


Abstract
The Smoothened receptor (Smo) mediates hedgehog (Hh) signaling critical for development, cell growth, and migration, as well as stem cell maintenance. Aberrant Hh signaling pathway activation has been implicated in a variety of cancers, and small-molecule antagonists of Smo have entered human clinical trials for the treatment of cancer. Here, we report the biochemical characterization of allosteric interactions of agonists and antagonists for Smo. Binding of two radioligands, [3H]3-chloro-N-[trans-4-(methylamino)cyclohexyl]-N-{[3-(4-pyridinyl)-phenyl]methyl}-1-benzothiophene-2-carboxamide (SAG-1.3) (agonist) and [3H]cyclopamine (antagonist), was characterized using human Smo expressed in human embryonic kidney 293F membranes. We observed full displacement of [3H]cyclopamine by all Smo agonist and antagonist ligands examined. N-[(1E)-(3,5-Dimethyl-1-phenyl-1H-pyrazol-4-yl)methylidene]-4-(phenylmethyl)-1-piperazinamine (SANT-1), an antagonist, did not fully inhibit the binding of [3H]SAG-1.3. In a functional cell-based β-lactamase reporter gene assay, SANT-1 and N-[3-(1H-benzimidazol-2-yl)-4-chlorophenyl]-3,4,5-tris(ethyloxy)-benzamide (SANT-2) fully inhibited 3-chloro-4,7-difluoro-N-[trans-4-(methylamino)cyclohexyl]-N-{[3-(4-pyridinyl)phenyl]methyl}-1-benzothiophene-2-carboxamide (SAG-1.5)-induced Hh pathway activation. Detailed “Schild-type” radioligand binding analysis with [3H]SAG-1.3 revealed that two structurally distinct Smoothened receptor antagonists, SANT-1 and SANT-2, bound in a manner consistent with that of allosteric modulation. Our mechanism of action characterization of radioligand binding to Smo combined with functional data provides a better understanding of small-molecule interactions with Smo and their influence on the Hh pathway.
The Hedgehog (Hh) signal transduction pathway is critical to development, differentiation, growth, and cell migration. Although Hh signaling is significantly curtailed in adults, it retains functional roles in stem cell maintenance (Lai et al., 2003; Machold et al., 2003; van den Brink et al., 2004) and tissue repair (Ito et al., 1999; Miyaji et al., 2003; Watkins et al., 2003; Karhadkar et al., 2004). Aberrant Hh signaling pathway activation has been implicated as the cause of certain cancers, including basal cell carcinomas and medullo-blastomas (Hahn et al., 1996; Dahmane et al., 1997; Reifen-berger et al., 1998; Xie et al., 1998; Zurawel et al., 2000), and in supporting the tumor microenvironment in numerous other cancers (Berman et al., 2003; Watkins et al., 2003; Karhadkar et al., 2004; Dierks et al., 2007) via both Hh ligand-independent and ligand-dependent pathways. Small-molecule antagonists of the Smoothened receptor (Smo) have entered human clinical trials for the treatment of advanced basal cell carcinoma and metastatic colorectal cancer (Von Hoff et al., 2008)
Hh polypeptide secretion activates the Hh pathway through two membrane-associated receptors, Patched (Ptch) and Smo. Hh binds to Ptch, the 12-transmembrane spanning receptor, alleviating suppression of Smo, a seven-transmembrane protein that is structurally similar to G protein-coupled receptors. Smo activation stimulates the Gli family of transcription factors to induce the expression of specific genes, including Gli and Ptch, forming both a negative and a positive feedback loop.
Because of the important role the Hh pathway plays in tumorigenesis and stem cell maintenance, small-molecule regulators of the Hh pathway have attracted considerable interest as potential therapeutic agents. Cyclopamine, a plant-derived steroidal alkaloid, has been shown to directly bind to and inhibit the Smo receptor (Chen et al., 2002a). Various screening efforts using cellular Hh pathway or biochemical assays directed at the Smo receptor have identified potent small-molecule agonists and antagonists with apparently competitive binding behavior for Smo (Chen et al., 2002b; Frank-Kamenetsky et al., 2002; Williams et al., 2003). The 12-pass transmembrane protein Ptch is inhibitory to Smo. Under physiological conditions, activation of the pathway is caused by binding of the endogenous morphogen Hh to Ptch, which leads to internalization of Ptch and consequently alleviates the inhibitory affect of Ptch on Smo. The exact mechanism for Ptch inhibition of Smo has not been elucidated. In addition, no endogenous “orthosteric” agonist for Smo has been identified. In fact, it has been proposed that Ptch may translocate an endogenous sterol (such as cyclopamine), which inhibits activation and is relieved when Hh binds Ptch (Bijlsma et al., 2006).
Here, we report the biochemical characterization of allosteric interactions of small-molecule agonists and antagonists for Smo. Binding of two radioligands, [3H]SAG-1.3 (an agonist) and [3H]cyclopamine (an antagonist), was characterized using human Smo expressed in HEK293F membranes. “Schild-type” radioligand binding analysis with [3H]SAG-1.3 revealed that the Smo antagonists SANT-1 and SANT-2 bound in a manner consistent with that of allosteric modulation. Both SANT-1 and -2 seem to bind in an allosteric manner exhibited by their partial competition of [3H]SAG-1.3 binding. In contrast, SANT-1 and -2 fully inhibited SAG-1.5-induced Hh pathway activation in a β-lactamase reporter gene assay in cells. Our results provide biochemical evidence that suggests SANT-1 and SANT-2 are allosteric inhibitors of [3H]SAG-1.3 binding to Smo. The mechanism of action characterization of radioligand binding to Smo combined with functional data supports the idea that functionally active small molecules interact with Smo in both competitive and allosteric manners. A deeper understanding of small-molecule interactions with Smo can positively influence the search for Smo-binding, functionally active chemical starting points.
Materials and Methods
Materials. SAG-1.3 was synthesized for radiolabeling by the Discovery Medicinal Chemistry Department of GlaxoSmithKline (Research Triangle Park, NC). The radiolabeling of [3H]SAG-1.3 (specific activity, 70 Ci/mmol) was performed by GE Healthcare (Chalfont St. Giles, Buckinghamshire, UK). [3H]Cyclopamine (specific activity, 20 Ci/mmol) was obtained from American Radiolabeled Chemicals (St. Louis, MO). SYTO-63 and BODIPY-cyclopamine were purchased from Invitrogen (Carlsbad, CA) and Toronto Research Chemicals Inc. (North York, ON, Canada), respectively. The CellSensor cell line and culture reagents Dulbecco's modified eagle's medium (DMEM)/GlutaMAX, Opti-MEM, newborn calf serum, nonessential amino acids, HEPES, and blasticidin were purchased from Invitrogen. Penicillin/streptomycin was purchased from Mediatech (Manassas, VA). Assay plates and compound dilution plates were purchased from Greiner Bio-One (Monroe, NC). Unlabeled cyclopamine and tomatidine were purchased from BIOMOL Research Laboratories (Plymouth Meeting, PA); 9-cyclohexyl-N-[4-(4-morpholinyl)phenyl]-2-(1-naphthalenyloxy)-9H-purin-6-amine (purmorphamine) and 3-keto-N-(aminoethyl-aminocaproyl-dihydrocinnamoyl)cyclopamine (cyclopamine-KAAD) were from Calbiochem (San Diego, CA); and SANT-1 was from Tocris (Ellisville, MO). SAG-1.3, SAG-1.5, SANT-2, Compound 1, GDC-0449, and N-{4-chloro-3-[5-(dimethylamino)-1H-benzimidazol-2-yl]phenyl}-2,3-dihydro-1,4-benzodioxin-6-carboxamide (Z′′′′) were synthesized by the Medicinal Chemistry Research Department of GlaxoSmithKline Research Laboratories (Durham, NC) and WuXi Pharma (Shanghai, People's Republic of China). Phenylmethylsulfonyl fluoride, pepstatin, Complete EDTA-free protease tablets, and Triton X-100 were purchased from Roche Diagnostics (Indianapolis, IN). Leupeptin was purchased from Calbiochem (San Diego, CA). All other components of the membrane preparation buffers and all other standard reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Generation of Recombinant BacMam Virus. The Smo Bac-Mam virus was constructed and amplified based on published methods (Fornwald et al., 2007). The coding region for the Smo gene, cloned in-house from human ovary, aligns with National Center for Biotechnology Information sequence NM_005631.3.
Transduction of HEK293F with Smo BacMam Virus. The cell line used for transductions, HEK293F, was purchased from Invitrogen. HEK293F cells were subcultured twice weekly in FreeStyle 293 expression medium (Invitrogen) and maintained in 3-liter sterile, plastic Erlenmeyer flasks on shaking platform set at 90 rpm in a humidified 37°C incubator with 5% CO2. Cells were seeded on day 0 at 6 × 105 cells/ml. The next day, the transduction was performed at a cell density of 1.0 to 1.2 × 106 cells/ml using 40 Smo BacMam virus particles per cell; the culture was supplemented with 2 mM sodium butyrate. Approximately 24 h later, transduced cells were harvested by centrifugation at 400g for 30 min at 4°C, washed once with a balanced salt solution, repelleted, and the pellet was flash-frozen in liquid nitrogen. The cell pellets were stored at -80°C until processed for membranes.
Membrane Preparation. HEK293F/Smo_BacMam cell pellets were resuspended at 4°C in 10 volumes of ice-cold buffer A (50 mM HEPES, pH 7.4, 1 mM EDTA, 25 μg/ml bacitracin, and 100 μM leupeptin) plus 1 mM phenylmethylsulfonyl fluoride and 2 μM pepstatin per gram of cell pellet, homogenized in a Waring Blender for 15 s, placed on ice for 5 min, and homogenized for an additional 15 s. To remove large particles, a low-speed centrifugation (500g for 30 min at 4°C) was performed, followed by high-speed centrifugation (48,000g for 45 min at 4°C), resuspension in buffer A plus 2 μM pepstatin, and a final high-speed centrifugation (48,000g for 45 min at 4°C). A Dounce homogenizer was used to resuspend the final pellet using ice-cold buffer A. The membrane suspension was passed through a 23-gauge needle, aliquoted, and stored at -80°C. Total protein concentration of the membrane preparation was determined with a Coomassie Plus reagent kit from Pierce Chemical (Rockford, IL) using bovine serum albumin as the standard.
[3H]SAG-1.3 and [3H]Cyclopamine Radioligand Binding Assays. Radioligand binding assays were performed under the same conditions, independently of the radioligand used. Membranes were diluted in buffer B (50 mM HEPES and 3 mM MgCl2, pH 7.2, at 23°C) to a concentration of 0.01 to 0.02 mg protein/ml. Assays were initiated by the addition of 200 μl of membrane suspension to 100 μl of radioligand at 1 to 3 times radioligand Kd and various concentrations of inhibitors in buffer B plus a cocktail of protease inhibitors (EDTA-free; Roche Diagnostics) and 0.02% bovine serum albumin to reduce nonspecific binding. Binding assays were performed in duplicate in polypropylene 96-well plates (Costar; Corning Life Sciences, Acton, MA). Nonspecific binding was defined in the presence of 1 to 10 μM cyclopamine ([3H]SAG-1.3 assays) or 10 μM SANT-2 ([3H]cyclopamine assays). Competition assays were performed at 23°C for 5 h to allow adequate time for the two ligands to reach equilibrium for binding. The separation of bound from free radioligand was accomplished by rapid vacuum filtration of the incubation mixture over glass fiber filters (Inotech Biosystems, Rockville, MD) presoaked for 2 h in 0.3% polyethylenimine, pH 13.0, using a cell harvester (Inotech Biosystems). Filters were washed 2 times with 0.3 ml of ice-cold phosphate-buffered saline, pH 7.0, containing 0.01% Triton X-100. Radioactivity on the filters was quantified using a TopCount liquid scintillation counter (PerkinElmer Life and Analytical Sciences, Boston, MA). Radioactive saturation experiments were performed with the addition of labeled [3H]SAG-1.3 over a range of concentrations (0.1–100 nM). Isotopic unlabeled saturation studies were carried out using [3H]cyclopamine or [3H]SAG-1.3 at two fixed concentrations in the presence of varied concentrations of unlabeled cyclopamine (0.001–10 μM) or SAG-1.3, respectively. Mode of inhibition (MOI) studies were performed using similar conditions including double titrations of both radioligand and unlabeled Smo ligands.
BODIPY-Cyclopamine Whole Cell Binding Assay. HEK MS RII cells were transduced at a density of 3 × 105 cells/ml with 17 Smo BacMam virus particles/cell in DMEM containing 0.5% fetal bovine serum and seeded at 15,000 cells/well in 384-well poly-d-lysine-coated plates followed by overnight (>20-h) incubation at 37°C and 5% CO2. Compound stocks were prepared in dimethyl sulfoxide, except for BODIPY-cyclopamine, which was prepared in 100% ethanol, according to the manufacturer's instructions. On day 2, DMEM media were removed with a Power Washer 384 (Tecan, Durham, NC); cells were fixed by adding 3.7% (v/v) formaldehyde (VWR, Bridgeport, NJ) to the plates using a Multidrop 384 (Thermo Fisher Scientific, Waltham, MA) and incubated for 20 min. After aspirating, the fixation buffer, 2 μM SYTO-63, 25 nM BODIPY-cyclopamine, and indicated concentrations of compounds (all diluted in Hanks' balanced salt solution without Ca2+/Mg2+; Invitrogen) were transferred to the assay plates with a Cybi-well (Cybio, Jena, Germany). Assay plates were incubated for 2 h, washed several times with buffer, and then imaged on an Opera instrument (PerkinElmer Life and Analytical Sciences). The fixing and washing protocol was optimized to eliminate nonspecific binding of the BODIPY-cyclopamine probe. On the Opera instrument, 635-nm and 488-nm excitation lasers were used to excite SYTO-63 and BODIPY, respectively, whereas 690/50- and 525/50-nm filters were used to collect the fluorescence signals. A primary 405/488/635-nm dichroic filter was used to filter laser light; a 650-nm long pass filter was used to split SYTO-63 and BODIPY emission wavelengths, sending wavelengths longer than 650-nm to the 690/50-nm filter and the remaining light to the 568-nm dichroic filter. The 568-nm dichroic filter was used to send wavelengths shorter than 568 nm to the 525/50-nm filter for detection of the BODIPY signal. Laser power and exposure times were optimized to avoid any cross talk between detection channels. The SYTO-63 signal was used to identify all cells as objects, whereas the BODIPY signal was used to quantify BODIPY-cyclopamine binding to Smo receptors in cells. Acapella software in the Opera imaging system was used to develop the analysis algorithms for quantification of the images.
β-Lactamase Reporter Gene Assay. CellSensor Gli-β-lactamase NIH3T3 cells were maintained at 20 to 80% confluence in DMEM/GlutaMAX media supplemented with 10% newborn calf serum, 0.1 mM nonessential amino acids, 25 mM HEPES, pH 7.3, 100 U/ml penicillin, 100 μg/ml streptomycin, and 5 μg/ml blasticidin. Assay medium was Opti-MEM supplemented with 0.5% newborn calf serum, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 10 mM HEPES, 100 U/ml penicillin, and 100 μg/ml streptomycin. Antagonist and agonist assays were carried out in black-wall, clear-bottomed, 384-well assay plates with 10,000 cells/well. For the antagonist assay, cells were plated using a Multidrop 384 (Thermo Fisher Scientific) at a volume of 20 μl/well in the presence of an agonist (EC80), followed by the addition of test compounds with a Cybi-well, and incubated for 40 to 48 h before detection. For the agonist assay, cells were plated, compounds were added as described above, and plates were incubated in the presence of test compounds for 40 to 48 h before detection. Final assay conditions included 1% dimethyl sulfoxide final concentration in a 25-μl total volume assay. After the 40- to 48-h incubation, 5 μl of a 6× LiveBlazer loading reagent (prepared as instructed by Invitrogen) was added to the assay plates using a Multidrop 384. Plates were then read using an Envision reader (PerkinElmer Life and Analytical Sciences) in bottom-read mode, with an excitation wavelength of 409 nm and emission wavelengths of 460 and 535 nm to measure the ratio of fluorescence intensity at 460 versus 535 nm as specified by Invitrogen.
Data Analysis. The binding rate constants (k+1 and k-1), apparent equilibrium dissociation constants (Kd), and the maximum number of binding sites (Bmax), EC50, IC50, and Ki analysis were performed using the nonlinear iterative curve-fitting computer program GraphPad Prism (GraphPad Software Inc., San Diego, CA). MOI analysis was performed using GraphPad Prism as described under Results.
Results
Characterization of [3H]SAG-1.3 Binding to HEK293 FSmo Cell Membranes. Binding of [3H]SAG-1.3 (2–4 nM) was observed at 23°C to membranes derived from HEK293FSmo cells and not to those from the HEK293F cells transfected with the vector control (Supplemental Fig. 1). Incubation of various amounts of HEK293FSmo-derived membrane homogenates with [3H]SAG-1.3 indicated that binding of the radioligand was linear with protein concentrations up to 12 μg/well (Supplemental Fig. 2). Based on the protein titration, a final membrane protein concentration of 2 to 4 μg/well was used in subsequent assays. Specific binding measured at this protein concentration was between 1 and 10% of the total amount of radioligand added, indicating that radioligand depletion was insignificant because of the low receptor concentration used (i.e., total [L] ∼ free ligand). Nonspecific binding was defined in the presence of 1 μM SAG-1.3 as well as 1 to 10 μM cyclopamine or cyclopamine-KAAD. Both yielded a common plateau of maximal inhibition. Cyclopamine was chosen for subsequent studies because it is structurally distinct from [3H]SAG-1.3. Under these conditions, [3H]SAG-1.3 binds to Smo-containing membranes with 70 to 80% specificity. Similar studies were performed to optimize binding of [3H]cyclopamine to HEK293FSmo membranes (data not shown).
Kinetics of [3H]SAG-1.3 and [3H]Cyclopamine Binding: Association/Dissociation. The time course of [3H]SAG-1.3 and [3H]cyclopamine binding to HEK293FSmo cell membranes both exhibited pseudofirst-order kinetics (Fig. 1, A and B). At 23°C, equilibrium was reached within 2 h and remained stable up to 5 h. After an incubation time of 5 min and longer, nonspecific binding (defined in the presence of 10 μM cyclopamine for [3H]SAG-1.3 and 10 μM SANT-2 for [3H]cyclopamine) remained constant. Two concentrations of [3H]SAG-1.3 and [3H]cyclopamine were analyzed using global fitting of the association kinetic model. Using this analysis, we derived a single best-fit estimate for both association (kon) and dissociation (koff) (Tables 1 and 2). For [3H]SAG-1.3, we derived an association rate constant of kon = 3.0 × 106 M-1·min-1 and a dissociation k-1 = 8.0 × 10-3·min-1 corresponding to an apparent half-time of 86 min. Nonlinear least-squares fitting of these data to a model assuming pseudo–first-order kinetics best fit the data compared with a biphasic model (F-test; p < 0.05) over the model of one binding site. The equilibrium dissociation constant (Kd = koff/kon) calculated from the kinetic data was 2.7 nM. The binding of [3H]cyclopamine displayed biphasic association kinetics. Fitting of these data to a model assuming the presence of two binding sites was preferred by the F-test (p < 0.05) over the model of one binding site. The fast and slow components of association exhibited half-times of 1.5 and 28 min, respectively. The data fit to pseudo–first-order kinetics resulted in an association rate constant of kon = 2.4 × 106 M-1 · min-1 and a dissociation koff = 2.0 × 10-2 · min-1, corresponding to an apparent half-time of 36 min. The equilibrium dissociation constant (Kd = koff/kon) calculated from the kinetic data was 12 nM.
Equilibrium binding of [3H]SAG-1.3 and [3H]cyclopamine to human smoothened receptors in HEK293F cell membranes Kd affinity and Bmax receptor density were determined using nonlinear curve fitting as described under Data Analysis in text. Data are the mean ± S.D. (n = 2–7).
Kinetic parameters of Smo ligand binding to human smoothened receptors in HEK293F cell membranes Kinetic rate values were determined using nonlinear curve fitting as described under Data Analysis in text. Data are the mean ± S.D. (n = 2–7).

A, determination of association and dissociation rates for [3H]SAG-1.3 to HEK293FSmo cell membranes. Aliquots of membranes were incubated with 4 and 14 nM [3H]SAG-1.3 at 23°C in the absence or presence of 10 μM cyclopamine (nonspecific) for varying lengths of time. Data using two radioligand concentrations were fit to the association kinetic model. Nonlinear least-squares fitting of this data to a model assuming pseudofirst-order kinetics best fit the data compared with a biphasic model by the F-test (p < 0.05) over the model of one binding site. The 95% confidence bands are shown by thin dashed lines. Individual replicates are plotted. The experiment was repeated two times with similar results. B, determination of association and dissociation rates for [3H]cyclopamine to HEK293FSmo cell membranes. Aliquots of membranes were incubated with 7 and 19 nM [3H]cyclopamine at 23°C in the absence or presence of 10 μM cyclopamine (nonspecific) for varying lengths of time. Nonlinear least-squares fitting of these data to a model assuming the presence of two binding sites was preferred by the F-test (p < 0.05) over the model of one binding site. The 95% confidence bands are shown by thin dashed lines. Individual replicates are plotted. The experiment was repeated two times with similar results.
Saturation of [3H]SAG-1.3 Binding to HEK293FSmo Cell Membranes. Saturation of [3H]SAG-1.3 binding to HEK293F smoothened cell membranes is demonstrated in Fig. 2. Iterative nonlinear analysis indicated (best fit) a single binding site of high affinity (Kd = 1.7 ± 0.54 nM; Bmax = 34 ± 2.3 pmol/mg protein) (Fig. 2A). Affinity and Bmax values derived from orthogonal homologous isotopic unlabeled saturation experiments yielded Kd = 5.9 ± 1.7 nM and Bmax = 40 ± 8 pmol/mg protein (Fig. 2B). The limit of detection for competing compounds using this radioligand was calculated from the density of receptors (Bmax) expressed in the membrane preparation. Based on the Bmax determined from saturation binding assays as well as the affinity of [3H]SAG-1.3, the limit of compound affinity that could be measured using this radioligand was 0.26 to 1.5 nM depending on the final assay volume (1–0.15 ml).
Homologous Isotopic Unlabeled Saturation of [3H]Cyclopamine Binding to HEK293FSmo Cell Membranes. Homologous isotopic unlabeled saturation of [3H]cyclopamine-specific binding to HEK293F smoothened cell membranes is demonstrated in Fig. 3. Iterative nonlinear analysis indicated a single binding site (Hill values of 0.9–1.1; Kd = 10 ± 2.5 nM; Bmax = 20 ± 0.9 pmol/mg protein). Based on the density of receptors determined from saturation binding assays, as well as the affinity of [3H]cyclopamine, the limit of compound affinity that could be measured using this antagonist radioligand was 0.4 to 2.6 nM, depending on the final assay volume (1–0.15 ml).
Pharmacology of [3H]SAG-1.3 and [3H]Cyclopamine Binding to Smoothened Receptors. The pharmacology of [3H]SAG-1.3 binding to Smo receptors in membranes derived from HEK293F cells was examined in competition experiments using small-molecule Smo agonists and antagonists. The SAG-1.3 and SAG-1.5 agonists are structurally related, whereas the purmorphamine agonist is structurally distinct. Likewise, the cyclopamine, cyclopamine-KAAD, and tomatadine (inactive) antagonists are structurally similar, whereas SANT-1, SANT-2, Z′′′′, Compound 1 (a Smo antagonist that is the racemate of a representative example #8 structure from Novartis (Basel, Switzerland) (Jain et al., 2007), and GDC-0449, Genentech's (South San Francisco, CA) clinical Smo antagonist (Von Hoff et al., 2008) (Fig. 4), represent distinct chemotypes. SAG-1.5 was the most potent agonist inhibitor of [3H]SAG-1.3 binding to Smo (Ki = 0.5 nM). SAG-1.3 bound with high affinity (Ki = 1 nM) followed by the antagonists GDC-0449, Z′′′′ (Compound 1), cyclopamine-KAAD, SANT-2, and cyclopamine (Fig. 5, A and B, and summarized in Table 3). It is noteworthy that GDC-0449 inhibited the binding of [3H]SAG-1.3 in a biphasic manner, with an apparent high affinity of 1.3 nM and low affinity of 2.3 μM (Fig. 5B). Purmorphamine and tomatidine (an inactive analog of cyclopamine) were weak inhibitors of binding (inactive up to 10 μM). SANT-1 only exhibited partial inhibition (40%) of [3H]SAG-1.3 binding, with a plateau from 0.04 to 10 μM, suggesting a possible allosteric interaction between SANT-1 and SAG-1.3.
Binding affinities and functional potencies of various smoothened ligands at wild-type human Smo receptors Radioligand and whole-cell binding data are the mean of 3 to 10 determinations ± S.E.M. Binding was performed in the presence of 1 to 4 nM [3H]SAG-1.3 or 7 to 10 nM [3H]cyclopamine. XC50 is EC50 for agonist compounds or IC50 for antagonist compounds. Agonists were examined for stimulation of reporter activity; antagonist activity was determined against a SAG-1.5 EC80. Reporter assay results represent the mean of 4 to 10 determinations ± S.D. Tomatidine is an inactive analog of cyclopamine.
Because of the differential response the agonists and antagonists exhibited against small-molecule agonist [3H]SAG-1.3 binding, we performed competition experiments with the same set of compounds using [3H]cyclopamine (an antagonist) as the radioligand (Table 3). GDC-0449 possessed the highest affinity for smoothened receptors labeled by [3H]cyclopamine, with a Hill slope of 1 and a 4-fold greater affinity against this ligand compared with the agonist radioligand [3H]SAG-1.3. Agonist compounds, SAG-1.3 and SAG-1.5, retained similar affinities for Smo defined by competition for [3H]cyclopamine binding to the Smo receptors, with Ki values of 3.7 and 2.3 nM, respectively (Fig. 5C; Table 3). Purmorphamine lacked significant inhibition up to the highest concentration tested. The binding profiles of cyclopamine, cyclopamine-KAAD, and the inactive analog tomatidine using [3H]cyclopamine were consistent with affinities measured using the agonist radioligand. The antagonist molecules Z′′′′, SANT-1, and SANT-2 showed high affinities for Smo labeled by [3H]cyclopamine (Fig. 5D; Table 3). In contrast to the partial inhibition of [3H]SAG-1.3 binding, SANT-1 completely inhibited binding of [3H]cyclopamine, with high affinity (Ki = 2.4 nM).
Mode of Inhibition Studies. The finding that SANT-1 only partially inhibited [3H]SAG-1.3 binding to Smo in competition assays suggests that various ligands may bind to Smo at distinct sites. To further investigate the MOI of these ligands, double titrations of both radioligand and compounds were performed for SAG-1.5, cyclopamine, SANT-1, and SANT-2. Compound displacement (specific binding) and apparent IC50 values were examined in the presence of varying concentrations of the radioligand probe. This radioligand binding method is comparable with Schild plot analysis (Ehlert and Authors, 1988) and is often referred to as Schild-type plots (Schetz and Sibley, 1997). Competitive binding interactions show a linear shift in apparent IC50 in relation to radioligand concentration, whereas allosteric antagonism can result in an incomplete inhibition of specific binding and or a nonlinear hyperbolic plot of IC50 values. In addition, the data can be quantified by fitting to a modified model based on the Cheng-Prusoff equation (Kenakin, 2006):  where Kd is the affinity value for the radiolabeled ligand, KB is the Kd for the unlabeled ligand, and α is a cooperativity term. For mutually exclusive ligands (i.e., competitive), the value of α in the equation is ∞. For mixed MOI (including allosteric) α is ≥1, but finite.
where Kd is the affinity value for the radiolabeled ligand, KB is the Kd for the unlabeled ligand, and α is a cooperativity term. For mutually exclusive ligands (i.e., competitive), the value of α in the equation is ∞. For mixed MOI (including allosteric) α is ≥1, but finite.
The Smo agonist SAG-1.5 and antagonist cyclopamine fully inhibited specific binding of [3H]SAG-1.3 to Smo receptors over the range of radioligand concentrations (1–40× Kd). Apparent IC50 values for SAG-1.5 shifted linearly with respect to radioligand concentration, with α values of 40 and 300 in two different experiments (Fig. 6, A and B). Cyclopamine also behaved in a competitive manner, with α>100 in three experiments (Fig. 6, C and D). The profile of these two compounds indicated an MOI consistent with competitive inhibition for binding to Smo labeled with [3H]SAG-1.3. SANT-1 competition resulted in only partial inhibition (<60%), along with a minor shift in the apparent IC50 values (7–16 nM; α= 4 and 4.5), consistent with hallmarks of an allosteric interaction with [3H]SAG-1 (Fig. 7, C and D). The effect of increasing [3H]SAG-1.3 concentrations on SANT-2 competition binding also revealed a profile consistent with allosteric inhibition. Incomplete inhibition of [3H]SAG-1.3 binding was observed only at [3H]SAG-1.3 concentrations above 2 nM. The relationship of apparent IC50 values (17–50 nM) to radioligand concentration was not linear, with calculated α values of 5 and 8 (Fig. 7, A and B). In addition, the data were fit to a more complex ternary model (Kenakin, 2006) to further quantify the interactions (data not shown). This analysis yielded affinity and α values that were consistent with the previous determinations, indicating a probe-dependent allosteric interaction for SANT-1 and SANT-2 for [3H]SAG-1.3 binding to Smo.
Whole Cell BODIPY-Cyclopamine Binding Assay. The potencies of known smoothened ligands and analogs for inhibition of fluorescent BODIPY-cyclopamine binding to smoothened receptors are summarized in Table 3. The rank order and potency of the compounds tested in the BODIPY-cyclopamine assay were similar to the affinities determined in both the [3H]SAG-1.3 and [3H]cyclopamine membrane binding assays. SANT-1 completely competed with BODIPY-cyclopamine, consistent with the complete inhibition in the [3H]cyclopamine binding assay.
β-Lactamase Reporter Gene Functional Assay. To understand the functional consequence of partial competition of [3H]SAG-1.3 by antagonists such as SANT-1 and 2, we also tested these agonists and antagonists in a β-lactamase reporter gene functional assay (Table 3). The purine derivative agonist purmorphamine exhibited weak activation of the system, consistent with previous observations in similar mouse reporter systems (Sinha and Chen, 2006). Both SAG-1.3 and SAG-1.5 displayed nanomolar-range potency stimulation of Gli-responsive reporter activity, with EC50 values of 0.4 and 7 nM, respectively. Because SAG-1.5 was competitive with [3H]SAG-1.3 for binding to Smo, yet was superior in potency than the structurally similar SAG 1.3 in β-lactamase reporter gene functional assay, an EC80 concentration of SAG 1.5 was used as the reporter activator for evaluation of functional inhibitors of Smo. The antagonist ligands SANT-1 and GDC-0449 were the most potent, with IC50 values of 20 and 45 nM, respectively. Additional antagonist ligands, such as cyclopamine-KAAD, SANT-2, Z′′′′, and Compound 1, retained similar rank order pharmacological potencies to the measured binding affinities determined using both agonist and antagonist radioligands. All of these antagonists, including SANT-1 and -2, exhibited complete inhibition of the response to the level of background (Fig. 8).

Saturation assays of [3H]SAG-1.3 binding to HEK293F cell membranes expressing human smoothened receptors. A, direct labeled saturation binding isotherm showing the total amount of [3H]SAG-1.3 bound, the amount of [3H]SAG-1.3 bound in the presence of 10 μM cyclopamine (nonspecific) and the specific (total-nonspecific) binding. The inset shows semilogarithmic transformation of the specific binding showing that full saturation has been obtained. B, homologous isotopic unlabeled saturation assay using two concentrations of [3H]SAG-1.3 (2 and 11 nM) in the presence of unlabeled ligand (0.003–10 μM). Saturation analysis best fit a single site model. Aliquots of HEK293F cell membranes expressing human smoothened receptors (1–2 μg/well) were incubated for 5 h at 23°C in binding buffer. The data shown are from a representative experiment. Each value is the average of two to three determinations (n = 2–7).

Homologous isotopic unlabeled saturation assay using two concentrations of [3H]cyclopamine (7 and 27 nM) in the presence of unlabeled ligand (0.006–30 μM). Global analysis of both data sets was performed to derive Kd and Bmax values. Aliquots of HEK293F cell membranes expressing human smoothened receptors (1–2 μg/well) were incubated for 5 h at 23°C in binding buffer. The data shown are from a representative experiment. Each value is the average of two to three determinations (n = 2).
Discussion
The Hh pathway plays an important role in development, stem cell maintenance, and tumorigenesis. The pathway activator Smo is being pursued as an attractive target for cancer, and small-molecule antagonists of Smo have entered clinical trials for the treatment of multiple cancers, including advanced basal cell carcinoma and metastatic colorectal cancer. A better understanding of the interactions and regulations of antagonists and agonists of Smo will facilitate the design and discovery of the next generation of small-molecule regulators of Smo and the Hh pathway.
The radioligand binding of a Smo agonist, [3H]SAG-1.5, has been characterized previously (Frank-Kamenetsky et al., 2002). We have extended their study by examining the binding of both an agonist, [3H]SAG-1.3, and an antagonist, [3H]cyclopamine, to Smo receptors expressed in HEK293F membranes. Equilibrium binding studies revealed that specific binding of both tritium-labeled SAG-1.3 and cyclopamine to human Smo receptors was saturable, reversible, of high affinity, and consistent with single-site binding. Binding kinetic studies revealed that [3H]SAG-1.3 dissociates from Smo receptors more slowly (t½ = 86 min) compared with cyclopamine (t½ = 36 min). Because of this, we performed mathematical analysis to show that when the radioligand dissociates more slowly than a competing ligand, binding is close to equilibrium conditions at t = 3.5/k-1 for the radioligand. In the absence of definitive rate constants for the competing ligands tested, we allowed the binding to equilibrate for 5 to 7 h, with no apparent detriment to membrane or ligand integrity. Binding of [3H]cyclopamine to smoothened receptors displayed a biphasic association (Fig. 1; Tables 1 and 2), which might indicate a ligand conformational change or binding site heterogeneity. Saturation binding studies with [3H]SAG-1.3 (classic labeled) and [3H]cyclopamine (isotopic unlabeled) demonstrated that each ligand binds to a single site with high affinity, yielding Kd values of 1.7 ± 0.6 and 10 ± 2.5 nM, respectively (Fig. 2). There was a 2-fold difference in the Bmax values determined for [3H]cyclopamine (isotopic) and [3H]SAG-1.3 (labeled) of 20 and 40 pmol/mg protein, respectively (Tables 1 and 2). Additional assays such as Schild-type binding and direct labeled [3H]cyclopamine saturation studies are warranted to determine whether these ligands bind to the same sites. Additional detailed kinetic and direct labeled saturation analysis with [3H]cyclopamine are required to further investigate the biphasic association and comparison of binding site number.

Smoothened receptor agonist and antagonist molecule structures. A, leiosamine agonist molecules (SAG-1.3 and SAG-1.5) and a purine derivative purmorphamine. B, antagonist chemotypes, including cyclopamine and active derivative cyclopamine-KAAD, as well as an inactive derivative tomatidine. Structurally distinct antagonist chemotypes that inhibit Hh signaling: SANT-1, SANT-2, Z′′′′, Compound 1, and the antagonist GDC-0449 (Genentech).
Several Smo agonists and antagonists have been reported previously. The affinity values for these ligands have been determined using a variety of techniques and often cannot be compared directly. Using both the agonist and antagonist radioligand probes, we have characterized and compared the binding affinity of three agonists and eight antagonists of Smo (Table 3). The pharmacology of most compounds using either [3H]SAG-1.3 or [3H]cyclopamine is similar. SANT-1 shows probe-dependent differences in competition, and they are discussed later. Using [3H]SAG-1.3, the potencies we have determined for SAG-1.5, SAG-1.3, and cyclopamine-KAAD are in agreement with those determined using [3H]SAG-1.5 to label murine Smo receptors (Frank-Kamenetsky et al., 2002). The affinities for most of the other compounds examined were in agreement with previously reported functional potencies, with the exception of purmorphamine, a hedgehog pathway activator. Purmorphamine has been reported to inhibit BODIPY-cyclopamine binding to Smo (IC50 of ∼1 μM) and purmorphamine-driven activation of Gli-dependent reporter was inhibited by cyclopamine (Sinha and Chen, 2006). It is noteworthy that, in our studies, purmorphamine weakly displaced BODIPY-cyclopamine (IC50 of ∼3.3 μM) binding but did not displace either [3H]SAG-1.3 or [3H]cyclopamine binding up to 10 μM. Two interpretations of these data may be that purmorphamine activates the Hh pathway indirectly without binding Smo or binds in an allosteric fashion to either [3H]SAG-1.3 or [3H]cyclopamine consistent with neutral cooperativity. Additional functional studies using Schild analysis of the interactions between cyclopamine and agonists such as purmorphamine and SAG-1.3 would help further the understanding of compound interactions and influence on the smoothened pathway.

Characterization of the pharmacological specificity of [3H]SAG-1.3 and [3H]cyclopamine binding in 293F cells expressing human smoothened receptor. Membranes prepared as described under Materials and Methods, were incubated with 3 nM [3H]SAG-1.3 (A and B) or 7 nM [3H]cyclopamine (C and D) and varying concentrations of competing compounds for 5 h at 23°C. The data shown are from representative experiments. Similar curves were obtained from multiple experiments (n = 3–10). The calculated Ki values for the compounds are shown in Tables 1 and 2.
Although the pharmacology of most ligands was similar using either [3H]SAG-1.3 or [3H]cyclopamine, we observed very different competition patterns for SANT-1, a previously identified small-molecule Smo antagonist. Although SANT-1 fully displaced [3H]cyclopamine binding and completely inhibited SAG-1.5-driven, Gli-dependent reporter activity, 10 μM SANT-1 inhibited only 40% of [3H]SAG-1.3 binding, suggesting potential allosteric interaction between SANT-1 and SAG-1.3. To further understand the mechanism of action for SAG-1.5, cyclopamine, SANT-1, and SANT-2 binding to Smo, we performed a series of double titration (Schild-type) experiments with [3H]SAG-1.3 and [3H]cyclopamine. Analysis of the interactions between [3H]SAG-1.3 and SAG-1.5 or cyclopamine yielded profiles consistent with competitive binding, with α values >>10. In addition, maximal inhibition to the level of nonspecific binding was obtained over the entire range of radioligand concentrations tested. It cannot be ruled out that cyclopamine may have a strong allosteric effect, reflected as competitive for [3H]SAG-1.3 binding. In contrast, our analyses clearly indicated an allosteric interaction for SANT-1 and SANT-2 toward SAG-1.3. Incomplete inhibition of specific [3H]SAG-1.3 binding (inability to reach level of nonspecific binding in the presence of high concentrations of SANT-1 or SANT-2), a hallmark of allosteric mode of inhibition, was observed for these antagonists. In addition, the plots for SANT-1 and SANT-2 each deviate from linearity, with calculated α values of <10 for each antagonist. Our data suggest that the interactions between these two antagonists and SAG-1.3 are not directly competitive. As has been observed with other receptors, the observed allosteric binding of SANT-1 and SANT-2 seems probe- or radioligand-dependent. That SANT-1 and SANT-2 bind smoothened receptors labeled with [3H]cyclopamine in a manner consistent with competitive antagonism illustrates this point. Further characterization of the allosteric effects could be performed through sensitive dissociation kinetic studies. In many cases, allosteric inhibitors have been shown to increase the rate of tracer dissociation (Kenakin, 2006). Although such assays may be helpful, the particular kinetics of a probe such as [3H]SAG-1.3 should be considered. The data presented demonstrate SANT-1 and SANT-2 modulate smoothened receptors labeled with [3H]SAG-1.3 in an allosteric manner.

Schild-type plot analysis of SAG-1.5 and cyclopamine inhibition of [3H]SAG-1.3 smoothened receptor binding. A and C, representative displacement curves of [3H]SAG-1.3 shown for a range of SAG-1.5 and cyclopamine concentrations. Curves generated in the presence of varying concentrations of radioligand from 1 to 40 times Kd. B and D, Schild-type plot transformations of IC50 apparent values representing the relative shift in competing compound potency. Note that α values greater than 10 are consistent with a competitive interaction between ligands. The data shown are a representative of several experiments (n = 2) in duplicate or triplicate.
It has been proposed that SANT-1 may not directly compete for cyclopamine binding because it failed to directly displace BODIPY-cyclopamine binding. Our competition experiments using both [3H]cyclopamine (membrane homogenate) and BODIPY-cyclopamine (whole cell) showed complete displacement by SANT-1. Therefore, we do not have any evidence that SANT-1, SANT-2, and cyclopamine have different binding sites. Because SANT-1 and SANT-2 completely inhibit SAG-1.5-induced Hh pathway activation, it seems that partial displacement of SAG-1.3 binding can nevertheless produce full functional inhibition. Site-directed mutagenesis studies using both binding and functional assays would supplement these data and enhance the understanding of smoothened receptor agonist and antagonist mechanism of action. Our results support the general bias observed in binding assays compared with functional assays. Because of this, we re-emphasize that the choice of ligand used in radio- or fluorescent binding studies should be considered, because each probe might specifically identify different sets of allosteric and competing compounds. The subtle differences in binding of allosteric ligands such as SANT-1 and SANT-2 may result in unique phenotypes because they may change the way smoothened receptors interact with accessory proteins. In addition, the specific cellular phenotype can influence the action of such ligands and is worthy of further investigation.

Schild-type plot analysis of SANT-1 and SANT-2 inhibition of [3H]SAG-1.3 smoothened receptor binding. A and C, representative displacement curves of [3H]SAG-1.3 shown for a range of SANT-1 and SANT-2 concentrations. Curves generated in the presence of varying concentrations of radioligand from 1 to 40 times Kd. B and D, Schild-type plot transformations of IC50 apparent values representing the relative shift in competing compound potency. Note that α values less than 10 and incomplete inhibitions are hallmarks of an allosteric binding interaction. The data shown are a representative of several experiments (n = 2) in duplicate or triplicate.
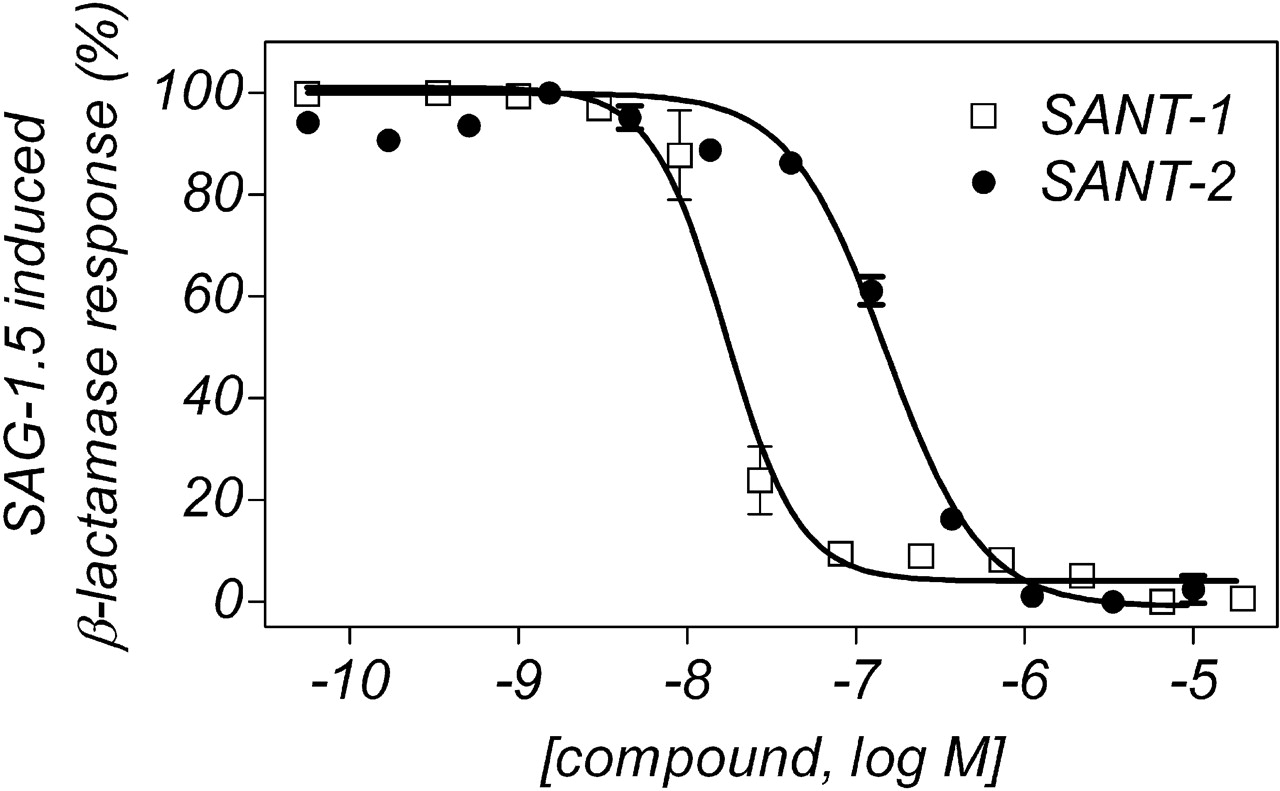
Inhibition by SANT-1 and SANT-2 of the SAG-1.5 induced β-lactamase response. CellSensor NIH3T3 cells stably expressing the Gli-β-lactamase reporter were exposed to compounds for 40 to 48 h in the presence of an EC80 concentration of SAG-1.5. Zero percent and 100% correspond to the basal level of fluorescence and to the SAG-1.5 induced fluorescence level, respectively. Data are representative of more than three separate experiments all performed in two to six replicates.
In conclusion, we have characterized radioligand binding of both an agonist and an antagonist of Smo. Using both probes, we have compared the binding of multiple antagonists and agonists to Smo. Our detailed biochemical mechanism of action analyses indicate SANT-1 and SANT-2 are allosteric inhibitors of [3H]SAG-1.3 binding. Our data have important implications on the understanding of Smo regulation and discovery of additional small-molecule Smo regulators.
Acknowledgments
We are grateful to Da-Yuan Wang for the generation of the Smo BacMam, to Kathleen Gallagher for cloning of the Smo cDNA, and to Terry Kenakin and Thomas Meek for helpful discussions and suggestions.
Footnotes
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.109.152090.
-
ABBREVIATIONS: Hh, Hedgehog; Smo, Smoothened; Ptch, Patched; SAG-1.3, 3-chloro-N-[trans-4-(methylamino)cyclohexyl]-N-{[3-(4-pyridinyl)-phenyl]methyl}-1-benzothiophene-2-carboxamide; HEK, human embryonic kidney; SANT-1, N-[(1E)-(3,5-dimethyl-1-phenyl-1H-pyrazol-4-yl)methylidene]-4-(phenylmethyl)-1-piperazinamine; SANT-2, N-[3-(1H-benzimidazol-2-yl)-4-chlorophenyl]-3,4,5-tris(ethyloxy)benzamide; SAG-1.5, 3-chloro-4,7-difluoro-N-[trans-4-(methylamino)cyclohexyl]-N-{[3-(4-pyridinyl)phenyl]methyl}-1-benzothiophene-2-carboxamide; DMEM, Dulbecco's modified Eagle's medium; Compound 1, 6-methyl-N-{2-[(1,3-thiazol-2-ylmethyl)amino]-2,3-dihydro-1H-inden-5-yl}-4′-(trifluoromethyl)-2-biphenylcarboxamide; GDC-0449, 2-chloro-N-[4-chloro-3-(2-pyridinyl)phenyl]-4-(methylsulfonyl)benzamide; MOI, mode of inhibition.
-
↵
 The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material. - Received February 19, 2009.
- Accepted March 18, 2009.

- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}