


Abstract
We evaluated the pharmacological profile of ritobegron [KUC-7483; (−)-ethyl 2-[4-(2-{[(1S,2R)-2-hydroxy-2-(4-hydroxyphenyl)-1-methylethyl]amino}ethyl)-2,5-dimethylphenyloxy]acetate monohydrochloride] and its effects on the bladder in cynomolgus monkeys by in vitro and in vivo experiments. In vitro, ritobegron decreased the resting tension of the isolated bladder in a concentration-dependent manner (EC50 8.2 ± 2.3 × 10−7 M; maximal relaxation 88.7 ± 3.7%). The β3-adrenoceptor (AR) antagonist 3-(2-allylphenoxy)-1-[(1S)-1,2,3,4-tetrahydronaphth-1-ylamino]-(2S)-2-propanol hydrochloride (SR58894A) produced a rightward shift of this concentration-response curve without altering the maximal response (pKB value 6.56 ± 0.35). In isolated atria, ritobegron increased the atrial rate only at high concentrations (EC50 6.5 ± 1.2 × 10−5 M). Ritobegron had no effect on tracheal contraction at concentrations from 10−9 to 10−4 M, and even at the highest concentration tested, 10−3 M, the maximal relaxation it induced was only 26.7 ± 8.1%. Tests of the selectivity of ritobegron for the bladder gave values of 79.3- and 1200-fold higher versus atria and trachea, respectively. In the in vivo study ritobegron significantly decreased intravesical pressure (ED50 1.44 mg/kg) without affecting either mean blood pressure or heart rate. In conclusion, ritobegron displayed potent and selective β3-AR agonistic activity and relaxed the monkey isolated bladder, and in vivo it decreased intravesical pressure without affecting cardiovascular parameters. These results suggest that ritobegron may be a promising potential agent for the treatment of overactive bladder.
Introduction
Overactive bladder (OAB) is a common condition that is characterized by urgency, often accompanied by increased daytime frequency, nocturia, and sometimes urge incontinence (Abrams et al., 2002). Antimuscarinic drugs have been widely used for the treatment of OAB. However, these drugs can have severe side effects (dry mouth, constipation, and blurred vision) and have the potential to cause voiding difficulty in a patient with a poorly contractile bladder. Thus, there is an urgent need for new therapeutic drugs with alternative mechanisms of action.
The urinary bladder is innervated by both the sympathetic and parasympathetic nervous systems, and sympathetic activation contributes to urine storage by relaxing the bladder smooth muscle via the activation of β-adrenoceptors (ARs) (Andersson, 1999). β-ARs are currently classified into β1-, β2-, and β3-AR subtypes (Bylund et al., 1994), and there are species differences in the subtypes involved in the relaxation of the mammalian bladder. For example, in cats (Nergardh et al., 1977) and guinea pigs (Li et al., 1992) relaxation of the bladder is mediated mainly via the β1-AR; in rabbits (Anderson and Marks, 1984; Levin et al., 1988; Yamazaki et al., 1998) relaxation of the bladder is mediated mainly via the β2-AR; in rats (Yamazaki et al., 1998) and pigs (Yamanishi et al., 2002) relaxation of the bladder is mediated mainly via both the β2-AR and β3-AR; and in ferrets (Takeda et al., 2000a) and dogs (Yamazaki et al., 1998) relaxation of the bladder is mediated mainly via the β3-AR. Furthermore, in primates such as cynomolgus monkeys (Takeda et al., 2002a) and humans (Igawa et al., 1998, 1999; Yamazaki et al., 1998; Takeda et al., 1999) bladder relaxation is reportedly mediated via the β3-AR, with 97% of the total β-AR mRNA in humans being of the β3-AR subtype (Yamaguchi, 2002; Nomiya and Yamaguchi, 2003). This evidence indicates that the cynomolgus monkey is an appropriate species for the evaluation of the effects of selective β3-AR agonists on bladder function.
Ritobegron [KUC-7483; (−)-ethyl 2-[4-(2-{[(1S,2R)-2-hydroxy-2-(4-hydroxyphenyl)-1-methylethyl]amino}ethyl)-2,5-dimethylphenyloxy]acetate monohydrochloride] is a phenoxyacetic acid derivative (Tanaka et al., 2001) that was synthesized and developed by Kissei Pharmaceutical Co. Ltd. as a novel selective β3-AR agonist. In this study, we evaluated its pharmacological profile and its effects on the bladder in the cynomolgus monkey by means of in vitro and in vivo experiments.
Materials and Methods
Animals.
This study was conducted according to guidelines approved by the Laboratory Animal Committee of Kissei Pharmaceutical Co. Ltd. Male and female cynomolgus monkeys (2–5 kg; Toyota Tsusho Corporation, Tokyo, Japan) were used in this study. All monkeys were housed individually at a stable temperature and humidity under a 12-h light/dark cycle, and they were maintained with free access to water and standard laboratory food until the day of the experiment.
In Vitro Experiments.
Monkeys were anesthetized with ketamine (10 mg/kg i.m.) and sacrificed by rapid exsanguination. The heart, trachea, and urinary bladder were then isolated. After removal of the fat and mucosa, the ventricles were removed from the heart, and the atria were prepared for experimentation. The trachea was cut into approximately 10 rings, each 2 mm in length, and they were appropriately prepared. The urinary bladder was opened longitudinally. After removal of the mucosa, a bladder preparation approximately 10 × 3 mm was prepared. Atria were suspended in a 20-ml organ bath, and other preparations were suspended in a 10-ml organ bath, each time containing Krebs' solution. This bath solution was maintained at 37°C and continuously gassed with a mixture of 95% oxygen and 5% carbon dioxide. Each preparation was connected to a force-displacement transducer (SB-1T; Nihon-Kohden, Tokyo, Japan) for continuous recording on a rectigragh (Recti-Horiz-8K; NEC San-ei, Tokyo, Japan). The preparations were allowed to equilibrate for 60 min after the establishment of an initial resting tension of 10 mN. After the basal tone had stabilized, we evaluated experimentally induced effects on bladder resting tension. In the case of the trachea, carbamoylcholine chloride (CCh) was added to induce contraction, then concentration-response curves were obtained for each preparation by cumulative addition of the appropriate drug to the bathing fluid.
To test the antagonistic potencies of β-AR antagonists against ritobegron, appropriate antagonists [(±)-2-hydroxy-5-[2-[[2-hydroxy-3-[4-[1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl]phenoxy]propyl]amino]ethoxy]-benzamide methanesulphonate (CGP-20712A), (±)-1-[(2,3-dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)amino]-2-butanol hydrochloride (ICI-118551), and/or 3-(2-allylphenoxy)-1-[(1S)-1,2,3,4-tetrahydronaphth-1-ylamino]-(2S)-2-propanol hydrochloride (SR58894A)] were added to the bath 60 min before the addition of ritobegron. Concentration-response curves for ritobegron were thus obtained in the presence of the antagonist. Only one agonist concentration curve was generated per preparation. All experiments were conducted in the presence of 10−6 M phentolamine (to block α-ARs), 5 × 10−7 M desipramine, and 3 × 10−5 M hydrocortisone (to block neuronal and extraneuronal uptake of catecholamines).
In Vivo Experiments.
Monkeys were initially anesthetized with ketamine (10 mg/kg i.m.). Then, after tracheal intubation, they were connected to a respirator (SN-480-5; Shinano Seisakusyo, Tokyo, Japan; 10 ml/kg; 20 strokes/min) and anesthetized with 1.5% enflurane. A cannula (PE-90; Nihon Becton Dickinson, Tokyo, Japan) filled with heparin-physiological saline solution (20 U/ml) was inserted into the right femoral artery, the other end being led to a transducer amplifier (1829; NEC San-ei, Tokyo, Japan) for blood pressure measurement via a pressure transducer (DT-XX; Nihon Becton Dickinson). Heart rate was measured via a tachometer (1321; NEC San-ei) connected to the transducer amplifier. A cannula (PE-90; Nihon Becton Dickinson) filled with physiological saline solution was inserted into the duodenum for intraduodenal drug administration. Through a midline abdominal incision, the ureter on each side and the proximal urethra were ligated, and a polyethylene catheter (PE-50; Nihon Becton Dickinson) was inserted into the urinary bladder via the top of the bladder dome, then connected through a three-way connector to a pressure transducer and a syringe filled with saline. The initial bladder pressure was adjusted to approximately 5 cmH2O by instillation of warmed saline (37°C) in 5-ml increments. Blood pressure, heart rate, and intravesical pressure were recorded continuously on a rectigragh. At the end of each experiment, isoproterenol (0.1 mg/kg i.v.) was administered to obtain the maximal intravesical pressure response.
Analysis of Data.
In the in vitro experiments, drug effects on the isolated bladder and trachea were expressed as a percentage of the maximal relaxation response to 10−5 M forskolin, whereas drug effects on atria were expressed as the differences before and after drug treatment. The EC50 value was calculated for each agonist from its concentration-response curve. Bladder selectivities versus atria and trachea were calculated by comparison with the relevant EC50 value. The pKB value was then calculated by using the following formula: pKB = log (CR − 1) − log [antagonist], where CR is the ratio of the EC50 values obtained in the presence and absence of antagonist. In the in vivo experiments, drug effects on intravesical pressure were expressed relative to the maximal response to isoproterenol (0.1 mg/kg i.v.) (see legend to Fig. 6). Drug effects on blood pressure and heart rate were assessed as the difference before and after drug administration.
All results are expressed as mean ± S.E. Statistical analysis was performed by using one-way analysis of variance followed by Dunnett's multiple comparison test. A probability less than 0.05 was accepted as significant. The SAS system (version 4.1; SAS Institute, Cary, NC) was used as the resource text for the statistical analysis.
Drugs.
Ritobegron (KUC-7483) is a prodrug, so in the in vitro studies we used the active form [KUC-7322; (−)-2-[4-(2-{[(1S,2R)-2-hydroxy-2-(4-hydroxyphenyl)-1-methylethyl]amino}ethyl)-2,5-dimethylphenyloxy]acetic acid], but in the in vivo study we used KUC-7483 itself. Ritobegron, its active form (KUC-7322), (R,R)-5-[2-[[2-(3-chlorophenyl)-2-hydroxyethyl]-amino]propyl]-1,3-benzodioxole-2,2-dicarboxylate (CL316,243), and SR58894A all were synthesized in our laboratory. The following drugs were obtained from commercial sources: (−)-isoprenaline (+)-bitartrate, phentolamine hydrochloride (isoproterenol), CCh, hydrocortisone 21-hemisuccinate, and desipramine hydrochloride (Sigma-Aldrich, St. Louis, MO); dimethyl sulfoxide (DMSO), 1 N NaOH, and arabic gum (Nacalai Tesque, Kyoto, Japan); sodium heparin (Aventis Pharma Japan, Tokyo, Japan); forskolin (Wako Pure Chemicals, Osaka, Japan); and CGP-20712A and ICI-118551 (Funakoshi, Tokyo, Japan). The Krebs' solution was of the following composition: 118.1 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 25.0 mM NaHCO3, 1.2 mM KH2PO4, and 11.1 mM glucose, pH 7.4. For the in vitro study, the active form (KUC-7322) was dissolved in distilled water containing two equivalents of NaOH. SR58894A was dissolved in 10% DMSO, whereas forskolin and hydrocortisone 21-hemisuccinate were dissolved in 100% DMSO, and the other drugs were dissolved in distilled water. The reported concentrations are the calculated final concentrations in the bath solution. For the in vivo study ritobegron was suspended in 0.5% arabic gum in distilled water; the other drugs were dissolved in saline.
Results
Effect of Ritobegron on Monkey Bladder.
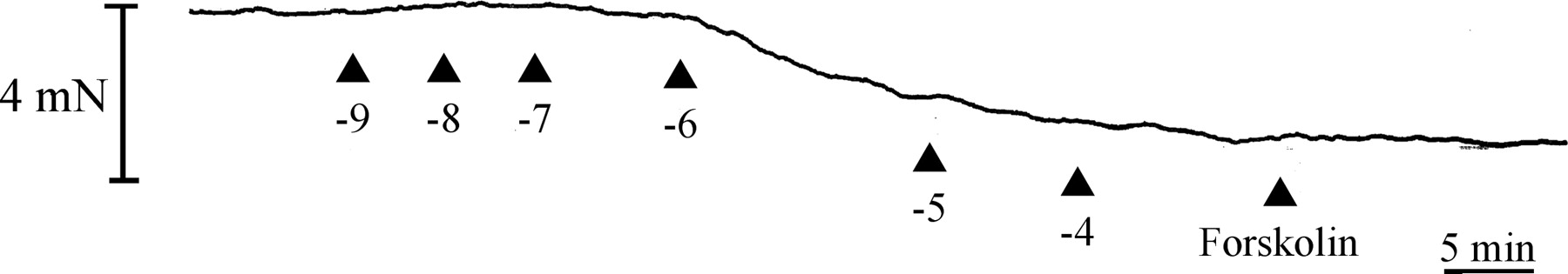
The chemical structures of ritobegron (KUC-7483) (A), KUC-7322 (B), CL316,243 (C), and isoproterenol (D) are shown in Fig. 1. KUC-7322 is the active form of ritobegron. Ritobegron decreased the resting tension of the isolated bladder in a concentration-dependent manner (Figs. 2 and 3). Isoproterenol (nonselective β-AR agonist) and CL316,243 (selective β3-AR agonist) also decreased the resting tension of the isolated bladder, each in a concentration-dependent manner (Fig. 3).

Chemical structures of ritobegron (KUC-7483) (A), KUC-7322 (B), CL316.243 (C), and isoproterenol (D). KUC-7322 is the active form of ritobegron.

Typical tracing of the effect of ritobegron on resting tension in isolated cynomolgus monkey bladder. Concentrations of ritobegron are shown as log M. Forskolin was applied at 10−5 M.

Effects of ritobegron, isoproterenol, and CL316,243 on resting tension in isolated cynomolgus monkey bladder. Data are expressed as a percentage of the maximal relaxation response to 10−5 M forskolin. Data represent the mean ± S.E. from five experiments.
The EC50 values obtained for ritobegron, isoproterenol, and CL316,243 were 8.2 ± 2.3 × 10−7, 1.9 ± 0.9 × 10−7, and 5.5 ± 3.1 × 10−6 M, respectively (Table 1). The rank order of their relaxing potencies was isoproterenol > ritobegron > CL316,243. The maximal relaxant effects of ritobegron, isoproterenol, and CL316,243 were 88.7 ± 3.7, 94.2 ± 4.3, and 73.9 ± 6.7%, respectively (Table 1).
EC50 values and maximal relaxation response to ritobegron, isoproterenol, and CL316,243 in isolated cynomolgus monkey bladder
Data represent the mean ± S.E. from five experiments.
Antagonism by Subtype-Selective β-AR Antagonists of Ritobegron-Induced Relaxation in Isolated Monkey Bladder.
In the isolated bladder, addition of the selective β1-AR antagonist CGP-20712A (10−7 M) plus the selective β2-AR antagonist ICI-118551 (10−7 M) had no effect on the relaxation induced by ritobegron (Fig. 4A). In the combined presence of CGP-20712A and ICI-118551, the β3-AR antagonist SR58894A (3 × 10−6 M) produced a rightward shift of the concentration-response curve for ritobegron without altering the maximal response (Fig. 4B). The pKB value obtained for SR58894A was 6.56 ± 0.35 (Fig. 4B).

Antagonism by subtype-selective β-AR antagonists of ritobegron-induced relaxation in cynomolgus monkey bladder. Data are expressed as a percentage of the maximal relaxation response to 10−5 M forskolin. Data represent the mean ± S.E. from seven experiments. CGP, CGP-20712A; ICI, ICI-118551; SR, SR58894A.
Effects of Ritobegron and Isoproterenol on Isolated Atrial Rate and Isolated Trachea Precontracted with CCh.
Both ritobegron and isoproterenol concentration-dependently increased atrial rate (Fig. 5A). The EC50 values for ritobegron and isoproterenol were 6.5 ± 1.2 × 10−5 and 2.2 ± 0.2 × 10−9 M, respectively (Table 2). Isoproterenol produced concentration-dependent relaxation of tracheas precontracted with 10−7 M CCh (Fig. 5B), the EC50 value being 1.6 ± 0.4 × 10−7 M. In contrast, ritobegron had no such effect between 10−9 and 10−4 M (Fig. 5B). Even at the highest concentration tested, 10−3 M, the maximal tracheal relaxation induced by ritobegron was only 26.7 ± 8.1%, indicating an EC50 value of 10−3 M or more (Table 2). Ritobegron was approximately 30,000 times less potent than isoproterenol in these two tissues (Table 2).

Effects of ritobegron and isoproterenol on isolated cynomolgus monkey tissues. Data are shown for atrial rate (A) and relaxation of CCh-precontracted trachea (B). Data for atrial rate are expressed as the difference before and after drug treatment, and data for trachea are expressed as a percentage of the maximal relaxation response to 10−5 M forskolin. Data represent the mean ± S.E. from five to seven experiments.
EC50 values in various tissues and bladder selectivities of ritobegron and isoproterenol in isolated cynomolgus monkey tissues
Data represent the mean ± S.E. from five experiments.
Bladder Selectivity of Ritobegron.
The selectivity of ritobegron for the bladder was 79.3-fold higher versus the atria and more than 1200-fold higher versus the trachea (Table 2). Ritobegron displayed much higher selectivity for the bladder than isoprotrenol (actually, isoproterenol did not display any bladder selectivity) (Table 2).
Effects of Ritobegron in Anesthetized Monkey.
The mean values obtained for intravesical pressure before the administration of ritobegron were not significantly different among the groups. Ritobegron, at a dose of 0.3 mg/kg, had no evident effect on intravesical pressure (Fig. 6A). However, from 1 to 10 mg/kg it significantly decreased intravesical pressure (Fig. 6A). The ED50 value obtained for ritobegron at 90 min after its administration was 1.44 mg/kg. Ritobegron had no effect on either mean blood pressure or heart rate (Fig. 6, B and C). At the end of the experiments, the effects of intravenous administration of isoproterenol (0.1 mg/kg) on mean blood pressure and heart rate were assessed in each treated group. Isoproterenol reduced mean blood pressure by 25.92 mm Hg (vehicle group), 35.83 mm Hg (0.3 mg/kg ritobegron group), 37.25 mm Hg (1 mg/kg ritobegron group), 38.87 mm Hg (3 mg/kg ritobegron group), and 33.59 mm Hg (10 mg/kg ritobegron group). Heart rate was increased by isoproterenol by 57.89, 44.12, 44.77, 36.55, and 58.03 beats/min, respectively, in those groups.

Effects of intraduodenal administration of ritobegron on intravesical pressure (A), mean blood pressure (B), and heart rate (C) in anesthetized cynomolgus monkeys. Data for intravesical pressure are expressed relative to the maximal response to isoproterenol (0.1 mg/kg i.v.) (i.e., that dose of isoproterenol would have reduced intravesical pressure from 100 to 0%). Data represent the mean ± S.E. from three animals. *, P < 0.05 versus vehicle (Dunnett's multiple comparison test).
Discussion
In the present in vitro and in vivo experiments we evaluated the pharmacological profile of the novel selective β3-AR agonist ritobegron and its effects on the cynomolgus monkey bladder.
It has been reported that in primates, such as cynomolgus monkey (Takeda et al., 2002a) and humans (Igawa et al., 1998, 1999; Yamazaki et al., 1998; Takeda et al., 1999), bladder relaxation is mediated via the β3-AR, and that in humans 97% of total β-AR mRNA is represented by the β3-AR subtype (Yamaguchi, 2002; Nomiya and Yamaguchi, 2003). For the above reason, in this study we used the cynomolgus monkey to evaluate the potential usefulness of ritobegron as a drug with beneficial effects on bladder function.
In the first in vitro experiment, we examined the relaxing effects of ritobegron, isoproterenol, and CL316,243. Although all three drugs decreased the resting tension of the isolated bladder, the slopes of their concentration-response curves were different. It has been reported that β3-AR agonists [(R*,R*)-(±)-4-[2-[(2-(3-chlorophenyl)-2-hydroxyethyl)amino]propyl]phenoxyacetic acid (BRL37344) and CL316,243] induce responses with a slower onset than that to isoproterenol (Roberts et al., 1999). Therefore, it seems reasonable that the slopes of the curves differed depending on the drug. The maximal relaxation to ritobegron was equivalent to that induced by isoproterenol, and both agents displayed full agonistic activities. In contrast, CL316,243 had a relatively weak relaxing effect, its EC50 value being approximately seven times higher than that of ritobegron. Moreover, CL316,243 displayed evidence of partial agonistic activity, with the maximum relaxing effect even at the highest concentration being only 73.9%. These results are consistent with previous reports that classic selective β3-AR agonists, such as CL316,243 and BRL37344, exhibit partial agonistic activities in both the monkey and human bladder at the same concentration as the maximal concentration used in this study (1 × 10−4 M) (Igawa et al., 1999, 2001; Takeda et al., 2002a). The above results suggest that in humans ritobegron might produce sufficient bladder relaxation and be more potent than classic β3-AR agonists.
In the second in vitro experiment we tried, by using subtype-specific β-AR antagonists, to determine which β-AR subtypes might be involved in ritobegron-induced bladder relaxation. The relaxing effect of ritobegron was not antagonized by either CGP-20712A (10−7 M) or ICI-118551 (10−7 M), even though at that concentration they occupy virtually all β1- or β2-AR, respectively (Takeda et al., 2002a). Thus, neither β1- nor β2-AR would seem to be involved in ritobegron-induced bladder relaxation in the cynomolgus monkey. In contrast, in the combined presence of CGP-20712A and ICI-118,551 the selective β3-AR antagonist SR58894A effectively antagonized the ritobegron-induced bladder relaxation, with the pKB value being 6.56. This pKB value is comparable with the pA2 value of 6.24 obtained for isoproterenol-induced relaxation of the human bladder, an effect that is known to be mediated predominantly through the β3-AR (Igawa et al., 1999). On those grounds, ritobegron-induced cynomolgus monkey bladder relaxation would seem to be mediated via the β3-AR.
In view of the above interpretation, and because monkey bladder relaxation has previously been reported to be mediated via β3-AR (Takeda et al., 2002a), we performed the third in vitro experiment to estimate the β3-AR-subtype selectivity of ritobegron. To this end, bladder selectivity was evaluated by using the pharmacologically characterized atrium (β1-AR subtype) and trachea (β2-AR subtype). The bladder selectivity of ritobegron was 79.3-fold higher and more than 1200-fold higher versus atria and trachea, respectively. Thus, ritobegron is highly selective for the β3-AR subtype, which is found in the monkey and human bladder. Unfortunately, we do not have data obtained by using recombinant human β-AR; to elucidate the effect on the human bladder would require such experimentation.
Finally, in the in vivo experiment we established that in anesthetized monkeys ritobegron significantly decreased intravesical pressure without affecting either mean blood pressure or heart rate. These results are consistent with previous reports that selective β3-AR agonists improve bladder functions in rats with minimal effects on the cardiovascular system (Takeda et al., 2000b, 2002b; Kaidoh et al., 2002). Moreover, when we measured the plasma concentration of ritobegron in cynomolgus monkeys that had received on intraduodenal administration of 1, 3, or 10 mg/kg the maximum plasma values we obtained were 3.7 × 10−7, 1.3 × 10−6, and 4.4 × 10−6 M, respectively (data not shown). Comparing these plasma concentrations to the concentrations used to obtain the in vitro results suggests that the latter results are well in line with our in vivo results. Actually, the concentration achieved in the plasma after administration of 1 mg/kg ritobegron (3.7 × 10−7 M) was enough to induce relaxation of the isolated bladder. In addition, 4.4 × 10−6 M (the plasma concentration achieved by giving 10 mg/kg ritobegron) did not induce a β1- or β2-AR-mediated effect in the present isolated tissues. Therefore, even at the highest dose used here, 10 mg/kg ritobegron, the heart rate elevation and blood pressure reduction attributed to β1- and β2-AR (Brodde, 1988; Ferro et al., 1993) did not occur in our in vivo study. Thus, we demonstrated that, because of its high selectivity for β3-AR, ritobegron decreased intravesical pressure in our monkeys without affecting cardiovascular parameters.
There is compelling in vivo evidence to support selective β3-AR agonists improving the urine-storage function in rat models in which bladder overactivity is induced by bladder-outlet obstruction (Woods et al., 2001), intravesical infusion of prostaglandin E2 (Takeda et al., 2002b), or cerebral infarction (Kaidoh et al., 2002). In a cystometric study in rats, selective β3-AR agonists significantly prolonged micturition interval and increased bladder capacity without affecting voiding functions (Takeda et al., 2000b). Collectively, these pieces of evidence predict a useful role for β3-AR agonists in the treatment of OAB.
In conclusion, our data indicated that ritobegron has potent and selective β3-AR agonistic activity and it relaxes the isolated monkey bladder via the β3-AR. Moreover, in vivo ritobegron decreased intravesical pressure without affecting cardiovascular parameters. To judge from these results, ritobegron shows promise as a potential agent for the treatment of OAB.
Authorship Contributions
Participated in research design: Maruyama, Tatemichi, Goi, and Yamazaki.
Conducted experiments: Maruyama, Tatemichi, Goi, and Yamazaki.
Performed data analysis: Maruyama, Tatemichi, Goi, and Yamazaki.
Wrote or contributed to the writing of the manuscript: Maruyama, Tatemichi, Maruyama, Hoyano, Yamazaki, and Kusama.
Footnotes
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
ABBREVIATIONS:
- OAB
- overactive bladder
- AR
- adrenoceptor
- CCh
- carbamoylcholine chloride
- DMSO
- dimethyl sulfoxide
- KUC-7483
- (−)-ethyl 2-[4-(2-{[(1S,2R)-2-hydroxy-2-(4-hydroxyphenyl)-1-methylethyl]amino}ethyl)-2,5-dimethylphenyloxy]acetate monohydrochloride
- KUC-7322
- (−)-2-[4-(2-{[(1S,2R)-2-hydroxy-2-(4-hydroxyphenyl)-1-methylethyl]amino}ethyl)-2,5-dimethylphenyloxy]acetic acid
- CL316,243
- (R,R)-5-[2-[[2-(3-chlorophenyl)-2-hydroxyethyl]-amino]propyl]-1,3-benzodioxole-2,2-dicarboxylate
- SR58894A
- 3-(2-allylphenoxy)-1-[(1S)-1,2,3,4-tetrahydronaphth-1-ylamino]-(2S)-2-propanol hydrochloride
- CGP-20712A
- (±)-2-hydroxy-5-[2-[[2-hydroxy-3-[4-[1-methyl-4-(trifluoromethyl)-1H-imidazol-2-yl]phenoxy]propyl]amino]ethoxy]-benzamide methanesulphonate
- ICI-118551
- (±)-1-[(2,3-dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)amino]-2-butanol hydrochloride
- BRL37344
- (R*,R*)-(±)-4-[2-[(2-(3-chlorophenyl)-2-hydroxyethyl)amino]propyl]phenoxyacetic acid.
- Received January 11, 2012.
- Accepted April 13, 2012.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}