mock cells. The uptake of folate increased rapidly, being in proportion to time up to 10 min in hPCFT/HCP1-HEK293 cells, whereas it was far slower in mock cells (Fig. 3A). Therefore, we, decided to evaluate the initial rate of uptake at 2 min. As shown in Fig. 3B, kinetic analysis indicated that the transport of folate mediated by hPCFT/HCP1 was saturable with a Km of 1.67 μM and a Vmax of 70.0 pmol/min/mg protein.

Functional characterization of hPCFT/HCP1 transiently expressed in HEK293 cells. A, pH dependence of the uptake of [3H]folate (10 nM) was evaluated at 37°C for 2 min in hPCFT/HCP1-transfected cells (closed circle) and mock cells (open circle). The uptake buffer contained 10 mM 2-(N-morpholino) ethanesulfonic acid for pH 6.0 and below and 10 mM HEPES for pH 6.5 and above. B, effect of ionic conditions in the medium on the uptake of [3H]folate (10 nM) at 37°C and pH 5.5 for 2 min was evaluated in hPCFT/HCP1-transfected cells by using, for both pre-incubation and uptake periods, the regular buffer containing NaCl, the regular buffer added with valinomycin, those in which NaCl was replaced with KCl (KCl buffer), potassium gluconate, or mannitol, and also the KCl buffer added with nigericin (10 μg/ml). Data are represented as the means ± S.E. (n = 4).
Under the condition of pH 5.5 and 2-min uptake period for regular transport assays, the uptake of folate in hPCFT/HCP1-HEK293 cells was typically 0.56 pmol/mg protein or, by multiplying the protein content of 0.2 mg of protein/well, 0.112 pmol/well. That was only approximately 5% of the initial amount of 2.5 pmol/well (0.25 ml/well of 10 nM solution), indicating that the initial concentration was practically maintained during the transport assay. Folate uptake in mock cells was only approximately 20% of that in hPCFT/HCP1-HEK293 cells, indicating that hPCFT/HCP1-specific uptake was the predominant uptake component in the latter and allowing its reliable evaluation as a difference in uptake between them. When unlabeled folate was added at 100 μM, which was approximately 60-fold higher than the estimated Km of folate transport, to evaluate the nonspecific uptake, the uptake of [3H]folate (10 nM) in hPCFT/HCP1-HEK293 cells was comparable with that in mock cells, as expected (data not shown).

Kinetic analyses of folate transport mediated by hPCFT/HCP1 transiently expressed in HEK293 cells. A, time courses of the uptake of [3H]folate (10 nM) were evaluated at 37°C and pH 5.5 in hPCFT/HCP1-transfected cells (closed circle) and mock cells (open circle). B, concentration-dependent uptake of [3H]folate was evaluated at 37°C and pH 5.5 for 2 min in hPCFT/HCP1-transfected cells and mock cells. The hPCFT/HCP1-specific uptake was calculated by subtracting the uptake in the latter from that in the former and was used for kinetic analysis. The Km and Vmax are 1.67 ± 0. 10 μM and 70.0 ± 2.7 pmol/min/mg protein, respectively, as the computer-fitted parameters with S.E. Solid line, computer-fitted profile. Eadie-Hofstee plot is shown in inset, where v and s represent the uptake rate (picomoles per minute per milligram of protein) and concentration (micromolar), respectively. C, inhibitory effect of folate derivatives on the hPCFT/HCP1-specific uptake of folate (10 nM) was evaluated in the presence of varied concentrations of folinate (closed square), MTX (open square), 5-methyltetrahydrofolate (closed circle), and tetrahydrofolate (open circle). The values of IC50 are 1.2, 2.0, 3.0, and 5.9 μM, respectively. The uptake rate is shown as the relative one normalized by the rate in the absence of inhibitors (0.35 pmol/min/mg protein). Data are represented as the means ± S.E. (n = 4).
Affinities of Folate Derivatives for hPCFT/HCP1. We compared the affinities of several folate derivatives, tetrahydrofolate (THF), 5-methyltetrahydrofolate (5-MTHF), MTX, and folinate for hPCFT/HCP1 (Fig. 3C). They were all found to be potent inhibitors in the following order of decreasing IC50 (increasing affinity): THF (5.9 μM) > 5-MTHF (3.0 μM) > MTX (2.0 μM) > folinate (1.2 μM). These IC50 values were comparable with the Km of folate transport, indicating comparable affinities, although those of THF and 5-MTHF were somewhat greater. Thus, hPCFT/HCP1 was suggested to have a characteristic of a folate transporter that prefers folate to reduced folates (THF and 5-MTHF) as substrates.
Effect of Various Compounds on hPCFT/HCP1-Mediated Folate Transport. To investigate the substrate specificity of hPCFT/HCP1, we examined the effect of various compounds (0.2 or 2.0 mM) on the hPCFT/HCP1-mediated uptake of folate (10 nM) using hPCFT/HCP1-HEK293 cells. As shown in Fig. 4, it was found that sulfobromophthalein and 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid are potent inhibitors of hPCFT/HCP1, whereas the inhibitory effects of several organic anions, such as estrone sulfate, probenecid, furosemide, lactate, α-ketoglutarate, succinate, p-aminohippurate, taurocholate, and urate, are only minimal or insignificant. It was also found that p-aminobenzoylglutamate, a chemical structural constituent of folates, is a potent inhibitor of hPCFT/HCP1. Therefore, p-aminobenzoylglutamate moiety may play a role in recognition by hPCFT/HCP1, although its affinity to hPCFT/HCP1 was suggested to be lower than that of folate because its inhibitory effect was minimal at 20 μM, a concentration approximately 10-fold higher than the Km of folate and the IC50 of MTX. Hemin was found to exhibit an inhibitory effect, as expected, but it was only minimal. Organic cations, such as tetraethylammonium and thiamine, and thiamine pyrophosphate (TPP), an inhibitor of RFC1 (Zhao et al., 2001), did not exhibit any inhibitory activity. Thus, only limited anionic compounds were suggested to have an affinity to hPCFT/HCP1, indicating its relatively narrow substrate specificity.

Effect of various compounds on folate uptake mediated by hPCFT/HCP1 transiently expressed in HEK293 cells. The uptake of [3H]folate (10 nM) was evaluated at 37°C and pH 5.5 for 2 min in hPCFT/HCP1-transfected cells and mock cells, and the hPCFT/HCP1-specific uptake was calculated by subtracting the uptake in the latter from that in the former. The uptake rate for control was 0.30 pmol/min/mg protein. Data are represented as the means ± S.E. (n = 4). *, significantly different from the value for control at p < 0.05. BSP, sulfobromophthalein; DIDS, 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid; PABG, p-aminobenzoyl-glutamate; PAH, p-aminohippurate.
Localization of hPCFT/HCP1 in MDCKII Cells. It has been reported that mouse PCFT/HCP1 is exclusively expressed in the duodenum and immunohistologically localized at the apical membrane (Shayeghi et al., 2005). To examine such membrane localization characteristic of hPCFT/HCP1, we generated a cDNA construct that expresses GFP-hPCFT/HCP1, stably transfected it in MDCKII cells, and determined the localization of expressed GFP-hPCFT/HCP1 by confocal laser scanning microscopy (Fig. 5A). GFP-hPCFT/HCP1 was mainly localized at the apical membrane of MDCKII cells that were cultured on the permeable membrane filter, which was inserted in a well and separating the apical and basal compartments (Transwell), to facilitate cellular polarization, whereas Na+,K+-ATPase was localized at the basolateral membrane. To confirm the functional localization, we compared the uptakes of MTX (10 nM) across apical and basal membranes by loading the uptake buffer containing MTX with the other side of compartment (Fig. 5B). In MDCKII cells expressing GFP-hPCFT/HCP1, the cellular accumulation of MTX was approximately 6-fold higher from the apical compartment than from the basal compartment. However, in mock cells, MTX uptakes were at similar low levels from the both compartments. Thus, GFP-hPCFT/HCP1 mediated MTX uptake in a polarized manner only through the apical membrane, indicating its functional localization at that membrane. All these results suggest that hPCFT/HCP1 is an apical protein as its mouse ortholog and, in the small intestine, presumably expressed at the brush-border membrane of epithelial cells.

Cellular localization of GFP-hPCFT/HCP1 stably expressed in MDCK cells. A, predominant apical membrane localization of GFP-hPCFT/HCP1 (green) is shown in GFP-hPCFT/HCP1-transfected MDCK cells cultured on the membrane insert of Transwell, whereas Na+,K+-ATPase, a marker exclusively localized at the basolateral membrane, is stained red by Alexia 546-labeled antibody bound to the monoclonal antibody bound to the ATPase. B, functional localization of GFP-hPCFT/HCP1 was evaluated by comparing the uptakes (37°C and pH 5.5 for 5 min) of [3H]MTX (10 nM) across apical and basal membranes in GFP-hPCFT/HCP1-transfected cells (closed bar) and mock cells (open bar) by loading the uptake buffer in the either side of compartment. Data are represented as the means ± S.E. (n = 4).
Effect of NSAIDs and DMARDs on hPCFT/HCP1-Mediated MTX Transport. To gain some information relevant to oral MTX therapy, we examined the effect of various compounds (0.2 or 2 mM), which include several drugs orally used with MTX for the treatment of rheumatoid arthritis, on the hPCFT/HCP1-mediated uptake of MTX (10 nM), using HEK293 cells stably expressing hPCFT/HCP1. As shown in Table 1, most of them that include many of the NSAIDs (ibuprofen, naproxen, salicylate, and so on) and DMARDs (aurothiomalate, azathioprine, and dexamethasone) did not inhibit MTX transport. However, indomethacin and diclofenac inhibited MTX transport significantly, and sulfasalazine (SSZ) did even more potently with an IC50 value of 60.4 μM (Fig. 6). These inhibitors, especially SSZ, might interfere with intestinal MTX absorption.
Effect of various compounds on MTX uptake mediated by hPCFT/HCP1 stably expressed in HEK293 cells
The uptake of [3H]MTX (10 nM) was evaluated at 37°C and pH 5.5 for 1 min in hPCFT/HCP1-transfected cells and mock cells, and the hPCFT/HCP1-specific uptake was calculated by subtracting the uptake in the latter from that in the former. The Km and Vmax for MTX transport were 3.67 ± 0.38 μM and 245.4 ± 19.1 pmol/min/mg protein, respectively, as the computer-fitted parameters with S.E. The uptake rate for control was 0.63 pmol/min/mg protein. Data are represented as the mean ± S.E. (n = 4).
Discussion
The mouse ortholog of PCFT/HCP1 (mPCFT/HCP1) was recently identified as a novel membrane transporter by a subtractive hybridization approach to isolate transporters that predominantly expressed in duodenum compared with ileum (Shayeghi et al., 2005). mPCFT/HCP1 was shown to mediate the transport of heme and zinc protoporphyrin when expressed in mammalian cells and was, therefore, suggested to be the intestinal heme transporter. However, in the present study, hPCFT/HCP1 transiently expressed in HEK293 cells failed to transport zinc protoporphyrin (Fig. 1). On the other hand, hPCFT/HCP1 mediated the transport of folate and MTX more efficiently. The Km of folate transport was as low as 1.67 μM, indicating quite high affinity (Fig. 3B). This Km value is in a range reported for folate transport in brush-border membrane vesicles from the small intestine of rat (0.42 μM), rabbit (1.5 μM), and human (1.69 μM) (Selhub and Rosenberg, 1981; Said et al., 1987; Itoh et al., 2001). Hemin could inhibit hPCFT/HCP1-mediated folate transport, but only moderately (29% inhibition at 100 μM; Fig. 4). In addition, it has been reported that the expression of mPCFT/HCP1 mRNA is not regulated by a mechanism linked to that of other proteins that are involved in iron metabolism, such as ferroportin and ferric reductase (Shayeghi et al., 2005). Therefore, the role of PCFT/HCP1 as a heme transporter, if any, does not seem to be its major one. It is most likely that PCFT/HCP1 works primarily as a folate transporter. These findings are in agreement with those reported for hPCFT/HCP1 expressed in X. laevis oocytes (Qiu et al., 2006).
The transport of folate mediated by hPCFT/HCP1 was clearly shown to be greater at lower extracellular pH, or greater inward H+ gradient across the plasma membrane, and also shown to be reduced when the H+ gradient was collapsed (Fig. 2). These suggest that H+ cotransport is the most likely mode of operation for hPCFT/HCP1. The H+ gradient required for hPCFT/HCP1 to operate is present in the small intestine because its luminal surface is maintained acidic at pH ranging from 6.0 to 6.8 by H+ supplied from epithelial cells by Na+/H+ exchangers at the brush-border membrane (Praetorius et al., 2000). Folate has two carboxyl groups at α and γ positions in its molecule, and both of them are almost completely dissociated in the pH range where its uptake was highly pH-sensitive because their pKa values are 3.1 to 3.5 and 4.6 to 4.8, respectively (Poe, 1977). Therefore, such pH-sensitive folate transport cannot be explained by facilitated or diffusive transport of its protonated form driven by physicochemical potential across the membrane. However, alternatively, allosteric modification of the transporter by protonation of protonatable residues exposed to the extra-cellular space might be a possible underlying mechanism of pH-sensitive transport. On the other hand, it was shown that hPCFT/HCP1 did not require membrane potential for operation in folate transport, or the transmembrane gradient of Na+ or Cl–, either (Fig. 2B). It is likely that its negative charge is countered by the positive charge of H+. Contradicting to such a finding of potential-independent transport, which indicates nonelectrogenicity of transport, hPCFT/HCP1 expressed in X. laevis oocytes reportedly mediates the uptake of folate electrogenically by cotransport with proton, which is associated with influx of positive charge (Qiu et al., 2006). However, such an electrogenic folate transport has not been observed in the intestine. Said et al. (1987) demonstrated that pH-sensitive folate transport in the brush-border membrane vesicles of the human small intestine was not affected by manipulations of membrane potential using valinomycin, in the presence of an inwardly directed K+ gradient or membrane-permeable anions, suggesting potential independence of transport. In addition, it is well known that, in various tumor cell lines, folate transport is not influenced by depolarization of the plasma membrane by the replacement of Na+ with K+ in the uptake buffer (Kumar at al., 1997). Thus, our finding of nonelectrogenic nature of hPCFT/HCP1-mediated transport is consistent with the well known characteristic of the intestinal folate transport system. hPCFT/HCP1 might be, when expressed in X. laevis oocytes, functionally modulated to mediate folate transport electrogenically, although this issue needs to be further clarified in the future.
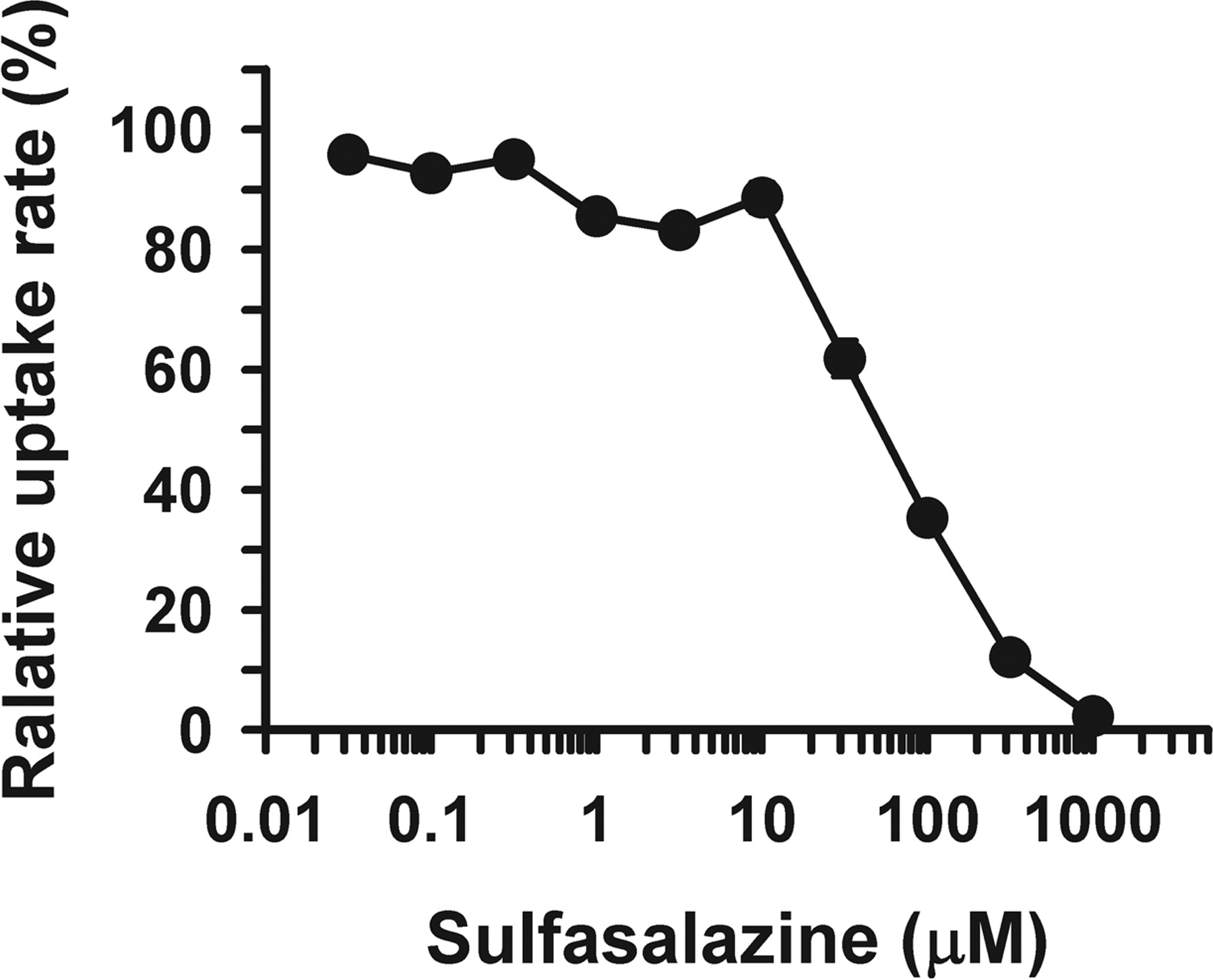
Effect of SSZ on MTX uptake mediated by hPCFT/HCP1 stably expressed in HEK293 cells. The uptake of [3H]MTX (10 nM) was evaluated at 37°C and pH 5.5 for 1 min in hPCFT/HCP1-transfected HEK293 cells and mock cells, and the hPCFT/HCP1-specific uptake was calculated by subtracting the uptake in the latter from that in the former. The uptake rate is shown as the relative one normalized by the rate in the absence of SSZ (0.75 pmol/min/mg protein). The IC50 is 60.4 μM. Data are represented as the means ± S.E. (n = 4).
An important feature of hPCFT/HCP1 is that it has a somewhat higher affinity for folate than reduced folates, such as tetrahydrofolate and 5-methyltetrahydrofolate (Fig. 3C). This is of physiological relevance because folate (oxidized form), but not reduced folates, is predominantly present in the intestinal lumen after food intake (Seyoum and Selhub, 1998). It is because reduced folates, which are unstable when exposed to air, heat, and acid, can be easily converted to oxidized form during food processing and, after intake, digestion in the gastrointestinal lumen. Contrasting to this sub-strate recognition feature of hPCFT/HCP1, it is known that RFC1 does not accept folate as a good substrate, whereas it can transport reduced folates and MTX efficiently. Thus, RFC1 may not be equipped for absorbing dietary folate in the intestine.
It has been reported that sulfobromophthalein and 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid are inhibitors of pH-sensitive folate transport in the small intestine (Perry and Chanarin, 1972; Schron et al., 1985). Consistent with that, we found that both of them inhibit folate transport mediated by hPCFT/HCP1 significantly (Fig. 4). Although Qiu et al. (2006) failed to observe inhibition of hPCFT/HCP1-mediated folate transport by these reagents in the X. laevis oocyte expression system, it may be because their concentration (20 μM) was too low, compared with 200 μM in the present study and earlier studies using intestinal preparations. Interestingly, TPP, an endogenous substrate of RFC1 (Zhao et al., 2001), did not show any inhibitory effect on folate transport mediated by hPCFT/HCP1. Thus, TPP is an inhibitor specific to RFC1 and, hence, would be useful to determine the contribution of RFC1, differentiating from that of hPCFT/HCP1, to folate transport in physiological systems.
It is of interest clinically to evaluate the effect of various drugs that are orally used together with MTX on the hPCFT/HCP1-mediated transport of MTX. NSAIDs are such drugs often prescribed with MTX to reduce inflammation associated with rheumatoid arthritis (Doan and Massarotti, 2005). Among various NSAIDs tested in the present study, we found that diclofenac and indomethacin inhibit hPCFT/HCP1-mediated MTX transport significantly (Table 1). It has been reported that several transporters such as OATs (OAT1, OAT2, and OAT3) and RFC1 can mediate influx transport of MTX and are inhibited by a more wide variety of NSAIDs, including ketoprofen, naproxen, diclofenac, indomethacin, and phenylbutazone (Khamdang et al., 2002; Nozaki et al., 2004). Particularly, OATs are known to be inhibited extensively by such NSAIDs. Thus, hPCFT/HCP1 seems to be less sensitive to NSAIDs than those transporters. SSZ, which is also used as an anti-inflammatory drug with MTX in therapy for rheumatoid arthritis (Verhoeven et al., 1998), was found to strongly inhibit hPCFT/HCP1-mediated MTX transport (Fig. 6). This drug is little absorbed in the small intestine after oral administration and releases 5-aminosalicylic acid as an active anti-inflammatory metabolite by bacterial degradation mainly in the colon (Klotz, 1985). Taking advantage of this feature, it was originally developed as a drug to treat ulcerative colitis. It has later been found to be effective also for rheumatoid arthritis. However, such disposition characteristics of SSZ indicates that it would be present at high concentrations in the small intestine and interfere with the absorption of simultaneously administered MTX. Because the dose of SSZ (mol. wt., 398.40) is as much as 500 mg, for rheumatoid arthritis, and higher up to 2 g, for ulcerative colitis, its concentration would be nominally approximately 5 mM or higher when taken with 200 ml of water and possibly close to that in the small intestine, much exceeding its IC50 of 60.4 μM. On the other hand, the concentration of MTX (mol. wt., 454.45) would be as low as approximately 20 μM for a typical dose of 2 mg for rheumatoid arthritis, being relatively closer to its Km of 3.67 μM. Indeed, there is a clinical study suggesting that combination therapy using MTX and SSZ cannot additively improve the pharmacological efficacy of MTX or SSZ alone (Haagsma et al., 1997). It has also been known that patients treated with SSZ have a risk of folate deficiency, typically associated with an increased total homocysteine concentration in plasma (Haagsma et al., 1999). This could also be due to malabsorption of folate by inhibition of its hPCFT/HCP1-mediated transport.
In conclusion, we clearly demonstrated that hPCFT/HCP1 functions as a proton-coupled folate transporter, which can also transport MTX and possibly reduce folates, rather than a heme transporter. The hPCFT/HCP1-mediated transport of folate was consistent with those of the well characterized carrier-mediated folate transport system in the small intestine, suggesting that it is the molecular entity of the transport system. These findings for hPCFT/HCP1 heterologously expressed in mammalian cells are mostly in agreement with those reported recently for hPCFT/HCP1 expressed in X. laevis oocytes. We additionally found that sulfasalazine is a potent inhibitor of hPCFT/HCP1, which would interfere with the intestinal absorption of MTX, when coadministered in therapy for rheumatoid arthritis, as well as folate.
Footnotes
-
This study was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology, Japan and by a Grant-in-Aid for Research in Nagoya City University.
-
Y.N. and K.I. contributed equally to this work.
-
A portion of this work was previously presented: Nakai Y, Inoue K, Hatakeyama M, Hayashi Y, and Yuasa H (2006) Molecular identification and functional characterization of a pH-sensitive folate transporter in human intestine, in 21st Annual Meeting of the Japanese Society for the Study of Xenobiotics; 2006 Nov 29–Dec 1; Tokyo, Japan. p 277, Japanese Society for the Study of Xenobiotics, Tokyo, Japan.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.107.122606.
-
ABBREVIATIONS: MTX, methotrexate; DMARD, disease-modifying antirheumatic drug; RFC, reduced folate carrier; PCFT, proton-coupled folate transporter; HCP, heme carrier protein; hPCFT, human proton-coupled folate transporter; NSAID, nonsteroidal anti-inflammatory drug; HEK, human embryonic kidney; PCR, polymerase chain reaction; GFP, green fluorescent protein; PBS, phosphate-buffered saline; BSA, bovine serum albumin; THF, tetrahydrofolate; 5-MTHF, 5-methyltetrahydrofolate; TPP, thiamine pyrophosphate; SSZ, sulfasalazine; mPCFT/HCP1, mouse ortholog of PCFT/HCP1; OAT, organic anion transporter.
- Received March 12, 2007.
- Accepted May 1, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}