


Abstract
Chronic obstructive pulmonary disease (COPD) is characterized by progressive airflow limitation caused by persistent inflammatory processes in the airways. An increased cholinergic tone mediates different pathophysiological features of COPD, such as bronchoconstriction and mucus hypersecretion, mostly through activation of the human muscarinic M3 receptor (hM3) subtype. Tiotropium bromide (Spiriva) is a well established muscarinic antagonist in the pharmacological management of COPD with a once-daily posology. The rationale behind the sustained bronchodilation obtained with tiotropium consists in its slow dissociation from hM3 receptors. In this study, we performed a comprehensive preclinical comparison of tiotropium with other long-acting muscarinic antagonists (LAMAs) currently in clinical development, namely aclidinium bromide and glycopyrrolate. The different muscarinic antagonists were characterized for their 1) affinity toward the different human muscarinic receptor subtypes expressed in Chinese hamster ovary cells and kinetics of receptor dissociation, 2) potency in inhibiting the agonist-induced activation of muscarinic receptors through measurement of second messengers, and 3) efficacy and duration of bronchoprotection, as tested in a model of acetylcholine-induced bronchoconstriction in anesthetized dogs over a period of 24 h. All of the tested LAMAs showed high affinity and potency toward the hM3 receptor (tiotropium, pA2 = 10.4; aclidinium, pA2 = 9.6; and glycopyrrolate, pA2 = 9.7). However, dissociation half-lives of the LAMAs from the hM3 receptor differed significantly (tiotropium, t½ = 27 h; aclidinium, t½ = 10.7 h; and glycopyrrolate, t½ = 6.1 h). In line with their kinetic properties at the hM3, the tested LAMAs provided different levels of bronchoprotection in the in vivo setting 24 h after administration (tiotropium = 35%, aclidinium = 21%, and glycopyrrolate = 0% at 24 h) when applied at equieffective doses.
The term COPD describes a clinical condition characterized by chronic airway obstruction caused by an abnormal inflammatory response of the respiratory system. The main causal factor in the pathogenesis of COPD is sustained inhalation of cigarette smoke (Rabe et al., 2007). Occasionally, other factors are involved, including a genetically induced deficiency of the enzyme α1-antitrypsin or exposure to noxious agents at work and in the environment. COPD is characterized by a predominantly neutrophilic inflammation of the airway walls, and airflow limitation is mainly a result of hyperplasia of mucosal glands, hypertrophy, and, in particular, constriction of the bronchial smooth muscle in the small airways (Barnes, 2000). COPD is associated with a significant level of morbidity and mortality (Rabe et al., 2007). The Global Burden of Disease Study has projected that COPD, which ranked sixth as the cause of death in 1990, will become the third leading cause of death worldwide by 2020 (Murray and Lopez, 1997).
National and international guidelines recommend that patients with COPD should receive bronchodilators as first-line maintenance treatment, because they increase expiratory flow by decreasing airway smooth muscle tone, thus leading to reduced lung hyperinflation (Rabe et al., 2007). The most frequently prescribed bronchodilators for COPD treatment are muscarinic antagonists and β2-agonists. In COPD, bronchoconstriction and mucus secretion are increased, and the airways become hyper-responsive to contractile agents. These changes are caused mostly by increased parasympathetic nerve activity (Barnes, 2004). Acetylcholine (ACh), released from parasympathetic nerve endings, activates postjunctional muscarinic M3 receptors present on airway smooth muscle and submucosal glands to induce bronchoconstriction and mucus secretion, respectively. Thus, anticholinergic bronchodilators have a particular value in the treatment of COPD because they block the effects of an increased vagal cholinergic tone. In clinical trials, muscarinic antagonists have been shown to produce an improvement in lung function, exercise endurance, and health-related quality of life and significant reductions of exacerbations, and—in summary—to improve the clinical course of COPD (Vincken et al., 2002; Tashkin et al., 2008). Importantly, muscarinic antagonists show a favorable side-effect profile. Thanks to the quaternary structure of modern anticholinergics, which prevents a substantial absorption from mucosal surfaces and penetration of the blood-brain barrier, and the inhalative administration route, which further limits systemic exposure, these drugs are virtually free of clinically relevant side effects, besides occasional reports of dry mouth (Gross, 2006).
Tiotropium (Spiriva), introduced recently for the treatment of COPD, is the first example of a long-acting muscarinic antagonist (LAMA): following a single dose of tiotropium, clinically relevant improvement in lung function lasts for more than 24 h, allowing once-daily dosing (Vincken et al., 2002). The mechanism behind its long duration of action relates to the slow rate of dissociation from its target, the human M3 muscarinic receptor (Disse et al., 1999). Long duration of action (preferably 24 h) is an important feature of drugs intended to treat chronic diseases, enabling both prolonged efficacy (Tashkin, 2005) and a simple, once-daily dosage regime that improves patient compliance (Tamura and Ohta, 2007). Other drugs are currently being evaluated in clinical trials for their ability to function as LAMAs with a potential for once-daily administration, i.e., aclidinium [also known as LAS34273 from Almirall Prodesfarma (Barcelona, Spain), currently in phase IIb trials] and glycopyrrolate [NVA-237, from Novartis (Basel, Switzerland), in phase III trials]. Currently, however, limited preclinical information is published concerning the pharmacological properties of these investigational drugs. Thus, in the present study, we have directly compared the pharmacology of the different LAMAs, namely tiotropium, aclidinium, and glycopyrrolate, in in vitro and in vivo models. To understand the antagonists' behavior at the molecular level, interaction with the different muscarinic receptor subtypes was analyzed in binding and functional assays. Given the rationale behind tiotropium's long duration of action, particular attention was given to the kinetics of dissociation from the muscarinic receptors. Efficacy and duration of bronchoprotection were tested in a pharmacological model of acetylcholine-induced bronchoconstriction in anesthetized dogs over a period of 24 h.
Materials and Methods
Chemicals and Reagents. [N-methyl-3H]Scopolamine methyl chloride ([3H]NMS, specific activity, 82 Ci/mmol) was obtained from PerkinElmer Life and Analytical Sciences (Waltham, MA). MgCl2, carbachol, muscarine, atropine sulfate, pirenzepine, N-methyl scopolamine bromide, EDTA, 3-isobutyl-1-methylxanthine, NaCl, and HEPES were obtained from Sigma-Aldrich (St. Louis, MO). Ipratropium bromide, tiotropium bromide, aclidinium bromide, and glycopyrrolate bromide were synthesized in the chemical laboratories of Boehringer Ingelheim (Biberach an der Riss, Germany). The tritiation of tiotropium was performed by RC Tritec AG (Teufen, Switzerland). [3H]Tiotropium was purified by high-performance liquid chromatography on a XBridge C-8 column (Waters GmbH, Eschborn, Germany) resulting in a radiochemical purity of ≥98%, and a specific activity of 65 Ci/mmol. All cell culture reagents were purchased from Invitrogen (Carlsbad, CA).
Cell Culture Techniques. Chinese hamster ovary (CHO) cells transfected with the cDNAs encoding the human M1 to M5 muscarinic acetylcholine receptors were purchased from PerkinElmer Life and Analytical Sciences. CHO cells were grown in Ham's F-12 medium supplemented with 10% fetal calf serum in the presence of the selection agent G418 (400 μg/ml). Cells were maintained at 37°C in humidified air containing 5% CO2.
Equilibrium Binding Experiments. Membrane isolation and purification from CHO cells stably expressing the human M1 to M5 receptors was performed as described previously (Casarosa et al., 2005). In brief, cells were suspended in buffer A (15 mM Tris-HCl, pH 7.5, 2 mM MgCl2, 0.3 mM EDTA, 1 mM EGTA), homogenized, and spun down for 30 min at 48,000g. The pellet was resuspended in buffer B (7.5 mM Tris-HCl, pH 7.5, 12.5 mM MgCl2, 0.3 mM EDTA, 1 mM EGTA, 250 mM sucrose), aliquoted, and stored at -80°C until use. Protein content was measured with the BCA kit (Thermo Fischer Scientific, Rockford, IL).
In all radioligand experiments, the binding buffer consisted of 10 mM HEPES, 1 mM MgCl2, pH 7.4. After the indicated incubation period, bound and free [3H]NMS were separated by rapid vacuum filtration using a Brandel Harvester (Gaithersburg, MD) on GF/B filters presoaked in 0.5% polyethylenimine and rapidly washed three times with ice-cold binding buffer. Filter disks were added to 3 ml of scintillation fluid (Ultima Gold from PerkinElmer Life and Analytical Sciences) in pony-vials, and radioactivity was quantified by use of liquid scintillation spectrometry on a Tri-Carb 2900TR Liquid Scintillation Analyzer (PerkinElmer Life and Analytical Sciences). In all experiments, total binding never reached 10% of that added, limiting complications associated with depletion of the free-radioligand concentration.
Saturation binding experiments were performed by incubating membranes (usually 5–10 μg/sample, adjusted according to the Bmax of the individual cell line), with a range of concentrations of [3H]NMS (4 pM to 8 nM) in a total volume of 4 ml, to avoid significant ligand depletion at the lower concentrations. Samples were incubated at room temperature for 2 h under gentle agitation before filtration. Nonspecific binding was determined for each radioligand concentration by coincubating in parallel a set of membranes with an excess of unlabeled atropine (10 μM).
To obtain affinity estimates of unlabeled antagonists, heterologous competition experiments against [3H]NMS were performed at equilibrium. Membranes were incubated in the presence of [3H]NMS (final concentration, approximately 0.1 nM) and different concentrations of unlabeled antagonist at room temperature with gentle agitation for 18 to 20 h before filtration. Competition displacement binding data were fitted to the equation described by Hill (1909), and IC50 values obtained from the inhibition curves were converted to Ki values by use of the method of Cheng and Prusoff (1973).
Kinetic Studies. To determine dissociation kinetic parameters according to the classical method, membranes expressing the different muscarinic receptor subtypes were first allowed to equilibrate with 0.45 nM [3H]NMS or 90 pM [3H]tiotropium at room temperature for at least 2 h (300 μl/sample). Subsequently, samples were added to test tubes already containing 3 ml of binding buffer with atropine 10-5M (time 0) to start dissociation. At the different time points, samples were filtered by use of a Brandel harvester, as described above. To monitor the potential membrane degradation occurring at later time points, some samples were added at time 0 to tubes containing 3 ml of binding buffer with the same concentration of radioligand present in the preincubation step (i.e., 0.45 nM [3H]NMS or 90 pM [3H]tiotropium), so that dissociation was not started and decrease in radioligand binding would reflect receptor breakdown. Dissociation data were fitted to a single-phase exponential decay function, and the Koff rate obtained was transformed into a t½ value (dissociation half-life) using the following equation: t½ = ln 2/Koff.
The dissociation kinetic parameters of unlabeled ligands were assessed with the use of the method of Motulsky and Mahan (1984). With this approach, the kinetics of an unlabeled ligand are determined via competition with a radioligand ([3H]NMS), whose kinetic parameters have already been determined for a given receptor. This method involves the simultaneous addition of both radioligand and unlabeled competitor to the membranes, so that at time 0 all receptors are unoccupied. Membranes were added at time 0 to tubes containing 0.3 nM [3H]NMS in the presence or absence of three different concentrations of unlabeled competitor (approximately 30-, 100-, and 300-fold Ki). At the different time points (up to 8 h), the degree of [3H]NMS bound to receptor was assessed by filtration harvesting, as described above. Dissociation rates for unlabeled antagonists were calculated with Prism (GraphPad Software Inc., San Diego, CA) using the equation for kinetics of competitive binding by fitting (least-squares) the data from the competition kinetic experiments to a two-component exponential curve, with all parameters fixed apart from k3 and k4. The kinetics of [3H]NMS, determined independently (average of three independent experiments) and used in these equations, are as follows: for hM1 receptor: kon = 3.22 × 108 M-1 min-1 and Koff = 0.019 min-1; for hM2 receptor: kon = 6.96 × 108 M-1 min-1 and Koff = 0.035 min-1; for hM3 receptor: kon = 5.96 × 108 M-1 min-1 and Koff = 0.013 min-1.
cAMP Assay. To determine the functional antagonistic potency of the different muscarinic antagonists at the hM2 receptor, changes in intracellular cAMP levels were determined with CHO-hM2 cells in suspension (5000 cells/well) with use of Lance technology (PerkinElmer Life and Analytical Sciences) and the 384-well plate format (Optiplate; PerkinElmer Life and Analytical Sciences), according to the manufacturer's protocol. In brief, cells were stimulated with a range of agonists' concentrations (either carbachol or muscarine, from 10-3 to 10-12 M) in Hanks' buffered saline solution supplemented with 5 mM HEPES, 0.1% bovine serum albumin, 500 mM 3-isobutyl-1-methylxanthine, and 1 μM forskolin in the absence or presence of at least six different concentrations of antagonists (usually from 10-5 to 10-10M) for 30 min at room temperature. A 15-min preincubation with antagonists was allowed to proceed before the addition of the agonist. Cells were lysed with use of Lance reagents, and after an additional 2 h, plates were read on an Envision plate reader (PerkinElmer Life and Analytical Sciences). The concentration of cAMP in the samples was calculated from a standard curve. The Gaddum equation was used to fit the data, and shifts in the dose ratio were plotted according to Schild.
Inositol Phosphate Accumulation Assay. Changes in intracellular levels of inositol phosphates were monitored in CHO-hM1 and CHO-hM3 cells by use of the IPone kit (Cisbio Bioassays, Bagnolssur-Cèze Cedex, France) according to the manufacturer's instructions. In brief, cells were plated 1 day in advance in 384-well white plates (Greiner Bio-One GmbH, Frickenhausen, Germany), with a density of 20,000 cells per well. On the day of the assay, cells were stimulated with a range of agonists' concentrations (either carbachol or muscarine, from 10-3 to 10-12 M) in a buffer consisting of 10 mM HEPES, 1 mM CaCl2, 0.5 mM MgCl2, 4.2 mM KCl, 146 mM NaCl, 5.5 mM glucose, and 50 mM LiCl (final pH 7.4) in the absence or presence of at least six different concentrations of antagonists (usually from 10-5 to 10-10M). A 15-min preincubation with antagonists was allowed to proceed before addition of the agonist. After 1-h stimulation at 37°C, cells were lysed using reagents provided by the kit, and after an additional hour, plates were read on an Envision plate reader. The concentration of inositol phosphates in the samples was calculated from a standard curve. The Gaddum equation was used to fit the data, and shifts in the dose ratio were plotted according to Schild.
In Vivo Experiments. Bronchoprotective effects of the muscarinic antagonists were investigated in a model of acetylcholine (ACh)-induced bronchoconstriction in anesthetized, ventilated beagle dogs. All animal experiments were conducted in accordance with the German Animal Welfare Law and received ethics committee approval. Male dogs weighing between 10 and 12 kg were randomly assigned to four groups, each consisting of four animals. The dogs were anesthetized with a bolus injection of propofol (10 mg/kg i.v.) followed by an infusion of 30 mg/kg i.v. propofol per hour (B. Braun Melsungen, Melsungen Germany) into the cephalic vein. The dogs were intubated and ventilated with volume-controlled pressure with a mixture of room air and oxygen (3:1) with a Siemens respirator at a rate of 15 strokes per minute. Optimal ventilation was ensured by regular measurement of acid-base status and oxygen saturation in the blood. Muscarinic antagonists were dissolved in water/ethanol (40:60, v/v) and administered by inhalation by use of the Respimat Soft Mist Inhaler (Boehringer Ingelheim Pharma GmbH, Ingelheim, Germany) connected to the tracheal tube of the ventilated dogs in a volume of 30 μl. Increases in airway resistance were induced by repeated intravenous injections of 10 μg/kg acetylcholine (Sigma-Aldrich). Experiments were performed as dose range-finding studies for 3 h before studies for 24 h. For the 3-h study setup, intravenous acetylcholine challenges were injected at: -45, -30, -15, 5, 10, 30, 60, 90, 120, 150, and 180 min after inhalation of the test compound. For the 24-h study setup, ACh was administered 30 and 15 min before and 5, 10, and 30 min and 6, 12, and 24 h after inhalation of the test compounds. Bronchoprotection was expressed as percentage of inhibition of the ACh-induced increase in airway resistance before the administration of the compound (predrug values).
Data Analysis. All experiments were analyzed by either linear or nonlinear regression analysis with the equations mentioned under the different assay methodology using Prism version 5.1 (GraphPad Software, San Diego, CA). Data are expressed as mean ± S.E.M.
Results
Determination of Ligand Affinity for the Human Muscarinic M1 to M5 Receptors. To analyze the pharmacological behavior of the different muscarinic antagonists in vitro, CHO cell lines selectively and stably expressing either of the human muscarinic M1, M2, M3, M4, and M5 receptors were used to ensure that measurements were made at a single receptor subtype. For each cell line, the Bmax values and affinities toward [3H]NMS, as determined in saturation experiments, are reported in Table 1.
Pharmacological characterization of the different CHO cell lines stably expressing human muscarinic receptor subtypes Dissociation constants for [3H]NMS (expressed in picomolars) and levels of receptor expression (expressed in picomoles per milligram of protein content of the membrane preparations) were determined in saturation binding assays, as described under Materials and Methods. The average of at least two independent experiments performed in triplicate is shown.
The antagonists' affinities for the different receptor subtypes were determined in heterologous competitive binding experiments against [3H]NMS. Because the time needed to reach equilibrium can change significantly, depending on the kinetic parameters of the unlabeled compound (Motulsky and Mahan, 1984), initial experiments were performed to decide how long incubation should proceed. This aspect is of particular importance for compounds with longer kinetics of receptor interaction, as shown for tiotropium (Disse et al., 1993), where too short incubation would result in underestimation of receptor affinity. As a model system, membranes expressing the hM3 receptor were used, and the different muscarinic antagonists, i.e., tiotropium, ipratropium (with fast dissociation from the hM3 receptor, see next paragraph), aclidinium and glycopyrrolate, were allowed to equilibrate in the presence of [3H]NMS for different times (Fig. 1). Ipratropium reached equilibrium after 2 h, as indicated by the absence of any further change in its IC50 over time. In contrast, tiotropium needed at least 20 h of incubation time to reach pseudo-equilibrium, as indicated by the shift in pIC50 values at shorter time points (pIC50 values after 2, 5, and 10 h were statistically different from that obtained at 20 h, P < 0.05). An intermediate behavior was observed for aclidinium and glycopyrrolate, which needed at least 5 h to reach pseudo-equilibrium in this experimental setting. Because the time needed to reach equilibrium is related to the receptor dissociation kinetics for each cold test compound (Motulsky and Mahan, 1984), these data suggest that, among the substances tested, tiotropium has the longest half-life at the hM3 receptor. Based on these results, an assay incubation time of approximately 20 h was used in all subsequent experiments, because this provided a satisfactory compromise between attaining equilibrium while avoiding membrane degradation.
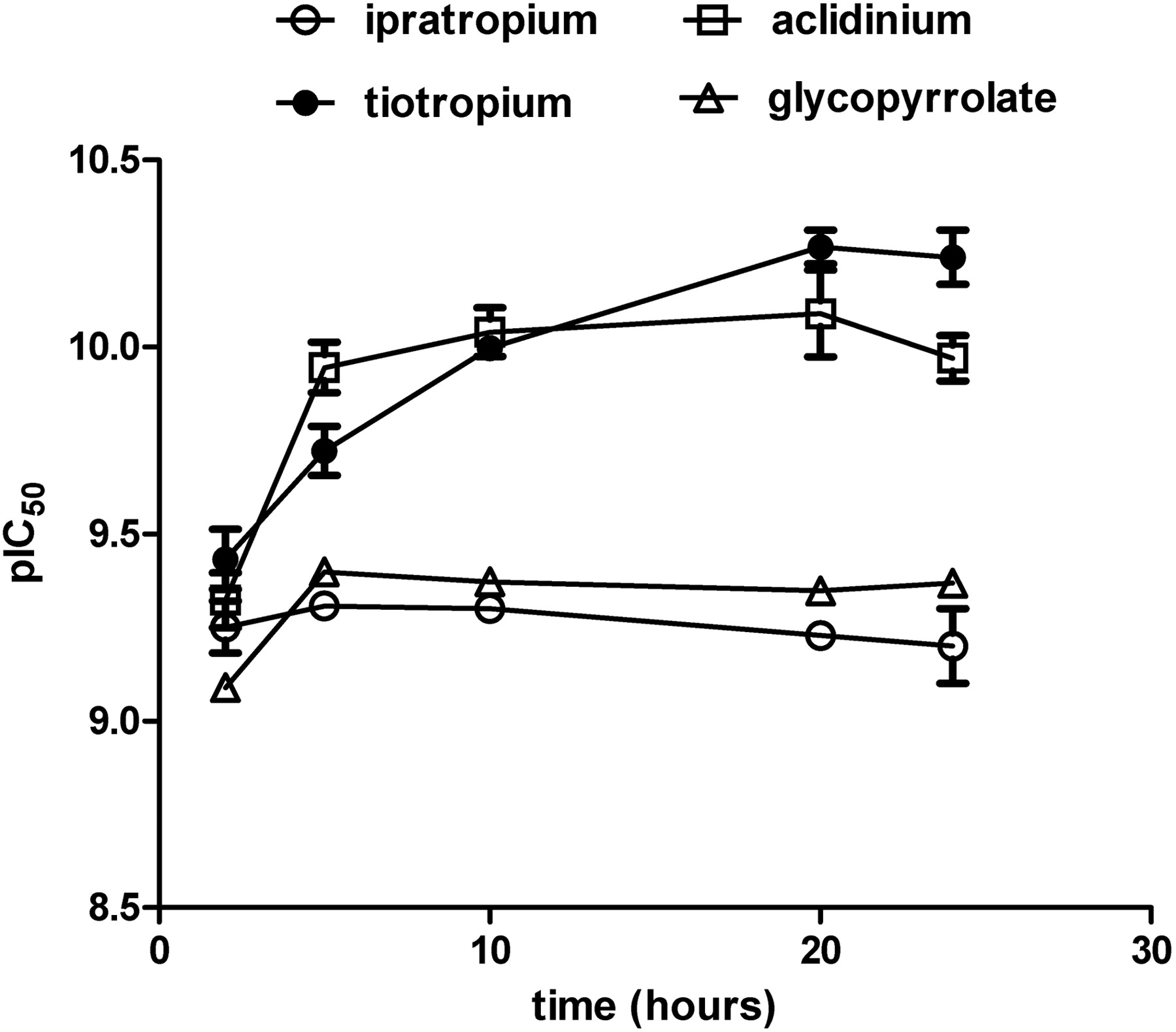
Shift of pIC50 values obtained in heterologous binding assays for different muscarinic antagonists over time. CHO-hM3 cell membranes were incubated with [3H]NMS and a range of competitor concentrations (ipratropium, ○; tiotropium, •; aclidinium, ▵; glycopyrrolate, □) at room temperature for different times (i.e., 2, 5, 10, 20, and 24 h) before harvesting through filtration. IC50 values were obtained by fitting a sigmoidal function. Data are shown as means ± S.E.M. for three experiments, each performed in triplicate.
Displacement curves were analyzed according to a model of orthosteric competition between [3H]NMS and the unlabeled competitors, so that IC50 values were transformed into Ki values according to the Cheng-Prusoff equation. To prove that this is the correct model for interpreting the data, the shifts in IC50 values in relation to the increase in amount of radioligand used in a given experiment were monitored for different ligands: the IC50 values shifted linearly in correlation with the increase of radioactive probe ([3H]NMS), as assumed in the model for competitive interaction (Cheng and Prusoff, 1973) (data not shown). With this method, the different muscarinic antagonists were tested, and the pKi values obtained under these conditions are summarized in Table 2.
Binding affinities of different muscarinic antagonists against the five human muscarinic receptor subtypes The equilibrium dissociation constants of the different muscarinic antagonists were determined in heterologous competition experiments against [3H]NMS. The pKi values shown are the average of at least three independent experiments performed in triplicate, and the means ± S.E. associated with the measurements were 0.1 or less.
Kinetics of Dissociation from Human Muscarinic Receptors. Another important parameter in describing the interaction taking place between ligand and receptor is the dissociation rate constant, Koff, which refers to the rate at which the drug receptor complex dissociates. It has previously been demonstrated for tiotropium that its slow dissociation from the target receptor hM3 is key to its long duration, which allows for once-daily administration of tiotropium (Disse et al., 1999). The dissociation rates of compounds are traditionally assessed directly by monitoring the specific binding of a radiolabeled form of the ligand of interest over time. As an alternative, Motulsky and Mahan (1984) introduced a method by which a kinetically characterized radioligand is added simultaneously with the unlabeled ligand to the receptor preparation of interest. Because the pattern of radioligand binding over time depends on both the concentration and kinetic parameters of the competitor, the dissociation rate of the unlabeled compound can be derived experimentally in this setting. This methodology has been used previously with success to assess the dissociation of ligands from the human M3 receptor (Dowling and Charlton, 2006).
The dissociation kinetic parameters for tiotropium were determined with the classical method (i.e., monitoring dissociation of [3H]tiotropium from hM3 receptors in time; Fig. 2A) and with the “Motulsky and Mahan” method (i.e., via competition kinetic experiments between unlabeled tiotropium and [3H]NMS, as explained under Materials and Methods; Fig. 2B). The dissociation half-life values of tiotropium obtained with the two different methods were in close agreement (Koff 0.028 ± 0.003 and 0.026 ± 0.005 h-1 derived with the classical and alternative method, respectively); therefore, the kinetic values of the other compounds were determined with the competition kinetic experiments, which do not require the preparation of a specific radioligand for each compound to be tested. The Koff and dissociation half-life values of the different muscarinic antagonists at the hM1, hM2, and hM3 receptors are reported in Table 3. The half-life ratio of each antagonist for dissociation from the hM3 and hM2 receptors is included in Table 3 as well.
Koff values (h–1) and dissociation half-lives (h) for the different muscarinic antagonists against the human M1, M2, and M3 receptor subtypes The dissociation constants were determined according to the method described by Motulski and Mahan (1984), by analyzing competition kinetics curves in the presence of [3H]NMS and different concentrations of unlabeled antagonist, as shown in Fig. 2B for tiotropium. Data shown are the mean Koff values ± S.E.M. of three independent experiments, each performed in triplicate, and the corresponding dissociation half-life (in hours).

Determination of the Koff parameter of tiotropium for the hM3 receptor by two independent methods. A, according to the traditional method, CHO-hM3 cell membranes were preincubated with [3H]tiotropium, and then at time 0 dissociation was started (•) by adding an excess of atropine, and the amount of receptor-bound [3H]tiotropium was monitored by filtering samples at the indicated time points. Data were best-fitted using a one-phase exponential decay. In some samples (○), dissociation was not started, and total binding was monitored at the different time points to rule out membrane degradation. The average of three independent experiments, each performed in triplicate, is shown. B, to determine the Koff value according to the method described by Motulski and Mahan (1984), competition kinetics curves were performed with CHO-hM3 membranes in the presence of [3H]NMS (approximately 0.45 nM) and tiotropium (•, 0; ▪, 0.3 nM; ○, 1 nM; □, 3 nM). Samples were incubated at room temperature for the indicated time points before filtration occurred. Data were fitted with Prism equations as described under Materials and Methods to calculate Koff values. Because the total binding somewhat varied from experiment to experiment, data are presented as mean ± S.D. from one representative of three independent experiments performed in triplicate. DR, drug receptor. D.P.M., desintegrations per minute.
Quantification of Functional Competitive Antagonism: Analysis of the Gq Pathway. The M1 to M5 receptors can be subdivided into two major functional classes according to their G-protein-coupling preference. The “odd-numbered” M1, M3, and M5 receptors selectively couple to G-proteins of the Gq/G11-family, whereas the “even-numbered” M2 and M4 receptors preferentially activate Gi/Go-type G-proteins (Wess et al., 2007). Given that the hM4 and hM5 receptors are expressed almost exclusively in the central nervous system and are basically absent in the lungs where predominantly hM3 and hM2 are expressed (Lee et al., 2001), the functional analysis of muscarinic antagonists was restricted to the hM1 to hM3 receptor subtypes.
The assays of choice to monitor receptor activation consisted in measurement of second messengers (i.e., inositol phosphates for Gq-coupled receptors and cAMP for the Gi-coupled receptors, respectively). Two different agonists belonging to different chemical classes, i.e., carbachol and muscarine, were tested. Both behaved as full agonists (α= 1) on the hM1 and hM3 receptors, with muscarine being slightly more potent (pD2 values of muscarine and carbachol are: 6.7 ± 0.1 and 6.0 ± 0.1 at the hM1 receptor; 7.3 ± 0.1 and 6.7 ± 0.1 at the hM3 receptor, respectively; n ≥ 11).
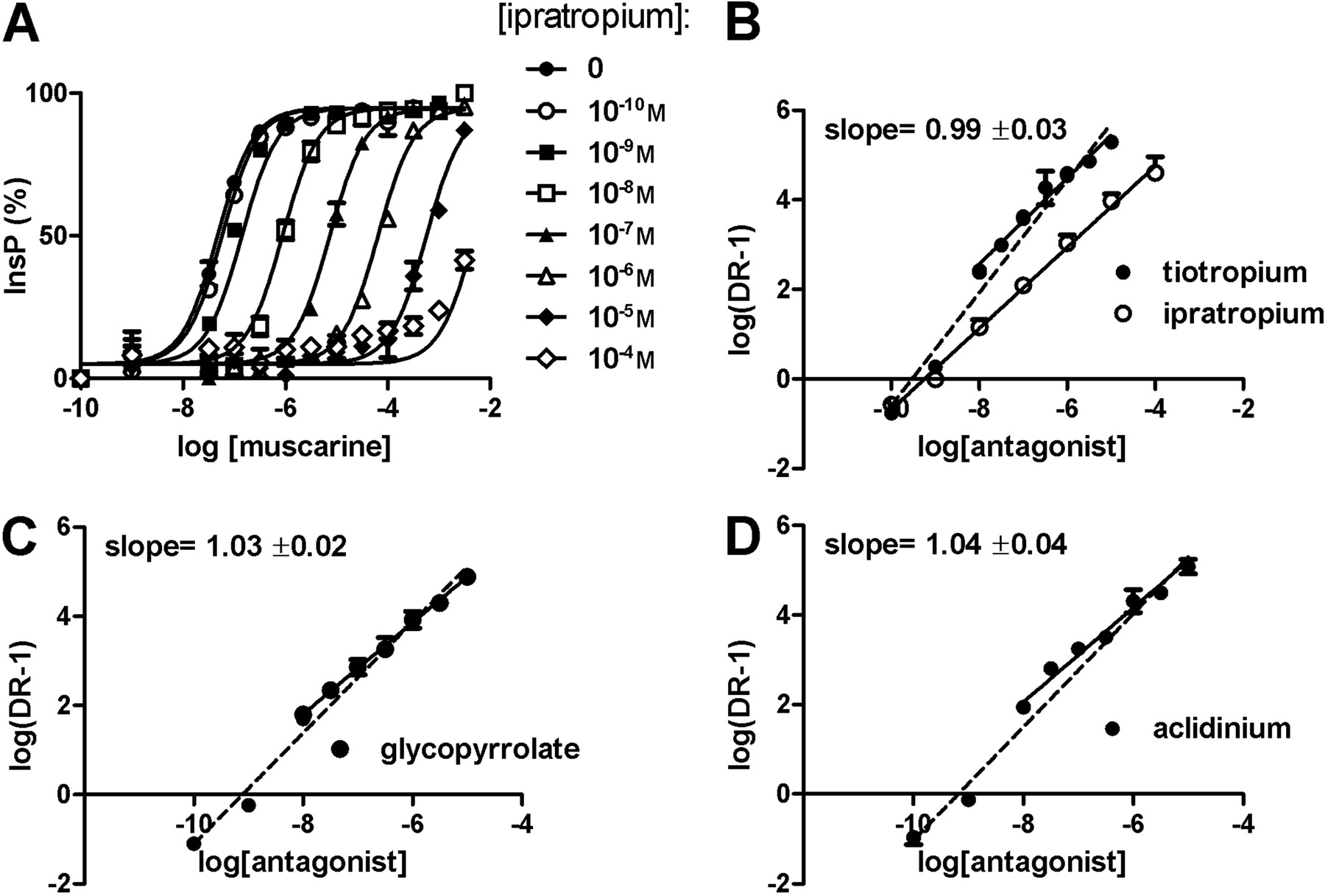
To measure the antagonism of the different anticholinergics, the shift in the agonist response curves induced by the presence of different antagonist concentrations were plotted according to Schild (e.g., in Fig. 3), and pA2 values were determined. All muscarinic antagonists tested showed a competitive and surmountable antagonistic behavior (exemplified for ipratropium in Fig. 3A). Most of the tested anticholinergic drugs (i.e., ipratropium, atropine, NMS, and pirenzepine) induced a parallel shift of the agonist concentration-response curve that could be perfectly fitted with the Schild equation, with a slope not significantly different from 1 (e.g., ipratropium, Fig. 3, A and B).
A different profile was observed for tiotropium, aclidinium, and glycopyrrolate, where fitting the dose ratios over a large range of antagonist concentrations (10-10-10-5 M) resulted in a regression with a slope different from 1. Slopes are 1.29 ± 0.05, 1.27 ± 0.07, and 1.27 ± 0.05 for tiotropium, aclidinium, and glycopyrrolate, respectively (see dashed lines in Fig. 3, B–D). A slope that is significantly greater than 1 could indicate a lack of antagonist equilibrium, as can be expected when compounds with slow receptor dissociation kinetics are tested. Because in conditions of nonequilibrium the speed of ligand association to the receptor is proportional to its concentration, the fractional antagonist receptor occupancy will be lower (compared with that at equilibrium) for the lower concentrations (Kenakin, 1984), and this causes the slope of the Schild regression to be more than 1. To solve this problem, these antagonists were tested at higher concentrations (i.e., from 10-8 to 10-5 M) with half-logarithmic concentration steps. With this adjustment, Schild regression turned out to be linear with unit slope (Fig. 3, B–D), allowing for correct estimation of pA2 values also for these muscarinic antagonists (Table 4).
Functional properties of the muscarinic antagonists at the human M1 and M3 receptors pA2 values of the different antagonists were determined in CHO-hM1 and CHO-hM3 cell lines with the inositol phosphate assay. Values were obtained experimentally by extrapolation of agonistic (muscarine and carbachol) concentration response shifts, according to Schild analysis, as shown in Fig. 3. Values shown are the average of at least three independent experiments, with each point determined in triplicate. S.E.M. values were 0.10 or less, and Schild slopes were not significantly different from unity.

Muscarinic antagonists inhibit agonist-induced inositol phosphates accumulation in CHO-hM3 cells. A, ipratropium induces a concentration-dependent rightward shift (concentrations tested from 10-10 to 10-4 M) in the agonist curve (here, muscarine), measured as inositol phosphates accumulation (InsP) in CHO-hM3 cells. B, Schild regression is shown for ipratropium (○, slope, 0.96 ± 0.03) and for tiotropium (•), with analysis of the concentrations from 10-8 to 10-5 M (slope, 0.99 ± 0.03). Data are the mean of at least three independent experiments (± S.E.M.). C and D, Schild regression is shown for glycopyrrolate (C) and for aclidinium (D), with analysis of the concentrations from 10-8 to 10-5 M. Data are the mean of three independent experiments (± S.E.M.). The dashed regression lines represent the plots of the 10-10 to 10-5 M concentration ranges. DR, drug receptor.
As predicted on the basis of receptor theory for competitive antagonists, pA2 values obtained for each muscarinic antagonist against the two different agonists are very similar. Furthermore, pA2 values are in close agreement with pKi values determined in the binding assays (compare Tables 2 and 4) as expected for competitive (i.e., orthosteric, surmountable, and reversible) antagonists.
Analysis of Gi/s-Pathway. Functional analysis of the antagonists' potencies against the hM2 receptor was measured with a cAMP assay. As with the hM1 and hM3 receptors, the two agonists muscarine and carbachol were tested in the presence of the adenylate cyclase activator forskolin to detect Gi-coupling (Fig. 4A). As expected on stimulation of a Gi-coupled receptor, like the hM2, both agonists induced a concentration-dependent inhibition of cAMP formation with identical potencies (pD2 values were 7.6 ± 0.1 for both muscarine and carbachol). Curiously, by further increasing the agonists' concentration, an inversion in the signaling profile was observed and an increase of cAMP accumulation was detected (pD2 values are: 5.4 ± 0.1 and 5.6 ± 0.1 for muscarine and carbachol, respectively). This behavior is consistent with the promiscuous coupling of the M2 receptor to different G-proteins, namely Gi and Gs, as shown by Mistry et al. (2005). Because of the considerable difference in the agonistic potencies for the two different pathways, the Gi-component could be isolated in each bell-shaped agonistic curve, and pA2 values could be accurately determined (Table 5). Figure 4C illustrates the effects of ipratropium on the Gi- and Gs-mediated responses.
Functional properties of the muscarinic antagonists at the human M2 receptors pA2 values of the different antagonists were determined in the CHO-hM2 cell line with the cAMP assay. To monitor the Gi signaling, cells were stimulated in the presence of the adenylate cyclase activator forskolin, whereas, to monitor the Gs component, cells were pretreated with PTX (overnight, 80 ng/ml). Values were obtained experimentally by extrapolation of agonistic concentration-response shifts, according to Schild analysis. Values shown are the average of at least three independent experiments, with each point determined in triplicate. S.E.M. values were below 0.10 and Schild slopes were not significantly different from unity.
Analysis of hM2 coupling to Gs was further investigated in the presence of pertussis toxin (PTX), which blocks Gi signaling (Fig. 4B). Under this condition, the receptor can just couple to Gs subunits, and therefore, the analysis of agonist and antagonist behavior reflects only their influence on this pathway. After PTX pretreatment, there is a slight increase in the potency and Emax of the Gs-mediated carbachol response (Fig. 4, compare B and A) as expected after removing the inhibitory component (i.e., Gi-coupling).
Therefore, for each antagonist, pA2 values were either determined in the presence of forskolin (monitoring their ability to antagonize the Gi-coupling of the hM2 receptor) or in cells pretreated with pertussis toxin (monitoring their ability to antagonize the hM2 receptor signaling through Gs-proteins). Interestingly, for each antagonist, the two pA2 values did not significantly differ from each other (Table 5), indicating that the antagonists' affinity for the receptor is not influenced by its coupling to different G-proteins.
In Vivo Bronchoprotection. Finally, we investigated the bronchoprotective effects of the muscarinic antagonists in two functional in vivo models of acetylcholine-induced bronchoconstriction in anesthetized dogs. Intravenous, repetitive administration of acetylcholine resulted in increased airway resistance, an effect mainly mediated by activation of the muscarinic M3 receptor that can by antagonized by anticholinergics blocking this receptor subtype.

The hM2 receptor shows promiscuous coupling to both Gi- and Gs-proteins. A, the muscarinic agonists carbachol and muscarine were added to CHO-hM2 cells, and changes in intracellular cAMP levels were monitored in the presence of the adenylyl cyclase activator forskolin (1 μM) These agonists caused an inhibitory response (because of Gi-coupling) at low concentrations and a stimulatory response (because of Gs-coupling) at high concentrations. The data were fit according to a bell-shaped dose-response curve using Prism. B, the muscarinic agonists carbachol and muscarine were added to CHO-hM2 cells that had been pretreated with pertussis toxin (overnight at 80 ng/ml), and changes in intracellular cAMP levels were monitored. Because PTX inactivated the Gαi subunits, only the stimulatory response caused by Gs-coupling remained present. C, ipratropium induces a concentration-dependent rightward shift (concentrations tested: from 10-10 to 10-5 M) of both the descending and the ascending part of the bell-shaped agonist-response curve (muscarine) of cAMP accumulation in CHO-hM2 cells in the presence of forskolin (1 μM).
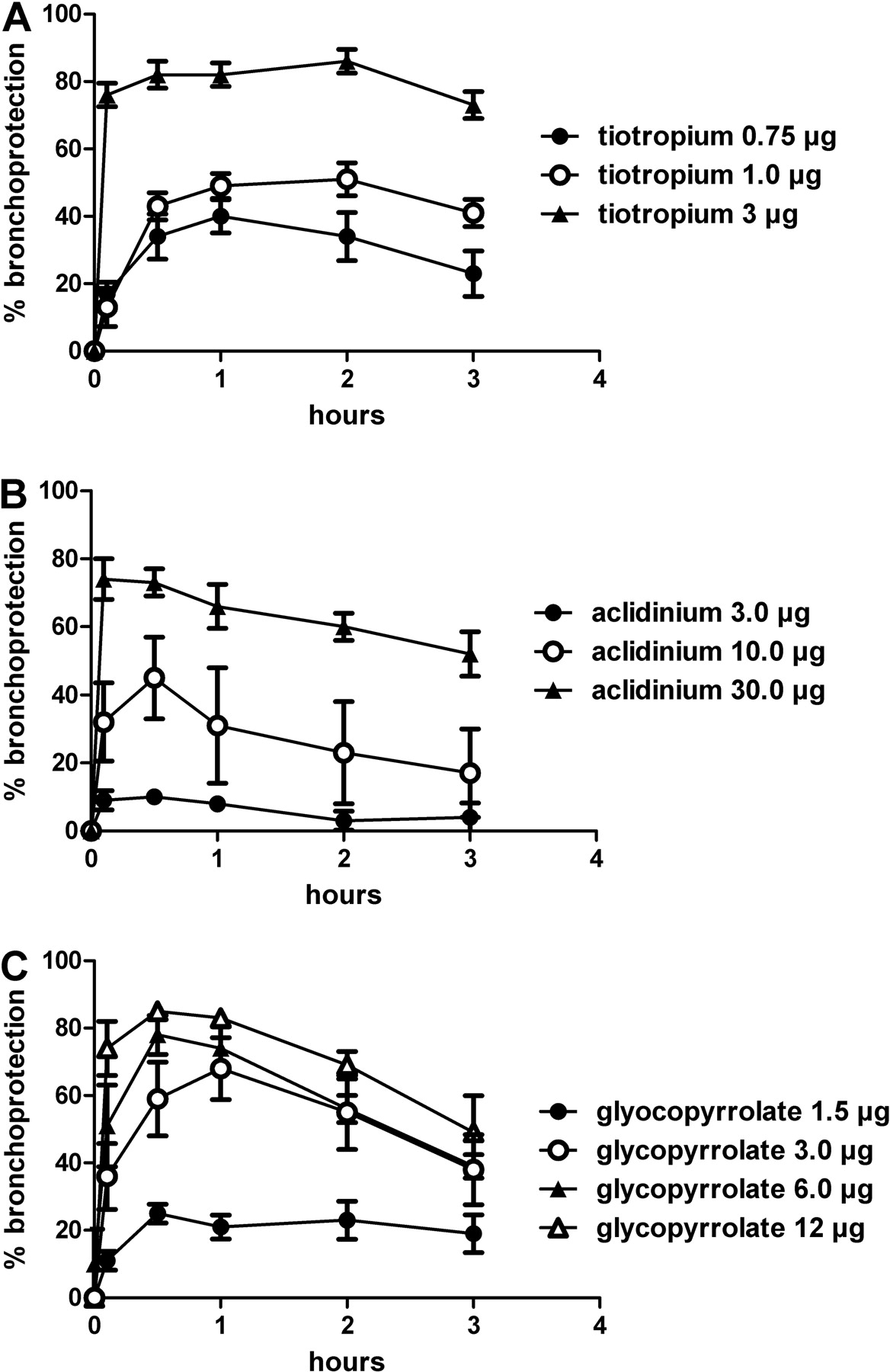
Dose-range studies were performed with each LAMA in anesthetized dogs for 3 h (Fig. 5). In this setup, the muscarinic antagonists were administered by inhalation with use of the Respimat Soft Mist Inhaler connected to the tracheal tube of the ventilated dogs, and their efficacies to protect the animals against acetylcholine-induced bronchospasms were recorded for 3 h. In this setting, the dose inducing 80% bronchoprotection was defined as the fully effective dose.
As shown in Figure 5A, tiotropium dose-dependently protected the dogs against acetylcholine-induced bronchoconstriction. The fully effective dose was determined as 3 μg/dog. At this dose, a bronchoprotection of 73% was still retained after 3 h.
Aclidinium, tested at inhaled doses of 3, 10, and 30 μg (Fig. 5B), dose-dependently inhibited the ACh-induced broncho-constriction. However, compared with tiotropium, a significantly higher dose (30 μg) was necessary to reach 80% bronchoprotection. At this dose, aclidinium retained a 52% bronchoprotection after 3 h. In addition, glycopyrrolate dose-dependently inhibited the cholinergic bronchoconstriction (Fig. 5C). At the fully effective dose of 12 μg, a 49% bronchoprotection was observed after 3 h.
As a next step, the muscarinic antagonists were investigated at their fully effective doses (3 μg for tiotropium, 30 μg for aclidinium, and 12 μg for glycopyrrolate) in the dog model over a time frame of 24 h to obtain information on their duration of action in a clinically more relevant time scheme (Fig. 6).
In line with the first series of experiments, all compounds induced an initial bronchoprotection of more than 80%. However, the level of bronchoprotection clearly differed among the anticholinergics after 24 h: tiotropium displayed a bronchoprotection of 35% after 24 h; in contrast, aclidinium retained a reduced activity (21%), whereas the activity of glycopyrrolate declined more rapidly. By 12 h after inhalation of glycopyrrolate, a complete loss of bronchoprotection was observed.
Discussion
An increased cholinergic tone is the major reversible component in the bronchoconstriction observed in COPD. Consequently, muscarinic antagonists are the most effective bronchodilators for COPD, with several studies indicating their superiority to β2-adrenoceptor agonists (Gross and Skorodin, 1984; Rennard et al., 1996).
Patient compliance is essential for the successful management of chronic diseases like COPD and asthma (Bender, 2002). Complicated or multiple treatment regimens negatively affect patient adherence; therefore, the simplification of dosing schemes (e.g., once-daily dosing) is an important strategy to improve compliance (Tamura and Ohta, 2007). Tiotropium, well established for the treatment of COPD, is the only LAMA on the market, with a once-daily dosage regime providing high patient compliance (Cramer et al., 2007). This successful strategy is being followed by several companies that, similarly, are developing other muscarinic antagonists with the potential for once-daily administration.
Here, we describe a comprehensive preclinical comparison of muscarinic antagonists with potential for once-daily administration, namely tiotropium and the investigational drugs aclidinium (LAS34273) and glycopyrrolate (NVA-237). Binding studies indicate that all tested LAMAs have a high affinity toward the muscarinic receptors under equilibrium conditions, with subnanomolar Ki values. Equilibrium binding experiments against [3H]NMS obey the Cheng-Prusoff equation, suggesting a competitive behavior of the tested LAMAs. These data are further supported by the functional in vitro experiments, where the competitive (i.e., orthosteric and reversible) behavior of the LAMAs was indicated by the parallel shift of the agonist curves, without depression in the maximal effect achieved. Importantly, binding affinities and functional potencies are in close agreement, as expected on the basis of receptor theory, with tiotropium being the most potent antagonist at the hM3 receptor, the primary pharmacological target for bronchodilation.
Even though the common parameter used in discussing antagonist action at a receptor is its affinity, the dynamic process of the drug-receptor interaction is better captured by parameters such as association and dissociation rates. Rate constants allow us to predict how the drug-receptor system responds to dynamic changes and how it behaves in time and, therefore, are more relevant parameters for understanding drug action in living systems. Of particular interest in this context is the dissociation rate of muscarinic antagonists from their target, the hM3 receptor, and its correlation to their duration of action in vivo. Indeed, achieving slow dissociation kinetics from the target receptor represents a novel approach in drug design for increasing drug duration of action and is the pharmacodynamic rationale behind the once-daily administration of tiotropium in clinical practice (Disse et al., 1999). Here, we confirm the long residence time of tiotropium at the hM3 receptor with two independent methodologies (Fig. 2). Importantly, our data indicate that tiotropium binding to the hM3 receptor is fully reversible, when dissociation is allowed to proceed long enough. Likewise, Koff values were calculated for aclidinium and glycopyrrolate; with dissociation half-lives of 10.7 and 6.1 h from the hM3 receptor, respectively, one would not predict a once-daily profile of these compounds in clinical practice, at least when only considering a pharmacodynamic rationale for the duration of action. However, duration of drug action can depend on pharmacokinetic factors as well, including absorption, tissue distribution, and clearance (Roberts, 2003). To obtain information on functional bronchoprotective properties, taking into account both pharmacodynamic and pharmacokinetic properties, the different LAMAs were tested in a model of acetylcholine-induced bronchoconstriction in anesthetized dogs. Because an increase in the cholinergic tone is considered the major reversible component in COPD (Barnes, 2004), this model is of high clinical relevance. First, dose-range-finding studies were performed for 3 h, and doses inducing 80% bronchoprotection were defined as fully effective doses. Comparing compounds at these doses would avoid ceiling effects at bronchoprotection levels close to 100%. Fully effective and thus initially equieffective doses of the respective antagonists were subsequently used in the dog model for studying a period of 24 h. In this experimental setting, we assume that the duration of the inhibitory effects of the different anticholinergics results from both the rate of dissociation from muscarinic receptors and from additional pharmacokinetic factors, e.g., the topical retention of the compound in the lung and its chemical and metabolic stability. All muscarinic antagonists induced a dose-dependent bronchoprotection upon ACh challenge, with a potency range of tiotropium > glycopyrrolate > aclidinium. Surprisingly, the potency for aclidinium was comparatively low, considering its high affinity for the hM3 receptor in vitro. Preliminary studies indicate that chemical instability of aclidinium in aqueous solutions might be responsible for this observation (unpublished data). A comparison of the duration of action of the muscarinic antagonists at equieffective doses showed that tiotropium induced the highest level of bronchoprotection (35%) after 24 h followed by aclidinium (21%), whereas glycopyrrolate failed to protect the air-ways for more than 12 h. These data are in agreement with a previous study testing glycopyrrolate in a guinea pig model of bronchoconstriction (Villetti et al., 2006), with the effect of glycopyrrolate reduced to one-third of the initial response after 16 h, whereas an equipotent dose of tiotropium was still fully effective after the same interval of time (Villetti et al., 2006).
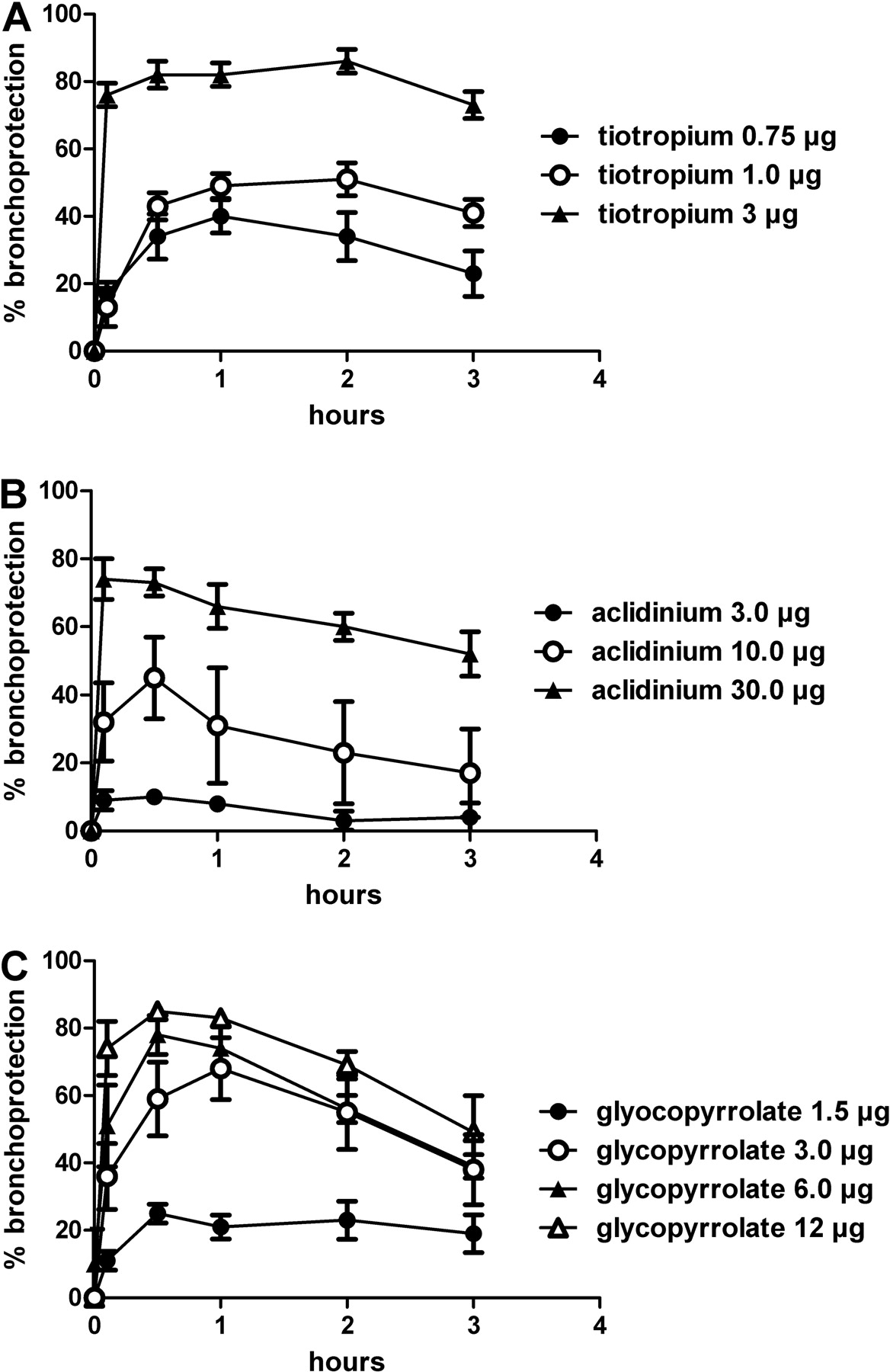
Dose finding of the different LAMAs in the dog model of ACh-induced bronchoconstriction. Protection afforded by intratracheally administered test compounds (A, tiotropium; B, aclidinium; and C, glycopyrrolate) of ACh-induced bronchoconstriction was monitored in anesthetized beagle dogs. ACh (10 μg/kg i.v.) was injected before (baseline value) and up to 180 min after administration of the test compounds. Each value represents the mean ± S.E.M. of data obtained from four animals.

Duration of action of the LAMAs at their fully effective doses in dogs. The different muscarinic antagonists (•, 3 μg of tiotropium; ○, 30 μg of aclidinium; ▴, 12 μg of glycopyrrolate) were administered intratracheally in anesthetized beagle dogs at time 0. Their ability to protect from ACh-induced bronchoconstriction was monitored for up to 24 h. Each value represents the mean ± S.E.M. of data obtained from four animals.
Recently, it was reported that glycopyrrolate can produce a long-lasting bronchoprotection, up to 30 h, in mildly asthmatic patients (Hansel et al., 2005). However, the prolonged effect of glycopyrrolate was achieved at doses of 1 to 2 mg, i.e., at doses at least 200 times higher than the effective dose of tiotropium (5 μg). The need to use such high doses cannot be rationalized solely on the basis of the lower affinity of glycopyrrolate at M3 receptors compared with tiotropium (10-fold difference; Table 2). We hypothesize that high doses of glycopyrrolate are needed not only to compensate for its lower potency but also, more importantly, to “accumulate” this compound in the lungs, thereby achieving a long duration of action (Villetti et al., 2006).
In conclusion, we performed a set of preclinical experiments to better understand the pharmacology of the different muscarinic antagonists and to predict, on this basis, their behavior in the clinical setting. In agreement with their Koff values at the hM3 receptor, tiotropium still induced approximately 40% bronchoprotection in the dog model after 24 h, whereas aclidinium was half as effective and glycopyrrolate completely failed to show bronchoprotective effects after that period. On this basis, we speculate that a putative glycopyrrolate- or aclidinium-based therapy for COPD might require more than once-daily administration to be as effective as the currently used therapy with tiotropium. Alternatively, the use of supramaximal effective doses might allow researchers to overcome their shorter duration of action, possibly through a mechanism implying the generation of a drug depot in the lungs. Whether such an approach will increase the risk of unwanted drug reactions, in relation to the inhalation of higher doses and therefore potentially a higher systemic exposure, and/or a higher degree of variation in drug absorption by different patient populations, remains to be seen.
Acknowledgments
We thank Helga Nickel and Angela Ostermann for excellent technical assistance.
Footnotes
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.109.152470.
-
ABBREVIATIONS: COPD, chronic obstructive pulmonary disease; [3H]NMS, [N-methyl-3H]scopolamine methyl chloride; hM, human muscarinic receptor; ACh, acetylcholine; LAMA, long-acting muscarinic antagonist; CHO, Chinese hamster ovary; PTX, pertussis toxin; ipratropium bromide, (1R,3r,5S,8r)-8-isopropyl-3-[(±)-tropoyloxy]tropanium bromide monohydrate; tiotropium bromide, 6β,7β-epoxy-3β-hydroxy-8-methyl-1αH,5αH-tropanium bromide di-2-thienylglycolate; LAS34273, aclidinium bromide, (3R)-3-{[hydroxydi(thiophen-2-yl)acetyl]oxy}-1-(3-phenoxypropyl)-1λ5-azabicyclo[2.2.2]octan-1-ylium bromide; NVA-237, glycopyrrolate bromide, 3-(α-cyclopentylmandeloyloxy)-1,1-dimethylpyrrolidinium bromide.
- Received February 16, 2009.
- Accepted May 26, 2009.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}