


Abstract
In neutrophils, growth-related protein-α (CXCL1) and interleukin-8 (CXCL8), are potent chemoattractants (Cytokine14:27–36, 2001; Biochemistry42:2874–2886, 2003) and can stimulate myeloperoxidase release via activation of the G protein-coupled receptors CXCR1 and CXCR2. The role of CXCR1 and CXCR2 in the pathogenesis of inflammatory responses has encouraged the development of small molecule antagonists for these receptors. The data presented herein describe the pharmacology of 2-hydroxy-N,N-dimethyl-3-{2-[[(R)-1-(5-methyl-furan-2-yl)-propyl]amino]-3,4-dioxo-cyclobut-1-enylamino}-benzamide (Sch527123), a novel antagonist of both CXCR1 and CXCR2. Sch527123 inhibited chemokine binding to (and activation of) these receptors in an insurmountable manner and, as such, is categorized as an allosteric antagonist. Sch527123 inhibited neutrophil chemotaxis and myeloperoxidase release in response to CXCL1 and CXCL8 but had no effect on the response of these cells to C5a or formyl-methionyl-leucyl-phenylalanine. The pharmacological specificity of Sch527123 was confirmed by testing in a diversity profile against a panel of enzymes, channels, and receptors. To measure compound affinity, we characterized [3H]Sch527123 in both equilibrium and nonequilibrium binding analyses. Sch527123 binding to CXCR1 and CXCR2 was both saturable and reversible. Although Sch527123 bound to CXCR1 with good affinity (Kd = 3.9 ± 0.3 nM), the compound is CXCR2-selective (Kd = 0.049 ± 0.004 nM). Taken together, our data show that Sch527123 represents a novel, potent, and specific CXCR2 antagonist with potential therapeutic utility in a variety of inflammatory conditions.
CXCR1 and CXCR2 are G protein-coupled receptors for a number of chemoattractant cytokines (chemokines). Both CXCR1 and CXCR2 are expressed on human neutrophils and mediate both neutrophil chemotaxis and myeloperoxidase release (Ahuja et al., 1996; Patel et al., 2001; Feniger-Barish et al., 2003). CXCR2 expression has also been demonstrated on monocytes and alveolar macrophages (Traves et al., 2004).
Interleukin-8 (IL-8) (CXCL8) is the most potent CXCR1 ligand, although granulocyte chemotactic protein-2 (CXCL6) binds to the receptor with nanomolar affinity (Wolf et al., 1998). CXCR2, on the other hand, is quite promiscuous, binding growth-related protein-α, -β, and -γ (CXCL1-3), epithelial-derived neutrophil attractant-78 (CXCL5), CXCL6, neutrophil-activating peptide-2 (CXCL7), and CXCL8. The receptors and/or their ligands are known to be elevated in a variety of inflammatory diseases, including rheumatoid arthritis, psoriasis, inflammatory bowel disease, acute respiratory distress syndrome, septic shock, pulmonary emphysema, and chronic obstructive pulmonary disease (Nickoloff et al., 1991; Woods et al., 2000; Kurdowska et al., 2002; Banks et al., 2003; Beeh et al., 2003). The putative role of CXCR1 and/or CXCR2 in inflammatory diseases has stimulated the development of small molecule antagonists with nanomolar affinity for either CXCR1, such as repertaxin [(R)(–)-2-(4-isobutylphenyl)propionyl methanesulfonamide] (Bertini et al., 2004), or CXCR2, such as SB-225002 [N-(2-hydroxy-4-nitrophenyl)-N′-(2-bromophenyl)urea], or SB-332235 [N-(2-hydroxy-3-sulfamyl-4-chlorophenyl)-N′-(2,3-dichlorophenyl)urea] (White et al., 1998). A dual CXCR1/CXCR2 antagonist, SB-468477 [N-(2-hydroxy-3-dimethylsulfonylamido-4-chlorophenyl)-N′-(2-bromophenyl)-N″-cyanoguanidine] (Traves et al., 2004), was reported to inhibit receptor binding with IC50 = 67 and 12 nM, respectively. More recently, a small molecule antagonist to murine CXCR2, Sch-N, was shown to inhibit smoke-induced pulmonary neutrophilia in mice (Thatcher et al., 2005). Details of its chemical synthesis (referred to as Compound 4 or Sch527123 [2-hydroxy-N,N-dimethyl-3-{2-[[(R)-1-(5-methyl-furan-2-yl)-propyl]amino]-3,4-dioxocyclobut-1-enylamino}-benzamide]) was published recently (Dwyer et al., 2006). The studies presented herein show that Sch527123 (Fig. 1a) is a potent, allosteric antagonist of both CXCR1 and CXCR2. As such, Sch527123 represents a novel small molecule antagonist for the treatment of inflammatory conditions arising primarily (or secondarily) from neutrophil infiltration.
Materials and Methods
Cell Culture and Isolation of Peripheral Mononuclear Cells. Ba/F3 cells stably transfected to express hCXCR1 and hCXCR2 (Hipkin et al., 2004) were propagated in RPMI 1640 supplemented with heat-inactivated FBS (10%), 1% penicillin-streptomycin, glutamine, and 1 ng/ml murine IL-3 (R&D Systems, Minneapolis, MN) under constant selection pressure with 500 μg/ml G418. Human neutrophils were isolated using one-step polymorph (Accurate Chemical & Scientific, Westbury, NY). The purity is over 95%, and 50 ml of blood normally yields 100 million neutrophils.
Cell Membranes. High-throughput screening-hCXCR1 and highthroughput screening-hCXCR2 cell membranes were purchased from Stratagene (La Jolla, CA). Ba/F3-hCXCR1, Ba/F3-hCXCR2, and human neutrophil membranes were prepared as described previously (Hipkin et al., 1997). Cells were pelleted by centrifugation and incubated in homogenization buffer (10 mM Tris-HCl, 5 mM EDTA, and 3 mM EGTA, pH 7.6) and 1 mM phenylmethylsulfonyl fluoride for 30 min on ice. The cells were then lysed with a Dounce homogenizer using a stirrer type RZR3 Polytron homogenizer (Caframo, Wiarton, ON, Canada) with 12 strokes at 900 rpm. The intact cells and nuclei were removed by centrifugation at 500g for 5 min. The cell membranes in the supernatant were then pelleted by centrifugation at 100,000g for 30 min. The membranes were resuspended in glygly buffer (20 mM glycylglycine, 1 mM MgCl2, and 250 mM sucrose, pH 7.2), aliquoted, quick-frozen, and stored at –80°C. Protein concentration in membrane preparations was determined using the Bradford method (Bradford, 1976).

Sch527123 (1a) and a schematic showing the synthesis of [3H]Sch527123 (1b).
[35S]GTPγS Binding Assay. The exchange of guanosine 5′-[γ-35S]triphosphate ([35S]GTPγS, triethylammonium salt; specific activity = 1250 Ci/mmol; PerkinElmer Life and Analytical Sciences, Boston, MA) was measured using a scintillation proximity assay (SPA) as described previously (Gonsiorek et al., 2003). For each assay point, 2 μg of membrane was preincubated for 15 min at room temperature with 200 μg of wheat germ agglutinin-coated SPA beads (WGA-SPA; GE Healthcare, Little Chalfont, Buckinghamshire, UK) in SPA binding buffer (50 mM Tris-HCl, 1 mM CaCl2, 5 mM MgCl2, 50 mM NaCl, 0.002% NaN3, 0.1% bovine serum albumin, and 10 μg/ml saponin, pH 7.6). The beads and membranes were transferred to a 96-well Isoplate (PerkinElmer Wallac, Gaithersburg, MD) and preincubated for 60 min with 10 μM GDP with the indicated concentrations of chemokine and/or Sch527123. The guanosine 5′-3-O-(thio)triphosphate exchange reaction was initiated by the addition of 0.1 nM [35S]GTPγS and was carried out for 60 min at room temperature. Membrane-bound [35S]GTPγS was measured using a 1450 Microbeta Trilux counter (PerkinElmer Wallac).
Synthesis of [3H]Sch527123. [3H]Sch527123 was prepared in two steps as shown in Fig. 1b; 3-amino-4,5,6-tribromo-2-hydroxy-N,N-dimethylbenzamide (1.08 mg) and 10% palladium on carbon (2.45 mg) were weighed into a 1-ml tritiation vessel and ethyl acetate (400 μl) and diisopropylethylamine (10 μl) was added. The vessel was attached to a IN/US systems Trisorber (Tampa, FL), frozen in liquid nitrogen, evacuated, and subjected to two freeze-thaw cycles. Carrier-free tritium gas (870 mCi) was then added to the vessel, and the reaction was vigorously stirred for 4 h at room temperature. Upon completion of the reaction, the vessel contents were diluted with ethanol and passed through a Millex-FH 0.45 μ-filter (Fisher Scientific, Pittsburgh, PA). The resulting solution was evaporated to dryness, the residue was redissolved in ethanol, and the evaporation process was repeated. A total of 73 mCi of 3-amino-4,5,6-tri-[3H]2-hydroxy-N,N-dimethylbenzamide at a radiochemical purity of 65% (HPLC System 1) was isolated from the reaction and used directly in the next step.
In a 1-ml “reactivial”, the crude 3H product (73 mCi) and 3-ethoxy-4-[1-(5-methyl-furan-2-yl)-propylamino]-cyclobut-3-ene-1,2-dione (5 mg) were dissolved in ethanol (20 μl) and diisopropylethylamine (2 μl). The reaction was heated at 65°C for 72 h, after which analysis by HPLC (HPLC System 2) showed that approximately 20% of the desired product had formed. The reaction was evaporated to dryness and purified by HPLC (HPLC System 3) to yield a batch of 10.6 mCi at a specific activity of 39 Ci/mmol. The radiochemical purity as determined by HPLC (HPLC Systems 2 and 4) was 95.2 and 97.8%.
The following HPLC systems were used: 1) SB Phenyl 3 × 150 mm, water:acetonitrile:formic acid (97:3:0.1) (Agilent Technologies, Palo Alto, CA) for 10 min followed by a step gradient to acetonitrile, 0.5 ml/min, 275 nm [HPLC eluate, Packard FloScint III cocktail (1:3); PerkinElmer Life and Analytical Sciences]; 2) SB Phenyl 3 × 150 mm, water:acetonitrile:formic acid (65: 35:0.1) (Agilent Technologies) for 15 min followed by a step gradient to acetonitrile, 0.5 ml/min, 295 nm [HPLC eluate, Packard FloScint III cocktail (1:3)]; 3) Nomura Develosil RP-AQ 9.4 × 250 mm, water:acetonitrile:formic acid (65:35:0.1), 6 ml/min, 295 nm; Phenomenex (Torrance, CA); and 4) Nomura Develosil RP-AQ 3 × 150 mm, water:acetonitrile:formic acid (65:35:0.1) for 15 min followed by a step gradient to acetonitrile, 0.5 ml/min, 295 nm [HPLC eluate, Packard FloScint III cocktail (1:3)].
Radioligand Binding Assays.125I-CXCL8 (specific activity = 2200 Ci/mmol) were obtained from PerkinElmer Life and Analytical Sciences. [3H]Sch527123 was synthesized as described above. Competition binding and saturation bindings assays and binding kinetics were performed using SPA technology (Cox et al., 2001). Membranes (2–6 μg per assay point) in SPA binding buffer were preincubated for 30 min at room temperature with WGA-SPA beads (1 μg of membrane protein/80 μg of beads), transferred to a 96-well Isoplate, and further incubated at room temperature with the radioligand and the indicated concentrations of radioactive competitors for 6 to 20 h. For experiments to determine ligand dissociation constants, membranes prebound to WGA-SPA beads were incubated with the indicated concentrations of [3H]Sch527123 in the absence or presence of excess unlabeled compound (total and nonspecific binding, respectively). The membranes and beads were further incubated with excess unlabeled compound (1–10 μM) or the appropriate diluent and counted sequentially every 30 s for up to 48 h. The dissociation rate(s) of specific binding (total–nonspecific binding) was calculated using Prism software (GraphPad Software, Inc., San Diego, CA). Ligand affinities (Ki) from competition binding experiments were calculated from binding IC50 using the Cheng-Prusoff equation (Cheng and Prusoff, 1973). Sch527123 was also tested in a variety of enzyme, receptor, and functional assays at MDS Pharma Services (King of Prussia, PA) according to established protocols.
Myeloperoxidase Assay. Neutrophils were cultured with 5 mM cytochalasin B with or without Sch527123 at 37°C for 10 min. Agonists were added, and the cells were incubated for an additional 1 h at 37°C. Tetramethylbenzidine substrate was added, and OD450 was read to determine myeloperoxidase activity in the supernatant. Myeloperoxidase release was normalized to total release after the addition of 1% Triton X-100.
Chemotaxis Assay. Chemotaxis experiments were performed as described previously (Hipkin et al., 2004). Recombinant cells were resuspended at 1 × 106/ml in assay buffer (phenol red free-RPMI 1640 supplemented with 2% FBS). Human neutrophils were resuspended at 2 × 106/ml in the same assay buffer containing 5% FBS. CXCL1 and CXCL8 used in this study were purchased from R&D Systems. CXCL1 binds only CXCR2 with high affinity, whereas CXCL8 binds both CXCR1 and CXCR2 with high affinity. Chemoattractants (30 μl) diluted in assay buffer were dispensed into the bottom wells of disposable microchemotaxis plates (ChemoTx 101-5 for BaF/3 cells and ChemoTx 101-3 for neutrophils (PMN); Neuroprobe, Gaithersburg, MD), which were then covered with filter. Cells were preincubated with Sch527123 in a CO2 incubator for 90 min. Cell aliquots (25 μl) were applied to each spot on the filter. After incubation (90 min for BaF/3 cells and 30 min for PMN in a CO2 incubator), the filters were removed. Migrated cells in the bottom wells were transferred to a Microlite luminometer plate (Thermo Electron Corporation, Waitham, MA), and 25 μl of ATPlite one-step (PerkinElmer Life and Analytical Sciences) were added to each well. After incubation at room temperature for 10 min, luminescence intensity was measured using a luminometer (Luminoskan; Thermo Electron Corporation). Data are represented as the mean of duplicate determinations.

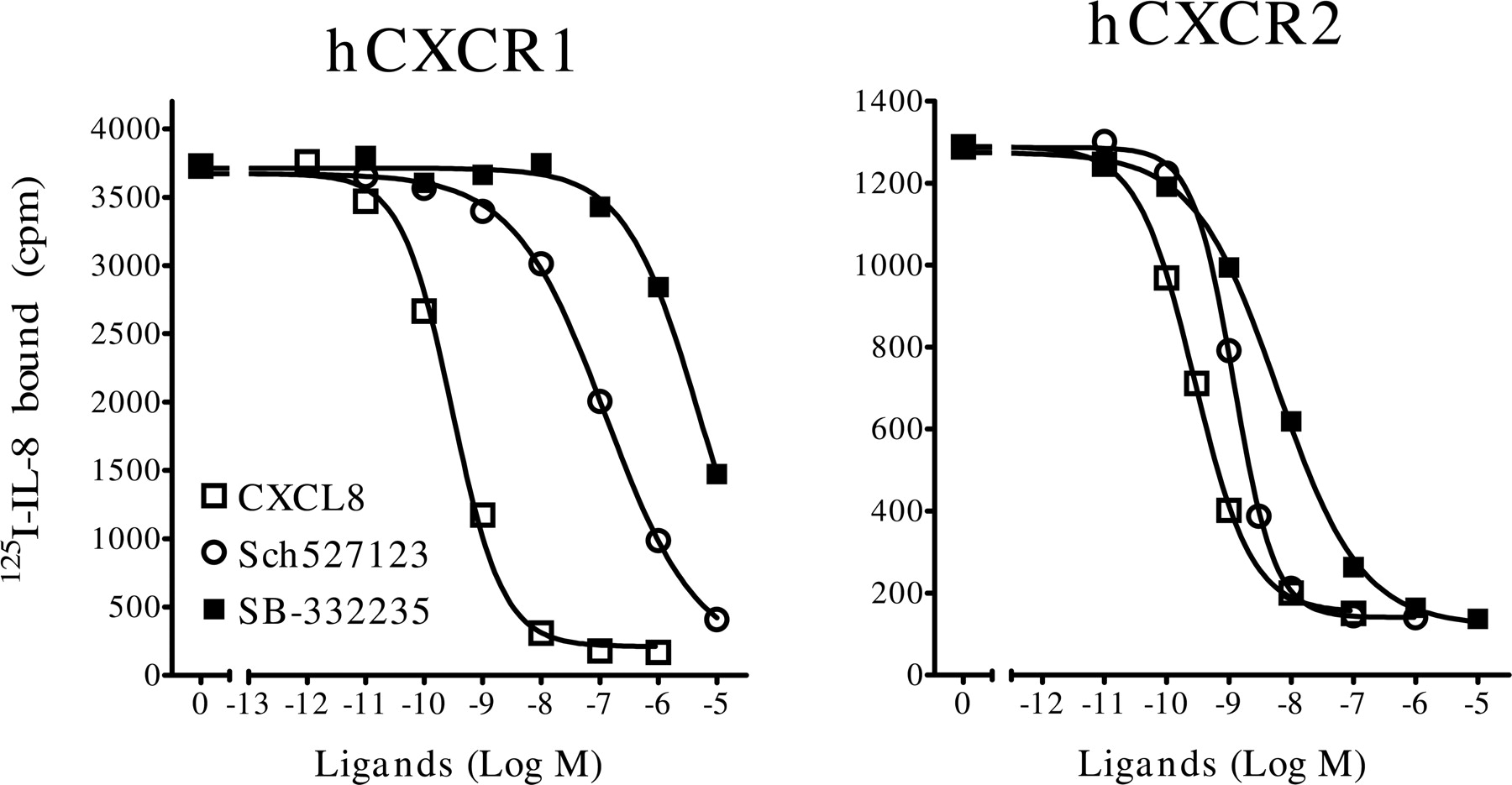
Competition binding assay with CXCL8, SB-332235, and Sch527123 at hCXCR1 and hCXCR2. Membranes from Ba/F3-hCXCR1 (left) and HTS-hCXCR2 cells (right) were incubated with the indicated concentrations of hCXCL8 (□), SB-332235 (○), or Sch527123 (▪) and 100 pM 125I-hCXCL8. Radioligand binding to the membranes was measured by WGA-SPA. Data show the mean binding of triplicate determinations from a representative experiment (n = 3–5).
Calcium Flux Assay. BaF3-hCXCR1 and BaF3-hCXCR2 cells were incubated at 37°C in a CO2 incubator for 30 to 45 min in culture media (1 × 106 cells/ml) containing 2.2 μM Fluo-3AM and 0.022% pluronic acid (Invitrogen, Carlsbad, CA), mixing briefly after 20 min. The cells were washed twice by centrifugation and suspended in Hanks' balanced saline solution, 25 mM HEPES, and 0.1% fetal calf serum (buffer A) at a density of 6 × 105 cell/ml. Cell suspension (40 μl; ∼25,000 cells) was added to each well of a 384-well polylysinecoated clear bottom plate (BD Biosciences, Bedford, MA). Plates were then centrifuged at room temperature at 400g for 1 min and then left undisturbed for 15 min. Compound or buffer (20 μl at 3× final desired concentration) was added, and the plates were incubated at 25°C for 20 min before placement into the stage of a fluorometric imaging plate reader (FLIPR384; Molecular Devices, Sunnyvale, CA). At this time, 20 μl of CXCL8 or buffer was added to give the indicated concentrations, and fluorescence measurements (excitation at 488 nm, emission at 510–570 nm) were recorded once per second for the first 60 s and then every 6 s for the next 120 s. Data were calculated as maximal fluorescence minus minimal fluorescence at baseline. Dose-response curves were fitted to the data points by nonlinear regression.
Results
Competition Binding Analyses at hCXCR1 and hCXCR2. Ba/F3-hCXCR1 or HTS-hCXCR2 membranes were incubated with 125I-CXCL8 and the indicated concentrations of CXCL8, SB-332235, or Sch527123 (Fig. 2). Receptor-bound radioligand was measured by scintillation proximity assay technology (as described under Materials and Methods).
The potency of SB-332235 to displace 125I-CXCL8 binding at CXCR2 (IC50 = 5.4 ± 1.2 nM, n = 2) was lower than that measured with Sch527123 (IC50 = 0.97 ± 0.05 nM, n = 2). The very steep Hill coefficient derived from the Sch527123 binding curve at CXCR2 (nH =–2.1 ± 0.16) suggests that the concentration of CXCR2 in the assay is too high. If compound bound:free » 10%, steep binding curves can occur (Kenakin, 1997). Sch527123 similarly inhibited the binding of 100 to 200 pM 125I-CXCL1 to CXCR2 (data not shown).
As expected, only high concentrations of the CXCR2-selective antagonist SB-332235 inhibited 125I-CXCL8 binding to CXCR1 (IC50 ≥ 10 μM). Sch527123 inhibited 125I-CXCL8 binding at CXCR1 (IC50 = 43 ± 3.6 nM, n = 10), albeit with less potency than was measured with CXCR2. The Hill coefficient for Sch527123 binding inhibition at CXCR1 was significantly less than unity (nH =–0.73 ± 0.03, n = 5) consistent with a noncompetitive interaction at the receptor between the compound and chemokine.
Effect of Sch527123 on hCXCR1 and hCXCR2 Signaling in Transfectants. The effect of Sch527123 on receptor signaling was first measured with [35S]GTPγS exchange assays in hCXCR1 or hCXCR2 membranes. Sch527123 inhibited [35S]GTPγS exchange in response to all relevant chemokines at both CXCR1 and CXCR2 (data not shown). For hCXCR1, [35S]GTPγS exchange was assessed in response to CXCL8 in the presence of 0, 30, 300, or 3000 nM Sch527123 as shown in Fig. 3 (left). CXCL8 stimulated a dose-responsive increase in [35S]GTPγS exchange with an EC50 = 0.5 ± 0.29 nM. Coincubation with Sch527123 decreased both the potency and efficacy of CXCL8 to stimulate [35S]GTPγS exchange. The effect of Sch527123 on CXCR1 signaling increased with drug concentration, although the potency of CXCL8 did not decrease predictably as the concentration of Sch527123 increased. For hCXCR2, [35S]GTPγS exchange was measured in response to CXCL1 in the presence of 0, 0.3, 1.0, or 10 nM Sch527123 as shown. CXCL1 stimulated a dose-responsive stimulation of [35S]GTPγS exchange with an EC50 = 26 ± 4.5 nM. As was the case with hCXCR1, Sch527123 decreased the potency and efficacy of CXCL1 to increase [35S]GTPγS exchange. Again, the effect of the compound on chemokine potency did not increase predictably as the levels of Sch527123 increased. Taken together, these data and the observation that the blunting of CXCR1 and CXCR2 activation by Sch527123 was not (consistently) surmountable imply that Sch527123 is a noncompetitive (allosteric) antagonist at both receptors.
We next assessed the effect of Sch527123 on chemokine stimulation of calcium signaling in cells transfected to express hCXCR1 (Ba/F3-hCXCR1) or hCXCR2 (Ba/F3-hCXCR2). Cells were loaded with Fluo-3AM and treated with CXCL8 in the presence or absence of Sch527123 or the compound SB-225002 (as described under Materials and Methods). As shown in Fig. 4, Sch527123 inhibited calcium signaling upon addition of CXCL8, with slightly more potency at hCXCR2 and then hCXCR1. In contrast, SB-225002 failed to inhibit CXCL8-stimulated calcium flux through hCXCR1 and was significantly less potent than Sch527123 in Ba/F3-hCXCR2 cells. Both of these findings are in agreement with our radioligand competition and GTPγS binding data.

Inhibition of chemokine-stimulated [35S]GTPγS exchange by Sch527123 in hCXCR1 or hCXCR2 membranes. Membranes from Ba/F3-hCXCR1 (left) and HTS-hCXCR2 cells (right) were incubated in SPA buffer containing 3 μM GDP with the specified concentrations of Sch527123 for 60 min before a 60-min incubation with the indicated concentrations of chemokine. The incubation continued for a another 60 min after the addition of 0.1 nM [35S]GTPγS. [35S]GTPγS binding to the membranes was measured by WGA-SPA. Data show the mean [35S]GTPγS binding of triplicate determinations from a representative experiment (n = 2).
Effect of Sch527123 on Cell Chemotaxis. The effects of Sch527123 on CXCR2- and CXCR1-mediated chemotaxis were examined using both recombinant cells (Ba/F3-hCXCR1 and Ba/F3-hCXCR2) and primary hPMN. Sch527123 inhibited chemotaxis (recombinant lines and neutrophils) to all relevant chemokines at CXCR1 and CXCR2 (data not shown). The data presented in Fig. 5 (left bottom) show that 1 nM Sch527123 reduced CXCL8 potency in stimulating Ba/F3-hCXCR2 chemotaxis. Coincubation with 3 nM Sch527123 both decreased chemokine potency and dramatically inhibited maximal cell chemotaxis. Cell movement to chemokine was essentially ablated with 10 nM Sch527123. For Ba/F3-CXCR1 chemotaxis (Fig. 5, left top), 30 nM Sch527123 had only a marginal effect on CXCL8 potency. Sch527123 (300 nM) significantly decreased chemokine potency and slightly decreased maximal cell movement. Effects of Sch527123 on CXCL1- and CXCL8-mediated human neutrophil chemotaxis are shown in Fig. 5 (middle). Sch527123 (3 nM) significantly inhibited the potency and efficacy of CXCL1-induced PMN chemotaxis, with almost complete inhibition of cell movement with 10 nM compound. These data are consistent with the pharmacology of the compound at CXCR2, which is selectively activated by CXCL1. Coincubation with 200 nM Sch527123 decreased only CXCL8 potency in stimulating PMN movement, a result that is very similar to the compounds effects in Ba/F3-CXCR1 chemotaxis. Similar data were generated with the CXCR2 antagonists SB-332235 and SB-225002 (data not shown). Taken together, these data suggest that directed neutrophil movement in response to low levels of CXCL8 is mediated by CXCR2, whereas PMN chemotaxis in response to higher concentrations of the chemokine occurs via activation of CXCR1.

Effect of Sch527123 and SB-225002 on chemokine-stimulated calcium flux in BaF-hCXCR1 and BaF-hCXCR2 cells. In brief, cells were loaded with Fluo-3AM/pluronic acid and preincubated with vehicle (○) Sch527123 (▪) or SB-225002 (□) for 20 min before stimulation with increasing concentrations of CXCL8 in vehicle-treated cells or 30 nM CXCL8 in cells pretreated with antagonist as described under Materials and Methods. Data were collected using a fluorometric imaging plate reader (FLIPR384) and represent the maximal fluorescence minus minimal fluorescence at baseline. Data show the mean calcium levels of triplicate determinations from a representative experiment (n = 2).

Effect of Sch527123 on chemokine-stimulated chemotaxis. Ba/F3 cells transfected to express either hCXCR1 (top left) or hCXCR2 (bottom left) or hPMN were pretreated with the indicated concentrations of Sch527123 and allowed to migrate to either CXCL8, CXCL1, C5a, or fMLP using a 96-well ChemoTx plate (5-micron pore size). Data of triplicate determinations from a representative experiment (n = 2) are expressed as the percentage of total cells migrating toward the respective chemokine.
Assessment of Sch527123 Selectivity. To assess Sch527123 selectivity, the effect of the compound on neutrophil chemotaxis to C5a and fMLP was tested. As can be seen in Fig. 5 (right), migration of neutrophils to these agents was unaffected by preincubation with 300 nM Sch527123, a concentration that effectively antagonizes chemotaxis through CXCR1 or CXCR2 in recombinants or human neutrophils. Neutrophil migration to leukotriene B4 was similarly unaffected by Sch527123 (data not shown). In addition, Sch527123 inhibited myeloperoxidase release from human neutrophils in response to CXCL8 without inhibiting the cell response to C5a, fMLP (Fig. 6), or leukotriene B4 (data not shown). The potency of Sch527123 to inhibit the CXCL8 response (≈30 nM) is consistent with the activity of the compound at CXCR1 (Figs. 2, 3, 4) and the observation that CXCL8 is more efficacious than CXCR2-selective chemokines (i.e., CXCL1) in stimulating myeloperoxidase release from neutrophils (data not shown). Lastly, Sch527123 (10 μM) had no significant activity (≥50% inhibition) in counterscreening against a large panel of G protein-coupled receptors, enzymes, and ion channels (MDS Pharma Services) according to established protocols (data not shown). Taken together, these data show that Sch527123 is a potent and specific antagonist of CXCR1 and CXCR2.

Effect of Sch527123 on myeloperoxidase release by hPMN in response to CXCL8, fMLP, or C5a. hPMN were pretreated for 5 min with the indicated concentrations of Sch527123 and stimulated with 50 nM CXCL8 (□), fMLP (▪), or C5a (trif) for 1 h at 37°C. Tetramethylbenzidine substrate was added, and OD450 was read to determine myeloperoxidase activity in the supernatant. Myeloperoxidase release was normalized to total release after the addition of 1% Triton X-100. Data show the mean of triplicate determinations from a representative experiment (n = 2).
Characterization of [3H]Sch527123 at hCXCR1 and hCXCR2. As an allosteric, binding affinities for Sch527123 derived from simple competitive binding assays with radiolabeled chemokine are not valid. To directly measure its affinity at CXCR1 and CXCR2, [3H]Sch527123 was synthesized (Fig. 1) by tritium gas reduction of 3-amino-4,5,6-tribromo-2-hydroxy-N,N-dimethylbenzamine, followed by coupling with 3-ethoxy-4-[1-(5-methyl-furan-2-yl)-propylamino]-cyclo-3-ene-1,2-dione. Saturation binding analysis with [3H]Sch527123 (specific activity = 39 Ci/mmol) demonstrated saturable, high-affinity binding in membranes expressing hCXCR1 (Kd = 3.9 ± 0.3 nM; Fig. 7, left) or CXCR2 (Kd = 0.049 ± 0.004 nM; Fig. 7, right). Whereas [3H]Sch527123 binding reached steady state ≤6 h in CXCR1 membranes, binding equilibrium was achieved only after prolonged incubation (≈24 h) in CXCR2 membranes. Binding competition with [3H]Sch527123 in membranes expressing CXCR1 and CXCR2 (Fig. 8) measured at 4 and 24 h shows that Sch527123 binds with high affinity to both CXCR1 and CXCR2, although the compound is CXCR2-selective. Because of the relatively low-specific activity of the trace, [3H]Sch527123 was used at concentrations (5–11 nM) in (great) excess of the compound affinities for CXCR1 and CXCR2. Therefore, binding affinities (Ki) generated from binding IC50 using this approach are suspect (at 24 h; CXCR1, Sch527123 Ki = 8.6 nM, SB-332235 Ki = 550 nM; CXCR2, Sch527123 Ki = 8 pM, SB-332235 Ki = 1.0 nM) due to the large correction in the ChengPrusoff equation (Cheng and Prusoff, 1973). Nonetheless, consistent with the binding assays with 125I-CXCL8 (Fig. 2), SB-332235 binds with lower affinity to both receptors relative to Sch527123. In addition, at either 4 or 24 h, CXCL8 does not effectively displace [3H]Sch527123 from CXCR1 or CXCR2, consistent with noncompetitive interaction at the receptors between chemokine and compound (see Fig. 3).

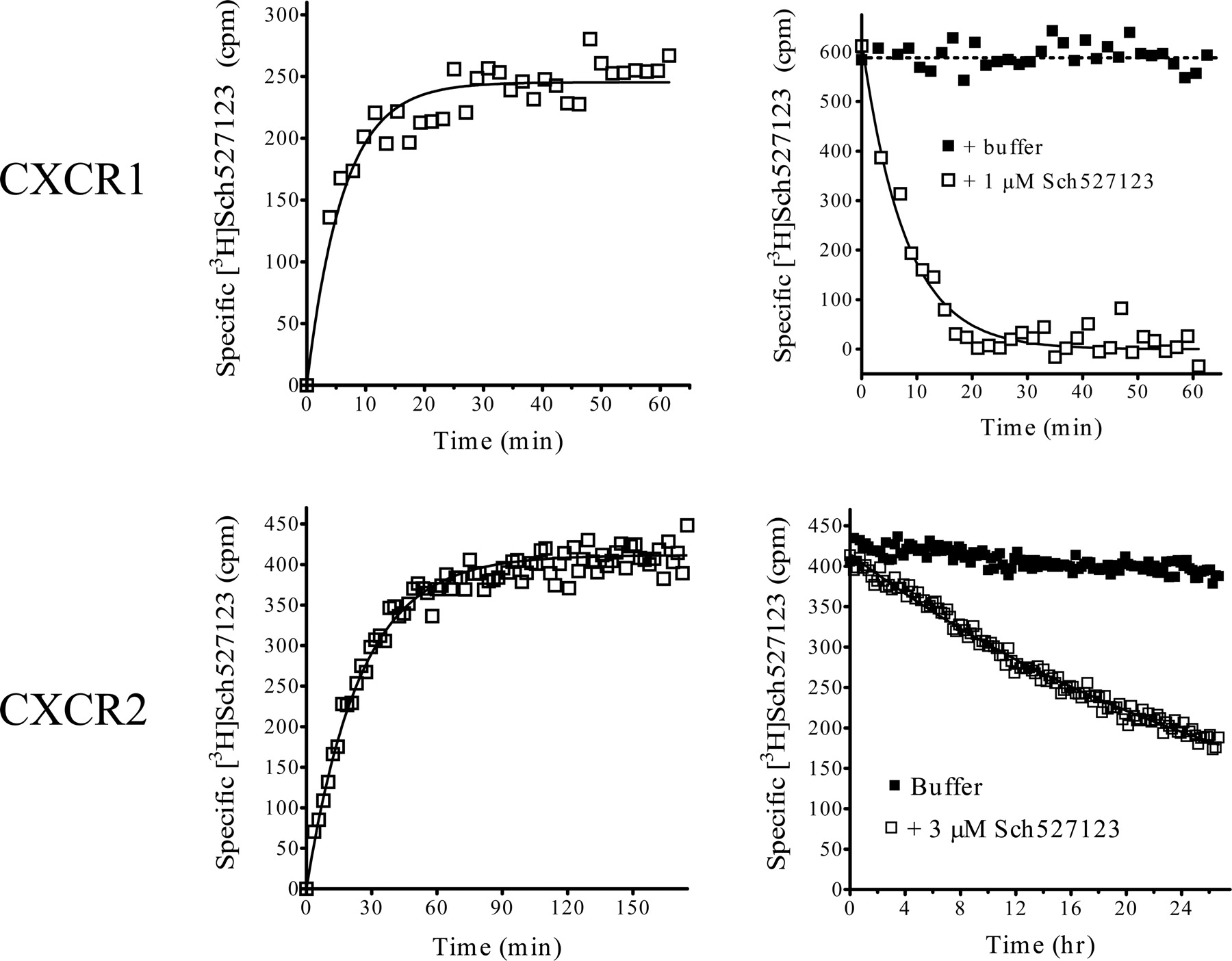
Saturation binding analyses with [3H]Sch527123 at hCXCR1 and hCXCR2. Membranes from HTS-hCXCR1 (left) and HTS-hCXCR2 (right) cells (4 μg/well) were incubated in SPA buffer at room temperature for the indicated times (as described under Materials and Methods) with the indicated concentrations of [3H]Sch527123 in the absence (total binding) or presence of 1 μM unlabeled compound (nonspecific binding). Radioligand binding to the membranes was measured by WGA-SPA scintillation at the indicated times. Data show the mean binding of triplicate determinations from a representative experiment (n = 2).
To determine the kinetic binding constants for [3H]Sch527123, membranes were incubated with 3 to 10 nM [3H]Sch527123 in the presence or absence of excess unlabeled compound, and the time course of specific binding was analyzed. The observed Kon values were linearly proportional to the concentration of [3H]Sch527123 for both CXCR1 and CXCR2 (data not shown). In CXCR1 membranes, an association binding constant (k1) value (1.64 × 107 ± 2.70 × 106 min–1 M–1, n = 3) was derived from an observed Kon = 0.25 min–1 (Fig. 9, top left). Binding at CXCR1 was reversible and dissociated fairly rapidly with a k2 = 0.141 ± 0.014 min–1 (t½ ∼5 min.) (Fig. 9, top right). Using these constants, the Sch527123 Kd for CXCR1 was calculated (k2/k1 = 8–9 nM), a value comparable with the Kd derived through saturation analyses (see above). With CXCR2 (Fig. 9, bottom left), the value for Sch527123 k1 = 2.08 × 107 ± 2.00 × 106 min–1 M–1 (n = 4) derived from the observed association rate constant (Kon = 0.12 min–1) was similar to that measured with CXCR1. The binding of [3H]Sch527123 to CXCR2 was also reversible (Fig. 9, bottom right), albeit at a much slower rate (k2 = 0.00058 ± 0.0001 min–1) with a t1/2 ≈ 22 h. Sch527123 Kd for CXCR2 calculated using its kinetic values (k2/k1 = 0.029 nM) was consistent with the affinity constant generated by saturation analyses (49 ± 4 pM; Fig. 7, left).

Competition binding analyses with [3H]Sch527123 at hCXCR1 and hCXCR2. Membranes from HTS-hCXCR1 (left panels) and HTS-hCXCR2 (right panels) cells (4 μg/well) were incubated in SPA buffer at room temperature for the indicated times with [3H]Sch527123 (5–11 nM) and the indicated concentrations of Sch527123, SB-332235, or CXCL8. Radioligand binding to the membranes was measured by WGA-SPA scintillation at the indicated times. Data show the mean binding of triplicate determinations from a representative experiment (n = 3–5).
Having established the binding constants for Sch527123 with CXCR1 and CXCR2 in transfectants, we initiated similar studies using human neutrophils, which express both receptors. Figure 10 illustrates the time course of [3H]Sch527123 dissociation in neutrophil membranes from two representative individuals (n = 3). [3H]Sch527123 dissociates with two very distinct rates. Approximately 60 to 70% total specific binding dissociates fairly rapidly (k2 = 0.077 ± 0.002 min–1) (regraphed in panels at right), whereas the remainder dissociates much more slowly (k2 = 0.00058 ± 0.00012 min–1). These dissociation rates are indistinguishable from the k2 values derived with recombinant CXCR1 and CXCR2, respectively (see Fig. 9). Using k1 values derived with recombinant membranes (or neutrophils; data not shown), calculation of binding affinities (k1/k2) generated two distinct binding affinities (Kd ∼5 nM and ∼0.04 nM), values that are indistinguishable from the CXCR1 and CXCR2 Kd values in recombinantly expressed receptor. Therefore, we conclude that [3H]Sch527123 binds with predicted affinities to CXCR1 and CXCR2 expressed on human neutrophils.
Discussion
The data presented herein illustrate that Sch527123 is a potent and specific allosteric antagonist of hCXCR1 and hCXCR2. The extraordinary potency of Sch527123 at hCXCR2 (49 pM) arises from its exceedingly slow dissociation rate from the receptor (approximately 24 h at room temperature). Although this dissociation will be predictably faster at 37°C, extension of CXCR2 coverage arising from slow receptor dissociation may well be therapeutically beneficial. The pharmacology of Sch527123 at CXCR1 is more “orthodox”, i.e., a low nanomolar affinity and more rapid receptor dissociation profile (approximately 5 min at room temperature). Therefore, whereas Sch527123 is a potent CXCR1 antagonist, it is undoubtedly CXCR2-selective. The fact that Sch527123 does not inhibit chemokine binding to CXCR1 and CXCR2 in a competitive manner is not unexpected, considering the size of the chemokines (8–10 kDa) and the multifaceted nature of chemokine-receptor interactions (Wu et al., 1996; Suetomi et al., 1999). Allosteric antagonism of chemokine receptors by small molecule compounds is probably the norm. For example, the CXCR1 antagonist repertaxin was described as an allosteric ligand that does not block chemokine binding but potently inhibits human neutrophil migration induced by CXCL8. A panel of structurally dissimilar antagonists were recently shown to interact with CCR5 in an allosteric fashion relative to its chemokine ligands (Watson et al., 2005). Indeed, CCR5 antagonist 873140 does not effectively inhibit 125I-CCL5 binding (Maeda et al., 2004; Watson et al., 2005), although CCR5 activation by the chemokine is blocked by the compound (Watson et al., 2005). Another CCR5 antagonist TAK-220 inhibits the binding of radiolabeled CCL3 (MIP-1α) and CCL5 but not CCL4 (Takashima et al., 2005). The effect of TAK-220 on CCR5 activation by CCL4 was not reported in this study. In contrast, Sch527123 potently inhibits CXCR1 and CXCR2 activation by all of their cognate ligands (data not shown) and inhibits the binding of 125I-CXCL8 to CXCR1 and CXCR2 (Fig. 2) and 125I-CXCL1 to CXCR2 (data not shown).

The kinetics of [3H]Sch527123 binding at hCXCR1 and hCXCR2. To measure the association rate of [3H]Sch527123 binding, membranes from HTS-hCXCR1 (top left) and HTS-hCXCR2 (bottom left) cells (2–4 μg/well) were incubated in SPA buffer at room temperature for the indicated times with 3 nM [3H]Sch527123 in the absence (total binding) or presence of 3 to 10 μM unlabeled compound (non-specific binding). Data show the mean binding of triplicate determinations measured at the indicated times from a representative experiment (n = 2). To measure the rate of [3H]Sch527123 dissociation, membranes from HTS-hCXCR1 (top right) and HTS-hCXCR2 (bottom right) cells (2–4 μg/well) were incubated in SPA buffer at room temperature for the indicated times with 3 to 10 nM [3H]Sch527123 in the absence (total binding) or presence of 1 to 3 μM unlabeled compound (nonspecific binding) before the addition of either buffer (▪) or 1 to 3 μM unlabeled compound (□). Data show the mean binding of triplicate determinations from a representative experiment (n = 2) measured sequentially at the indicated times.

[3H]Sch527123 dissociation kinetics in human neutrophil membranes. Human neutrophil membranes (6 μg/well) were incubated with 5 nM [3H]Sch527123 in the absence (total binding) or presence of 10 μM unlabeled compound (nonspecific binding) for 60 min. The incubation was then continued after the addition of 10 μM unlabeled compound (•) or diluent (□). Receptor-bound [3H]Sch527123 was measured sequentially at the indicated times (n = 3). Data (mean specific binding ± S.E.M. of triplicate determinations) from two representative experiments are shown. After the addition of Sch527123, data were analyzed using a two-phase exponential decay. The data from the initial 60 min were regraphed in the right panels.
Sch527123 could have clinical utility in disease processes that may be directly (or indirectly) mediated by cells expressing CXCR1 and/or CXCR2, i.e., rheumatoid arthritis, inflammatory bowel disease, acute respiratory distress syndrome, septic shock, pulmonary emphysema, psoriasis, and chronic obstructive pulmonary disease (Nickoloff et al., 1991; Woods et al., 2000; Kurdowska et al., 2002; Banks et al., 2003; Beeh et al., 2003). Indeed, the first definitive evidence of CXCR2 ligand expression in psoriatic tissue (CXCL7 and CXCL8) was published over 15 years ago (Baggiolini and Walz, 1989; Christophers et al., 1989), with the first evidence of receptor up-regulation published 4 years later (Schulz et al., 1993; Kulke et al., 1998). Studies implicating CXCR2 in mediating various pulmonary inflammations are many (for review, see Mukaida, 2003; De Boer, 2002; Hay and Sarau, 2001). Studies with Sch527123 in a variety of rodent and primate models of pulmonary inflammation (Thatcher et al., 2005; Chapman et al., 2007) support this hypothesis.
In conclusion, Sch527123 is a novel allosteric antagonist that binds with high affinity to both CXCR1 and CXCR2. Nonetheless, the concentrations of Sch527123 required to ablate PMN chemotaxis through CXCR1 (≥1 μM versus CXCL8; data not shown) are such that, therapeutically, Sch527123 would be predicted to be a de facto CXCR2 antagonist.
Acknowledgments
We thank Jay Fine and John Hey for advice and support.
Footnotes
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.106.118927.
-
ABBREVIATIONS: IL-8, interleukin-8 (CXCL8); CXCL5, endothelial-cell derived neutrophil activating protein 78; fMLP, formyl-methionyl-leucyl-phenylalanine; CXCL6, granulocyte chemotactic protein-2; CXCL1-3, growth-related protein-α,-β, and -γ;[35S]GTPγS, guanosine 5′-[γ-35S]-triphosphate, triethylammonium salt; IL-3, interleukin-3; MPO, myeloperoxidase; CXCL7, neutrophil-activating peptide-2; FBS, fetal bovine serum; RLU, relative light intensity unit; WGA-SPA, wheat germ agglutinin bead-scintillation proximity assay; Sch527123, 2-hydroxy-N,N-dimethyl-3-{2-[[(R)-1-(5-methyl-furan-2-yl)-propyl]amino]-3,4-dioxo-cyclobut-1-enylamino}-benzamide; repertaxin, (R)(–)-2-(4-isobutylphenyl)propionyl meth-anesulfonamide; SB-225002, N-(2-hydroxy-4-nitrophenyl)-N′-(2-bromophenyl)urea; SB-332235, N-(2-hydroxy-3-sulfamyl-4-chlorophenyl)-N′-(2,3-dichlorophenyl)urea; SB-468477, N-(2-hydroxy-3-dimethylsulfonylamido-4-chlorophenyl)-N′-(2-bromophenyl)-N″-cyanoguanidine; h, human; PMN, neutrophil(s); HPLC, high-performance liquid chromatography; 873140, 4-{[4-({(3R)-1-butyl-3-[(R)-cyclohexyl(hydroxy)methyl]-2,5-dioxo-1,4,9-triazaspiro[5,5]undec-9-yl}methyl)phenyl]oxy}benzoic acid hydrochloride; CCL5, regulated on activation normal T cell expressed and secreted; TAK-220, 1-acetyl-N-{3-[4-(4-carbamoylbenzyl)piperidin-1-yl]propyl}-N-(3-chloro-4-methylphenyl)piperidine-4-carboxamide.
- Received December 21, 2006.
- Accepted May 7, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}