


Abstract
(R)-6-[(3-{[4-(5-{[2-hydroxy-2-(8-hydroxy-2-oxo-1,2-dihydroquinolin-5-yl)ethyl]amino}pent-1-yn-1-yl)phenyl] carbamoyl}phenyl)sulphonyl]-4-[(3-methoxyphenyl)amino]-8-methylquinoline-3-carboxamide trifluoroacetic acid (GS-5759) is a bifunctional ligand composed of a quinolinone-containing pharmacophore [β2-adrenoceptor agonist orthostere (β2A)] found in several β2-adrenoceptor agonists, including indacaterol, linked covalently to a phosphodiesterase 4 (PDE4) inhibitor related to 6-[3-(dimethylcarbamoyl)benzenesulphonyl]-4-[(3-methoxyphenyl)amino]-8-methylquinoline-3-carboxamide (GSK 256066) by a pent-1-yn-1-ylbenzene spacer. GS-5759 had a similar affinity for PDE4B1 and the native β2-adrenoceptor expressed on BEAS-2B human airway epithelial cells. However, compared with the monofunctional parent compound, β2A, the KA of GS-5759 for the β2-adrenoceptor was 35-fold lower. Schild analysis determined that the affinities of the β-adrenoceptor antagonists, (2R,3R)-1-[(2,3-dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl) amino]-2-butanol (ICI 118551) and propranolol, were agonist-dependent, being significantly lower for GS-5759 than β2A. Collectively, these data can be explained by “forced proximity,” bivalent binding where the pharmacophore in GS-5759 responsible for PDE4 inhibition also interacts with a nonallosteric domain within the β2-adrenoceptor that enhances the affinity of β2A for the orthosteric site. Microarray analyses revealed that, after 2-hour exposure, GS-5759 increased the expression of >3500 genes in BEAS-2B cells that were highly rank-order correlated with gene expression changes produced by indacaterol and GSK 256066 in combination (Ind/GSK). Moreover, the line of regression began close to the origin with a slope of 0.88, indicating that the magnitude of most gene expression changes produced by Ind/GSK was quantitatively replicated by GS-5759. Thus, GS-5759 is a novel compound exhibiting dual β2-adrenoceptor agonism and PDE4 inhibition with potential to interact on target tissues in a synergistic manner. Such polypharmacological behavior may be particularly effective in chronic obstructive pulmonary disease and other complex disorders where multiple processes interact to promote disease pathogenesis and progression.
Introduction
Long-acting β2-adrenoceptor agonists (LABAs) produce clinical benefit in chronic obstructive pulmonary disease (COPD) by promoting a long-lasting increase in airway caliber. Although the rapid, cAMP-dependent inhibition of myosin phosphorylation is thought to account for this effect (Giembycz and Raeburn, 1991), evidence has recently become available that LABAs also act transcriptionally to protect the airways against agonist-induced bronchoconstriction (Holden et al., 2011). A logical extension of that discovery is that LABAs may also improve lung function in COPD through their ability to promote anti-inflammatory gene expression. Indeed, in airway epithelial cells, the LABAs formoterol, salmeterol, and indacaterol upregulate a variety of genes whose expression may impart clinical benefit in COPD beyond directly reducing airway smooth muscle tone (Kaur et al., 2008a,b; Rider et al., 2011; Nasreen et al., 2012; Komatsu et al., 2013; Moodley et al., 2013; Holden et al., 2014; BinMahfouz et al., 2015; Joshi et al., 2015b; Patel et al., 2015).
In individuals with severe, bronchitic COPD who suffer frequent exacerbations, the phosphodiesterase 4 (PDE4) inhibitor roflumilast is a recommended treatment option through its ability to improve airway caliber (www.goldcopd.org). However, roflumilast does not promote acute bronchodilation; instead, it increases lung function several hours after dosing (Grootendorst et al., 2003). This delayed onset of action suggests new gene induction, and subsequent suppression of airway inflammation may be required. Indeed, an anti-inflammatory effect of roflumilast is suggested from clinical trials where it reduced the frequency of COPD exacerbations, which is typically associated with a worse inflammatory status (Calverley et al., 2009; Kew et al., 2013). Furthermore, in severe, bronchitic disease, the addition of roflumilast to an inhaled corticosteroid/LABA combination therapy reduced exacerbations (Muñoz-Esquerre et al., 2015), attenuated inflammation in the small airways, arrested hyperinflation, and improved regional airflow distribution (De Backer et al., 2014). Thus, the remedial actions of a PDE4 inhibitor in COPD may also depend upon cAMP-dependent gene expression (Giembycz and Newton, 2014).
Polypharmacology is a subdivision of pharmacology devoted to single chemical entities that interact simultaneously with more than one molecular target over a similar concentration range, or to a cocktail of drugs that bind to different targets within a disease network (Boran and Iyengar, 2010; Jalencas and Mestres, 2013). Theoretically, polypharmacological methods could be particularly effective in treating complex pathologies such as COPD, where multiple, adverse processes interact to promote disease pathogenesis and progression (Morphy and Rankovic, 2005; Jalencas and Mestres, 2013). An approach receiving considerable interest in respiratory medicine is the development of inhaled, bifunctional ligands. These new medicines contain two pharmacophores joined covalently at linker atoms by a pharmacologically inert “spacer” (Shonberg et al., 2011; Hughes et al., 2012; Phillips and Salmon, 2012). Bifunctional ligands have several advantages over the monofunctional parent compounds in combination. Typically, their high molecular weight often translates into greater lung retention, low oral bioavailability, reduced systemic exposure, and an improved therapeutic ratio (Phillips and Salmon, 2012; Robinson et al., 2013). Moreover, the development of these compounds is simplified in terms of matched pharmacokinetics, formulation, and deposition characteristics (Shonberg et al., 2011). This latter property is particularly desirable if both pharmacophores are designed to interact synergistically at target tissues to deliver superior clinical outcomes. Clearly, this is the case for a LABA and PDE4 inhibitor, which can act on the same signaling pathway to enhance cAMP synthesis and block cAMP degradation, respectively.
(R)-6-[(3-{[4-(5-{[2-hydroxy-2-(8-hydroxy-2-oxo-1,2-dihydroquinolin-5-yl)ethyl]amino}pent-1-yn-1-yl)phenyl]carbamoyl}phenyl)sulphonyl]-4-[(3-methoxyphenyl)amino]-8-methylquinoline-3-carboxamide trifluoroacetic acid (GS-5759) is a novel, bifunctional ligand in which the quinolinone-containing pharmacophore responsible for β2-adrenoceptor agonism in several LABAs has been conjugated to a PDE4 inhibitor that is a close structural analog of 6-[3-(dimethylcarbamoyl)benzenesulphonyl]-4-[(3-methoxyphenyl)amino]-8-methylquinoline-3-carboxamide (GSK 256066) (Woodrow et al., 2009; Tralau-Stewart et al., 2011) by a pent-1-yn-1-ylbenzene spacer (Baker et al., 2011) (Fig. 1). This molecule has been optimized for inhaled delivery, with COPD being a primary indication (Tannheimer et al., 2014; Salmon et al., 2014). In the present study, we compared the pharmacology of GS-5759 with representative monofunctional pharmacophores in BEAS-2B human airway epithelial cells using gene expression as a therapeutically relevant output. To achieve this objective, stable transfection of a simple cAMP-response element (CRE) luciferase reporter was used as a model system to define the pharmacodynamics of GS-5759, while increases in gene expression that could contribute to the therapeutic activity of a LABA/PDE4 combination therapy were identified and validated by microarray profiling and reverse-transcription polymerase chain reaction (RT-PCR) respectively.

Structure of the bifunctional ligand GS-5759 and analogs thereof. Bifunctional ligands contain two pharmacophores joined covalently at linker atoms (black arrows) by an appropriately defined “spacer.” GS-5759 is composed of the quinolinone-containing β2-adrenoceptor agonist (pink; GS-709344; β2A) and a close structural analog of the PDE4 inhibitor, GSK 256066 (blue; denoted GSK 256066a), linked covalently by a pent-1-yn-1-ylbenzene spacer (black). β2A linked to 4-(pent-1-yn-1-yl)phenylaniline (GS-548834; β2A-S) is also shown together with a bifunctional ligand (GS-493163) in which the quinolone in GS-5759 has been methylsulphonylated to reduce inhibitory activity at PDE4. The asterisks indicate chiral centers.
Materials and Methods
Stable Generation of BEAS-2B Luciferase Reporter Cells.
Cells were transfected with 8 µg of plasmid DNA (pADneo2-C6-BGL) using Lipofectamine 2000 (Invitrogen, Burlington, ON, Canada) to generate 6×CRE BEAS-2B cells, as described previously (Chivers et al., 2004; Meja et al., 2004). Cells were cultured for 3 days under a 5% CO2/air atmosphere at 37°C in 24-well plastic plates (Corning Life Sciences, Lowell, MA) containing keratinocyte serum-free medium (SFM) supplemented with epidermal growth factor (5 ng/ml), bovine pituitary extract (50 μg/ml), penicillin (100 mg/ml), and streptomycin (100 IU/ml), and for a further 24 hours in keratinocyte SFM (Greer et al., 2013). At this time, cultures were confluent and were processed for the measurement of luciferase activity or gene expression.
Measurement of Luciferase Activity.
Confluent 6×CRE BEAS-2B reporter cells in 24-well plates were treated as indicated with the compound(s) of interest. Cells were lysed at 6 hours in 1×firefly luciferase lysis buffer (Biotium, Hayward, CA), and luciferase activity was measured by luminometry. Data are expressed as fold increase in luciferase activity using time-matched activity values in unstimulated cells as the denominator.
RNA Isolation, Reverse Transcription, and RT-PCR.
Confluent 6×CRE BEAS-2B reporter cells were treated as indicated, and total RNA was extracted (RNeasy Mini Kit; Qiagen Inc., Mississauga, ON, Canada) and reverse transcribed using a qScript cDNA synthesis kit according to the manufacturer’s instructions (Quanta Biosciences, Gaithersburg, MD). β-Adrenoceptor expression levels were determined with a commercially available TaqMan G-protein–coupled receptor array (Applied Biosystems, Foster City, CA) using an ABI 7900HT instrument (Applied Biosystems). The amplification conditions used were as stated in the G-protein–coupled receptor array protocol. Relative cDNA concentrations for β-adrenoceptor subtypes were determined by the comparative Ct (ΔΔCt) method and were expressed relative to the gene encoding the 18S ribosomal subunit (RPS18).
Genes upregulated by GS-5759, indacaterol, GSK 256066, and indacaterol and GSK 256066 in combination (Ind/GSK) were quantified by RT-PCR analysis of cDNA using the primer pairs shown in Supplemental Table 1 (designed using Primer Express software; Applied Biosystems). When possible, primers were designed to span an intron. Nine genes were selected from the array: suppressor of cytokine signaling 3 (SOCS3); complement component 5a receptor 1 (C5AR1); fibroblast growth factor receptor 2 (FGFR2); dual specificity phosphatase 1 (DUSP1; aka mitogen-activated protein kinase phosphatase-1); nuclear receptor subfamily 4, group A, member 2 (NR4A2); cluster of differentiation 200 (CD200); regulator of G-protein signaling 2 (RGS2); cyclin-dependent kinase inhibitor 1C (CDKN1C, aka kinase inhibitor protein 2 of 57 kDa); and cysteine-rich secretory protein LCCL (limulus clotting factor C, Cochlin, Lgl1) domain-containing 2 (CRISPLD2). These reactions were performed using an ABI StepOnePlus instrument (Applied Biosystems) on 2.5 µl of cDNA in 10-µl reactions using Fast SYBR Green chemistry (Invitrogen, Carlsbad, CA) according to the manufacturer’s guidelines. Transcript levels were determined from a cDNA standard curve (analyzed simultaneously) and are presented as a ratio to glyceraldehyde-3-phosphate dehydrogenase. Amplification conditions were as follows: 95°C, 20 seconds, followed by 40 cycles of 95°C, 3 seconds and 60°C, 30 seconds. Dissociation curves (95°C, 15 seconds; 60°C, 1 minute; 95°C, 15 seconds) were constructed to confirm primer specificity.
Microarray and Data Processing.
BEAS-2B cells in keratinocyte SFM without supplements were cultured for 1, 2, 6, and 18 hours with vehicle, GS-5759 (10 nM), or Ind/GSK (both at 10 nM; N = 4 at each time point). Total RNA was extracted and quantified (NanoDrop 2000; Thermo Scientific, Wilmington, DE). The quality of each sample was determined using the Agilent 2100 Laboratory-on-a-Chip System (Agilent, Santa Clara, CA) before being processed for gene profiling by Expression Analysis Inc. (Durham, NC). Each RNA sample was converted to a biotinylated cRNA target using a GeneChip 3′-in vitro transcription express kit (Affymetrix, Santa Clara, CA) and hybridized to an Affymetrix U133plus2.0 human GeneChip, which contains 54,612 probe sets. Chips were stained, washed, and fluorescence intensity scanned using the protocol described by Affymetrix. The array images were normalized using the probe logarithmic intensity error algorithm (http://media.affymetrix.com/support/technical/technotes/plier_technote.pdf) implemented in Expression Console (Affymetrix) and stored as .CEL files. At this time, data from all genomics samples were subjected to rigorous GeneChip level Affymetrix quality control procedures to detect potential outliers due to processing and instrumentation. Signals from the four replicates for each probe set in drug- and time-matched, vehicle-treated cells were averaged and analyzed by one-way analysis of variance. Probe sets with an unadjusted P value of <0.05 were considered significant. The relative expression patterning of probe sets was implemented in Transcriptome Analysis Console (Affymetrix) and visualized by generating volcano plots and heat maps. These microarray data have been deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus and are freely accessible through Gene Expression Omnibus accession number GSE84880 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE84880). All genes are described using Human Genome Nomenclature Committee symbols. Common names, if applicable, for those genes subjected to PCR validation are also provided (Supplemental Table 1). Functional classification of GS-5759–induced genes including associated gene ontology (GO) terms was performed with the Database for Visualization and Integrated Discovery (DAVID) bioinformatics resource (version 6.7); pseudogenes, hypothetical genes, and redundant sequences lacking annotation were excluded (Huang et al., 2009).
Measurement of PDE4 Activity.
Phosphodiesterase activity was determined at room temperature in 96-well plates by fluorescence polarization (BPS Bioscience, San Diego, CA). Reactions were conducted in a total volume of 50 µl of assay buffer containing human recombinant PDE4B1 (6.7 pM; BPS Bioscience), 2'-(6-[fluoresceinyl]aminohexylcarbamoyl) cyclic adenosine-3′,5′-monophosphate (100 nM; Biolog Life Science Institute, Bremen, Germany), and the inhibitor of interest. cAMP hydrolysis was allowed to proceed for 60 minutes and was terminated by the addition of 100 µl of “binding reagent” (BPS Bioscience). Plates were incubated for a further 60 minutes with gentle shaking, and fluorescence intensity was measured (λexcitation = 485 nm; λemission = 528 nm). The molar concentration of drug that inhibited enzyme activity by 50% (IC50) was determined by nonlinear, iterative curve fitting (vide infra) and converted into an equilibrium dissociation constant (KI) according to either Cheng and Prusoff (1973) or, for a tight-binding inhibitor where IC50 ≈ total enzyme concentration ([E]), Copeland et al. (1995) (eqs. 1 and 2, respectively). Thus: (1)
(1) (2)where [S] is the substrate concentration, and Km is the concentration of substrate at which the reaction rate is 50% of the maximum velocity.
(2)where [S] is the substrate concentration, and Km is the concentration of substrate at which the reaction rate is 50% of the maximum velocity.
Curve Fitting.
Monophasic E/[A] curves were fit by least-squares, nonlinear, iterative regression to the following form of the Hill equation (Prism 4; GraphPad Software Inc., San Diego, CA) (Motulsky and Christopoulos, 2003): (3)where E is the effect; Emin and Emax are the basal response and maximum response, respectively; p[A] is the negative log molar concentration of the compound of interest; p[A]50 is a location parameter equal to the negative log molar concentration of compound producing (Emax − Emin)/2; and n is the gradient of the E/[A] curve at the p[A]50 level.
(3)where E is the effect; Emin and Emax are the basal response and maximum response, respectively; p[A] is the negative log molar concentration of the compound of interest; p[A]50 is a location parameter equal to the negative log molar concentration of compound producing (Emax − Emin)/2; and n is the gradient of the E/[A] curve at the p[A]50 level.
Determination of Antagonist Equilibrium Dissociation Constants.
The affinities of (2R,3R)-1-[(2,3-dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl) amino]-2-butanol (ICI 118551) and propranolol were estimated by least-squares, nonlinear regression as described by Waud et al. (1978). GSK 256066 (10 nM) was present throughout to eliminate any contribution that PDE4 inhibition could make to the position or shape of GS-5759 E/[A] curves. Agonist E/[A] curves were generated in 6×CRE BEAS-2B reporter cells pretreated (30 minutes) with vehicle or β-adrenoceptor antagonist at the concentrations indicated. Each family of E/[A] curves was fit simultaneously to eq. 4, where [A] and [B] are the molar concentration of the agonist and antagonist, respectively; S is the Schild slope factor; and pA2 is the affinity of the antagonist when S = 1, which is equivalent to the pKB:
 (4)
(4)Determination of Agonist Equilibrium Dissociation Constants.
Agonist affinities were estimated in 6×CRE BEAS-2B reporter cells by fractional, irreversible, β2-adrenoceptor inactivation (Furchgott, 1966). Agonist E/[A] curves were generated in cells pretreated (60 minutes) with vehicle or the alkylating agent 5(2-(((1′-(4′-isothiocyanatephenylamino)thiocarbonyl)amino)-2-methylpropyl) amino-2-hydroxypropoxy)-3,4-dihydrocarbostyril (DCITC; 100nM) and then washed in SFM (Deyrup et al., 1998). Each pair of E/[A] curves was fit simultaneously to the operational model of agonism (eq. 5), which describes a theoretical relationship between pharmacological effect (E) and agonist concentration (Black and Leff, 1983). Algebraically, (5)where Em is the theoretical maximum response of the tissue; n is the slope of the relationship between the concentration of agonist-receptor complexes and response; and τ is the operational efficacy of the agonist, which is the reciprocal of the percentage of agonist-bound receptors required to give half maximum response. In these analyses, a common value of Em, KA, and n is assumed (Black and Leff, 1983; Leff et al., 1990). Only τ, which at submaximal responses decreases proportionally with the remaining fraction of noninactivated receptors, was allowed to vary between individual E/[A] curves (Black and Leff, 1983; Leff et al., 1990). Thus, for each experiment, a single estimate of Em, n, and KA was calculated as well as the operational efficacy (τ) of the agonist before receptor inactivation. To ensure that operational parameter estimates could be compared, these experiments were conducted in the presence of GSK 256066 (10 nM) to eliminate any contribution that PDE4 inhibition could make to the position or shape of E/[A] curves generated for GS-5759 and a close structural analog, (R)-6-[(3-{[4-(5-{[2-hydroxy-2-(8-hydroxy-2-oxo-1,2-dihydroquinolin-5-yl)ethyl]amino}pent-1-yn-1-yl)phenyl] carbamoyl}phenyl)sulphonyl]-4-[(3-methoxyphenyl)(methylsulphonyl)amino]-8-methylquinoline-3-carboxamide trifluoroacetic acid (GS-493163) (Fig. 1).
(5)where Em is the theoretical maximum response of the tissue; n is the slope of the relationship between the concentration of agonist-receptor complexes and response; and τ is the operational efficacy of the agonist, which is the reciprocal of the percentage of agonist-bound receptors required to give half maximum response. In these analyses, a common value of Em, KA, and n is assumed (Black and Leff, 1983; Leff et al., 1990). Only τ, which at submaximal responses decreases proportionally with the remaining fraction of noninactivated receptors, was allowed to vary between individual E/[A] curves (Black and Leff, 1983; Leff et al., 1990). Thus, for each experiment, a single estimate of Em, n, and KA was calculated as well as the operational efficacy (τ) of the agonist before receptor inactivation. To ensure that operational parameter estimates could be compared, these experiments were conducted in the presence of GSK 256066 (10 nM) to eliminate any contribution that PDE4 inhibition could make to the position or shape of E/[A] curves generated for GS-5759 and a close structural analog, (R)-6-[(3-{[4-(5-{[2-hydroxy-2-(8-hydroxy-2-oxo-1,2-dihydroquinolin-5-yl)ethyl]amino}pent-1-yn-1-yl)phenyl] carbamoyl}phenyl)sulphonyl]-4-[(3-methoxyphenyl)(methylsulphonyl)amino]-8-methylquinoline-3-carboxamide trifluoroacetic acid (GS-493163) (Fig. 1).
Determination of Receptor Reserve.
Receptor occupancy-response curves were constructed using KA values determined by fractional, irreversible, β2-adrenoceptor inactivation (vide supra). At each concentration of agonist and, therefore, at each level of response, fractional receptor occupancy (i.e., the ratio of agonist-occupied receptors, RA, to the total receptor population, Rt, in control cells) was determined (Furchgott, 1966) assuming the binding of ligand to the β2-adrenoceptor was a noncooperative process. Thus
 (6)
(6)Drugs and Analytical Reagents.
GS-5759, GS-493163, 5-[(1R)-2-{[5-(4-aminophenyl)pent-4-yn-1-yl]amino}-1-hydroxyethyl]-8-hydroxyquinolin-2(1H)-one bis-trifluoracetic acid (GS-548834), 8-hydroxy-5-((R)-1-hydroxy-2-methylaminoethyl)-1H-quinolin-2-one (GS-709344), GSK 256066, and indacaterol were synthesized “in house.” ICI 118551 and (±) propranolol HCl were purchased from Sigma-Aldrich (St. Louis, MO). (Z)-7-[(1R,2R,3R,5R)-5-chloro-3-hydroxy-2-[(E,4S)-4-hydroxy-4-(1-prop-2-enylcyclobutyl)but-1-enyl]cyclopentyl]hept-5-enoic acid (ONO-AE1-259), DCITC, and 4-(3-methoxyphenylamino)-8-methyl-6-(3-(methyl carbamoyl)phenylsulphonyl)quinoline-3-carboxamide (GSK 256066a) were generously donated by ONO Pharmaceuticals (Osaka, Japan), Dr. Stephen Baker (University of Florida, Tampa, FL), and Dr. Michael Pollastri (Northeastern University, Boston, MA), respectively. All drugs were dissolved in dimethylsulfoxide and diluted to the required working concentrations in SFM. The highest concentration of dimethylsulfoxide used in these experiments (0.2%, v/v) did not affect any output measured.
Statistics.
Data points, bars, and values in the text and figure legends represent the mean ± S.E. mean of N independent determinations. All CRE reporter and TaQman PCR data were analyzed by Student’s two-tailed t test or repeated measures, one-way analysis of variance as indicated followed by Tukey’s multiple comparison test when appropriate. In Schild analysis, to determine if S deviated significantly from unity, the family of E/[A] curves that made up an individual experiment was fit globally to eq. 4 under two conditions: one where S was constrained to a constant equal to 1, and the other where it was a shared value for all data sets. The F test was applied to establish the condition that gave the best fit, which was used for the analysis. The same approach was used to determine if slopes of lines of regression were significantly different from unity. To establish if microarray data were sampled from a Gaussian or non-Gaussian distribution, the D’Agostino and Pearson omnibus K2 normality test was applied. Rank-order correlations were then calculated using two-tailed parametric (Pearson’s r) and nonparametric (Spearman’s ρ) tests as appropriate. The null hypothesis was rejected when P < 0.05.
Results
Conjugating the quinolinone-containing orthostere GS-709344 (referred to as β2A), which confers β2-adrenoceptor agonism in several LABAs, including indacaterol, carmoterol, and abediterol (Montuschi and Ciabattoni, 2015), to a close structural analog of GSK 256066 [GSK 256066a; compound 12b in Ochiana et al. (2015)] by a pent-1-yn-1-ylbenzene spacer afforded the bifunctional compound GS-5759 (Fig. 1; Baker et al., 2011). Two additional ligands were also studied: GS-493163, in which the quinoline in GS-5759 is methylsulphonylated to reduce its PDE4 inhibitory activity, and GS-548834, in which β2A is linked to 4-(pent-1-yn-1-yl)aniline (referred to as β2A-S; Fig. 1).
β-Adrenoceptor Subtypes on BEAS-2B Cells.
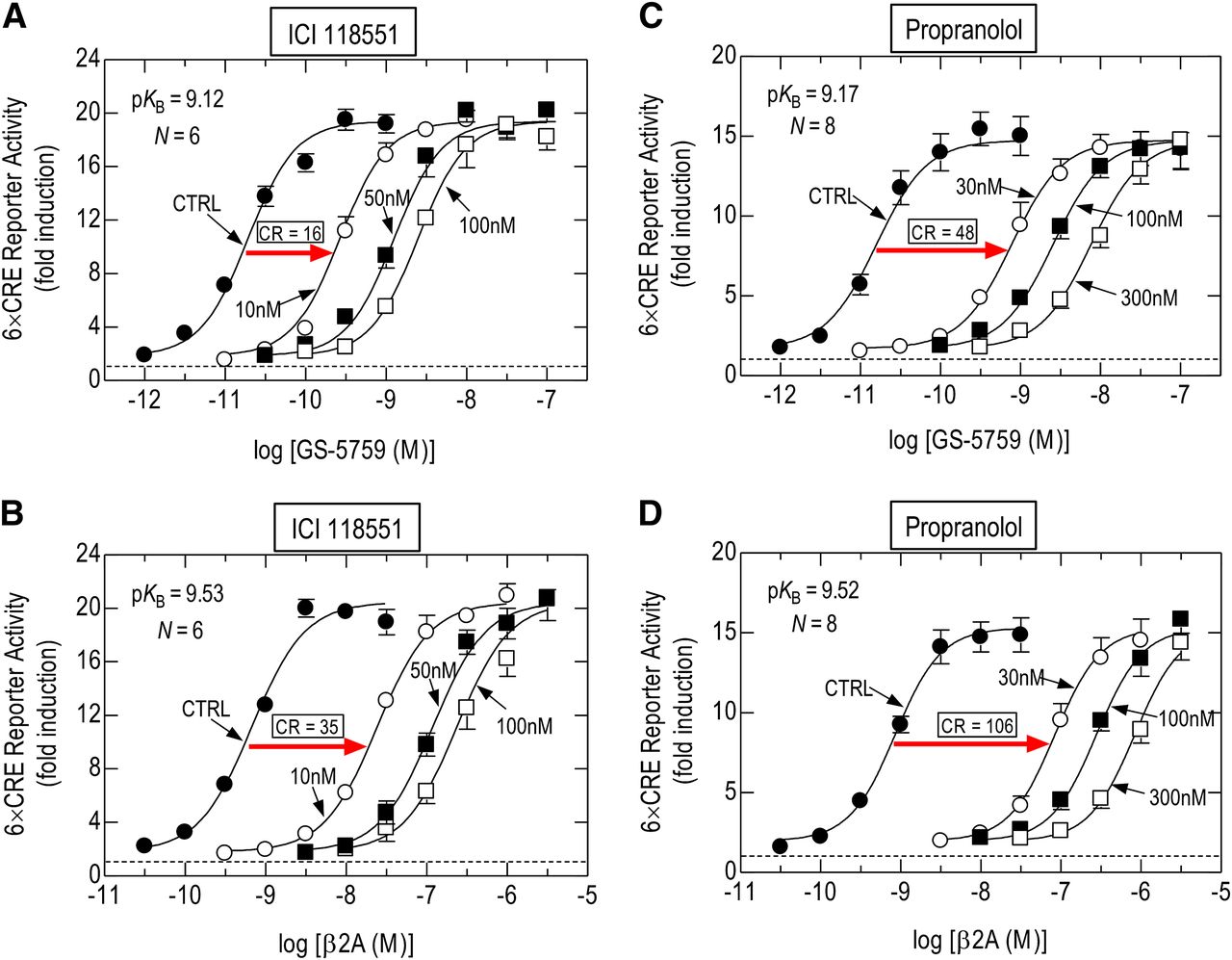
To define the pharmacology of indacaterol, β2A, and GS-5759, mRNA transcripts encoding β-adrenoceptor subtypes on BEAS-2B cells were quantified, and their functional significance was assessed. The relative mRNA abundance was ADRB2 (β2) > ADRB1 (β1) > >> ADRB3 (β3), with ADRB2 being expressed at a level that was 15.5- and >8800-fold higher than ADRB1 and ADRB3, respectively (Supplemental Fig. 1A). ADRB1 mRNA was present at a level comparable to transcripts that encode other functional, 7-transmembrane-spanning receptors (7-TMRs), such as the prostanoid EP2 receptor (Supplemental Fig. 1, A and B), suggesting that β1-adrenoceptors may be expressed on BEAS-2B cells and mediate CRE-dependent transcription. This was important to establish given indacaterol’s weak selectivity across β2-adrenoceptor subtypes (Battram et al., 2006), which could also apply to β2A and GS-5759 (Tannheimer et al., 2014). However, under conditions of PDE4 inhibition, pretreatment (30 minutes) of BEAS-2B cells with a selective β1-adrenoceptor antagonist, 2-hydroxy-5-[2-[[2-hydroxy-3-[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenoxy]propyl]amino]ethoxy]benzamide (CGP 20712A) (500 nM; pKI = 8.5–9.2) (Alexander et al., 2015), had no significant effect on either the shape or position of the E/[A] curves that described indacaterol-, β2A-, and GS-5759-induced, CRE-dependent reporter activation (Supplemental Fig. 1, B–D). In contrast, a selective β2-adrenoceptor antagonist, ICI 118551, produced concentration-related, parallel, dextral displacements of GS-5759 and β2A E/[A] curves under identical experimental conditions (Fig. 2, A and B). Enumeration of the Schild slope factor (S) indicated that this parameter did not deviate significantly from unity. Thus, ICI 118551 behaved as a surmountable, competitive antagonist. Accordingly, S was constrained to a value of 1 from which pKB values of 9.12 ± 0.04 and 9.53 ± 0.08 were derived with GS-5759 and β2A as agonists, respectively (Fig. 2, A and B). These values fall within the range expected for an interaction of ICI 118551 with the human β2-adrenoceptor (Alexander et al., 2015) and suggest that GS-5759 binds to the same site as β2A via its quinolinone-containing pharmacophore. Nevertheless, these pKB values were statistically different (by 2.6-fold), suggesting that ICI 118551 was less able to compete with GS-5759 than with β2A (Fig. 2, A and B). A similar discrepancy in affinity was obtained with propranolol, for which pKB values of 9.17 ± 0.05 and 9.52 ± 0.08 were derived with GS-5759 and β2A, respectively (Fig. 2, C and D).

Schild analysis of the antagonism of CRE reporter activation in BEAS-2B cells by ICI 118551 and propranolol. E/[A] Curves were constructed for GS-5759 and β2A in the absence and presence (60 minutes preincubation) of ICI 118551 (A and B) or propranolol (C and D) at the concentrations indicated. GSK 256066 (10 nM) was present throughout to eliminate any contribution that PDE4 inhibition could make to the position or shape of GS-5759 E/[A] curves. Each family of E/[A] curves was then fit simultaneously to eq. 4, from which pKB values were derived. Red arrows indicate concentration ratios (CRs) for each agonist at 10 nM ICI 118551 and 30 nM propranolol. The horizontal dashed line in each panel represents baseline luciferase activity. Data points represent the mean ± S.E. mean of N independent determinations. CTRL, Control.
Estimation of the Affinity of GS-5759 for PDE4.
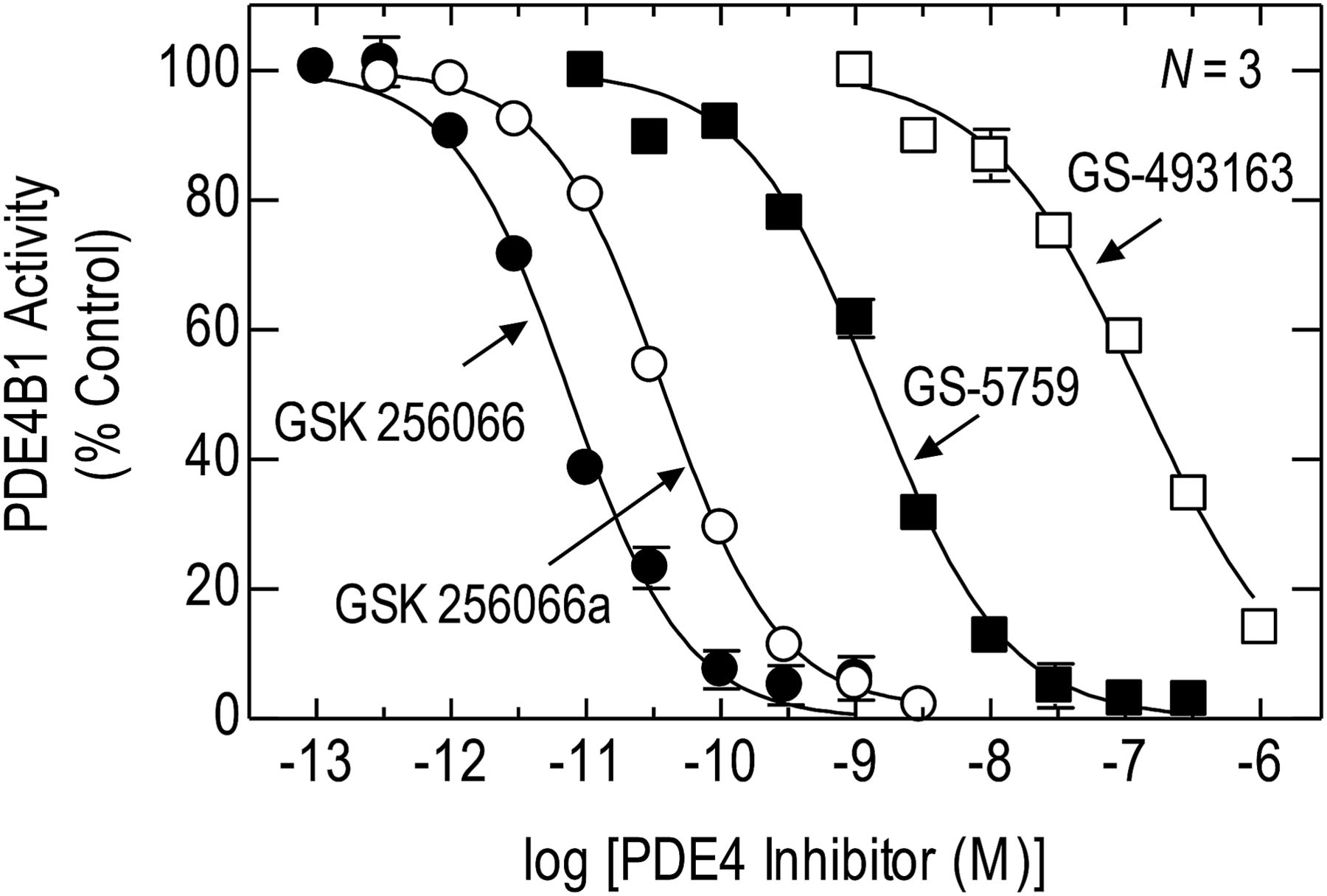
PDE4B is the most abundant PDE4 variant in BEAS-2B cells (BinMahfouz et al., 2015) and was used in the present study to compare the inhibitory activity of GS-5759 and comparator compounds. On human recombinant PDE4B1, GS-5759 inhibited cAMP hydrolysis in a concentration-dependent manner, with an IC50 value (1.38 nM) that was 224- and 36-fold less potent than GSK 256066 and GSK 256066a, respectively (Fig. 3; Table 1). Assuming competitive inhibition and that the Km of cAMP for PDE4B1 is 2 μM (Huston et al., 1997), the KI values for GS-5759, GSK 256066, and GSK 256066a were 1.15 nM, 2.63 pM, and 32.4 pM, respectively. Thus, the affinity of GS-5759 for PDE4B1 was 35-fold lower than the monofunctional parent compound, GSK 256066a, and 437-fold lower than GSK 256066 (Table 1). The 2-fold discrepancy between the IC50 and KI ratios of GSK 256066 (i.e., 224- vs. 437-fold) is explained by tight binding inhibition of PDE4B1 (Tralau-Stewart et al., 2011), resulting in an IC50 value that underestimates its potency (Materials and Methods; Table 1).
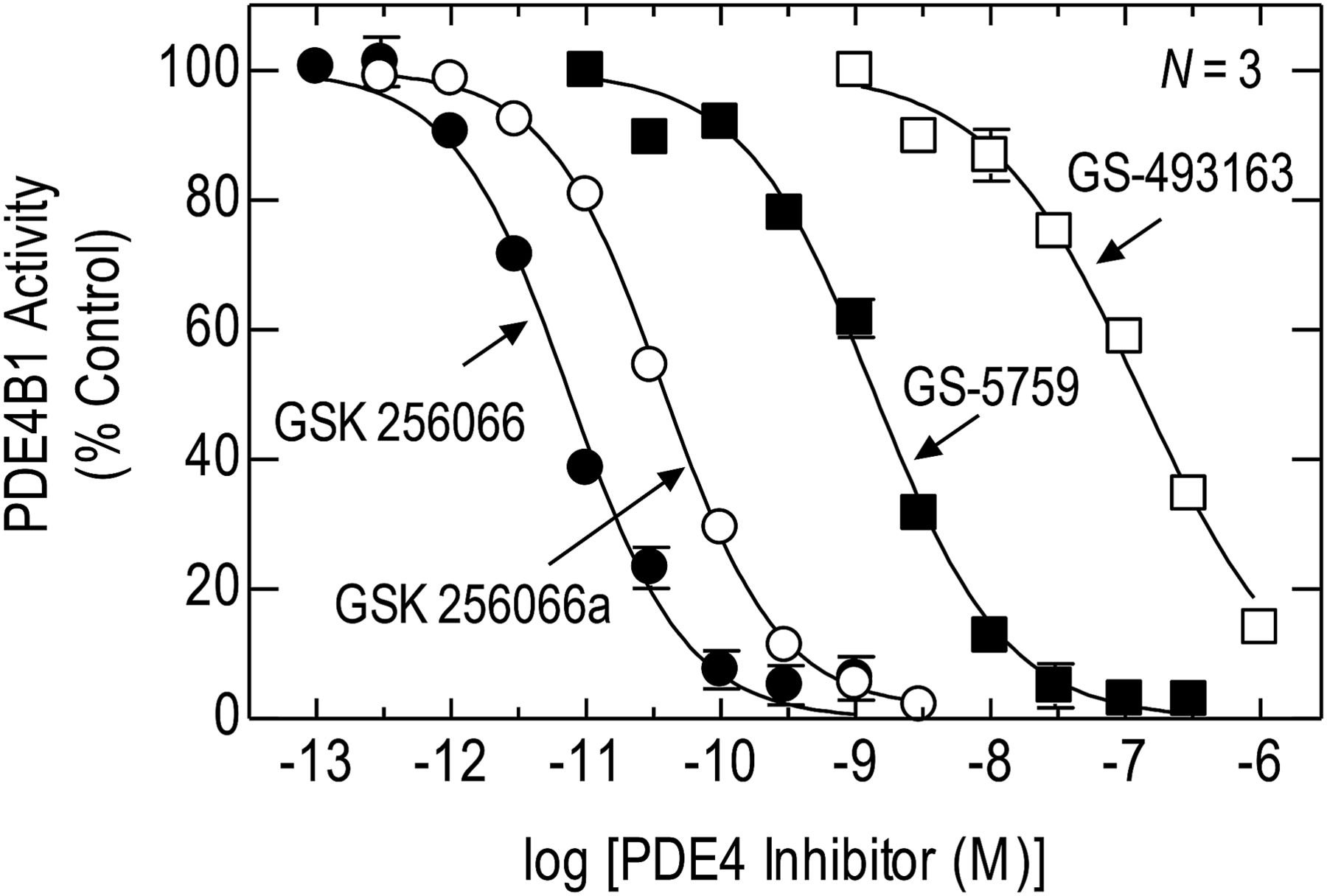
Effect of GSK 256066, GSK 256066a, GS-5759, and GS-493163 on PDE4B1 activity. IC50 values were determined at 60 minutes by nonlinear, iterative curve fitting and converted into KI values. See Materials and Methods and Table 1 for further details. Data represent the mean ± S.E. mean of N independent determinations.
Potency and equilibrium dissociation constants of test compounds at human recombinant PDE4B1 and the native β2-adrenoceptor in BEAS-2B cells
Effect of GS-5759 and Comparator Test Compounds on CRE-Dependent Reporter Activation.
On 6×CRE BEAS-2B reporter cells, GS-5759 and all comparator test compounds increased luciferase activity in a concentration-dependent manner, with a rank order of potency of GS-5759 > GSK 256066 > GSK 256066a > β2A-S > indacaterol > β2A (Fig. 4; Table 1). All ligands were equieffective activators of this construct, except GSK 256066 and GSK 256066a, which increased luciferase activity to a level that was only 4.0 and 5.1% of the maximal β2A-induced response, respectively (Fig. 4). There was no significant difference (P < 0.05) in the Hill coefficients between the monofunctional parent compound β2A (n = 2.04 ± 0.20), the parent compound attached to the spacer (β2A-S; n = 1.73 ± 0.15), and the bifunctional ligand GS-5759 (n = 1.91 ± 0.25). Further quantification of these data indicated that the addition of 4-(pent-1-yn-1-yl)aniline to β2A increased its potency by ∼8-fold, which was enhanced further (by ∼37-fold over the parent compound) following conjugation of the PDE4 inhibitor pharmacophore to form GS-5759 (Fig. 4, green and red arrows, respectively; Table 1).

Effect of GS-5759 and comparator test ligands on CRE reporter activation in BEAS-2B cells. Cells were treated with the indicated ligands for 6 hours, lysed, and E/[A] curves constructed from which p[A]50 and Emax values were derived (Table 1). Green and red arrows indicate the fold gain in potency of β2A afforded by adding the spacer to form β2A-S, and the spacer plus the PDE4 inhibitor to form GS-5759, respectively. The right-hand panel depicts the results obtained for GSK 256066 and GSK 256066a with an expanded y-axis. The vertical arrow in the right-hand panel and horizontal dashed lines represent maximum fold reporter activation and baseline luciferase activity, respectively. Data points represent the mean ± S.E. mean of N independent observations (see Table 2 for analyses of these data).
The higher potency of GS-5759 over β2A was unexpected. Although PDE4 inhibition could be a contributing factor, the magnitude of the difference and the higher potency of β2A-S over β2A suggested that other mechanisms were responsible. Indeed, β2A and β2A-S E/[A] curves constructed in the presence of a concentration of GSK 256066 (10 nM) that is predicted to abolish PDE4 activity in intact cells were displaced to the left by a factor of ∼2-fold relative to their respective control curves (Fig. 5, blue arrows). Thus, GS-5759 was still 20- and 2-fold more potent than β2A and β2A-S, respectively (Fig. 5, A and B, green arrows); this was despite its affinity for PDE4B1 being 35-fold lower than GSK 256066a (Table 1). Moreover, GSK 256066 (10 nM) produced a similar (2.5-fold) sinistral displacement of the mean GS-5759 E/[A] curve (Fig. 5, A and B, orange arrows; Table 2), suggesting that PDE4 inhibition did not contribute to reporter activation. To interrogate these findings further, the pharmacological behavior of GS-493163 was examined. This molecule was a relatively weak inhibitor of PDE4B1 with an affinity (KI = 132 nM) >100-, >4000-, and >5 × 105-fold lower than GS-5759, GSK 256066a, and GSK 256066, respectively (Fig. 3; Table 1). On 6×CRE BEAS-2B reporter cells, GS-493163 produced the same maximum response as GS-5759, although it was ∼6-fold less potent (Table 1). Moreover, GSK 256066 (10 nM) produced a 2-fold sinistral displacement of the mean GS-493163 E/[A] curve (Fig. 5C, orange arrow), suggesting again that PDE4 inhibition did not contribute to reporter activation ([A]50/KIPDE4 = 0.0018; Table 1). Nevertheless, GS-493163 was still ∼6- and ∼3-fold more potent than β2A alone and β2A in the presence of GSK 256066, respectively (10 nM; Fig. 5C, red and green arrows).

Effect of PDE4 inhibition on GS-5759-, β2A-, β2A-S-, and GS-493163–induced, CRE-dependent reporter activation in BEAS-2B cells. E/[A] curves were constructed for GS-5759 and β2A (A), GS-5759 and β2A-S (B), and GS-493163 and β2A (C) in the absence and presence of GSK 256066 (10 nM). The blue and orange arrows indicate the fold, sinistral displacements of agonist E/[A] curves produced by GSK 256066 as indicated. The red arrows in (A) and (C) depict the fold gain in potency of β2A imparted by linking the spacer plus PDE4 inhibitor pharmacophore to form GS-5759 and GS-493163, respectively. The red arrow in (B) shows the fold gain in potency of β2A-S by linking the PDE4 inhibitor to form GS-5759. The green arrows in each panel illustrate the difference in potency between the monofunctional compounds β2A and β2A-S determined in the presence of GSK 256066 (10 nM) and the bifunctional ligands GS-5759 and GS-493163. The horizontal dashed line in each panel represents baseline luciferase activity. Data points represent the mean ± S.E. mean of N independent observations (see Table 2 for analyses of these data).
Effect of the PDE4 inhibitor, GSK 256066, on the potency and maximum fold induction of test compounds on CRE-dependent reporter activation in BEAS-2B cells
Confluent cells were treated with test compounds for 6 hours in the absence and presence of GSK 256066 (GSK; 10 nM). Luciferase activity in cell lysates was then determined and expressed as a fold induction relative to time-matched, untreated cells. Data were calculated from the graphs in Fig. 5.
Estimation of the Affinity of GS-5759 for β2-Adrenoceptor–Mediated, CRE-Dependent Reporter Activation.
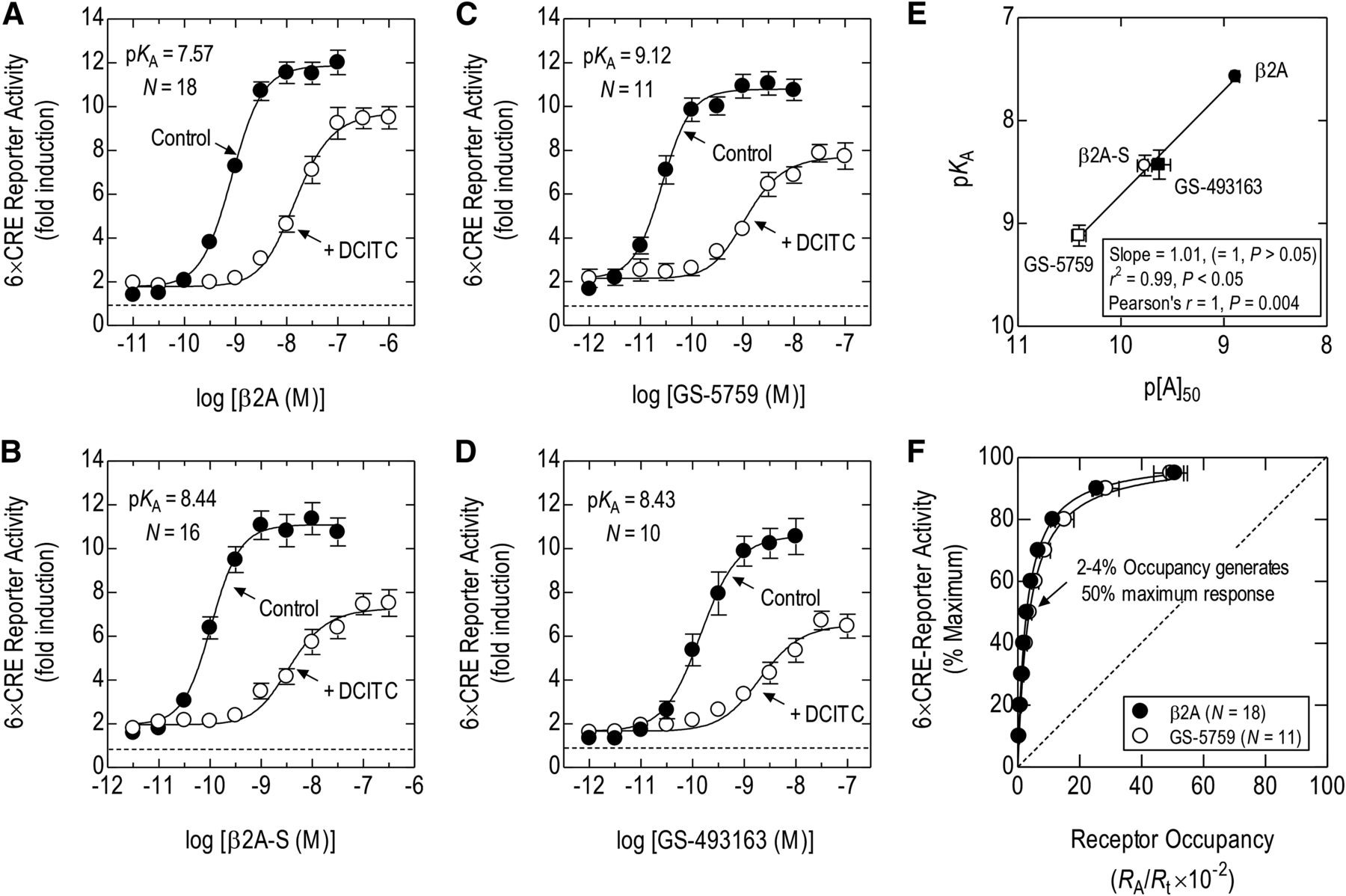
Operational model fitting of E/[A] curve data before and after treatment of cells with the alkylating agent DCITC was used to determine if the higher potency of GS-5759 relative to β2A was due to an increase in affinity for the β2-adrenoceptor. A saturating concentration of GSK 256066 (10 nM) was used in these experiments to eliminate any error that GS-5759 may introduce through its ability to inhibit PDE4. The addition of the spacer to β2A to form β2A-S was associated with a 7.5-fold decrease in KA (from 27 to 3.6 nM; Fig. 6, A and B; Table 3). Conjugating β2A-S to GSK 256066a to form GS-5759 increased affinity further over the β2A parent compound by a factor of 35.5-fold (KA = 0.76 nM; Fig. 6C; Table 3). The KA of GS-493163 (3.7 nM) for β2-adrenoceptor–mediated reporter activation was also higher than β2A (Fig. 6D). However, unlike GS-5759, the gain in affinity was relatively modest (7.3-fold) and of a magnitude similar to that effected by 4-(pent-1-yn-1-yl)aniline in β2A-S (Table 1). The relationship between potencies of β2A, β2A-S, GS-493163, and GS-5759 and their calculated affinities was linear with a rank-order correlation (Pearson’s r) and slope of unity (Fig. 6E).

Application of fractional, receptor inactivation to estimate pharmacodynamics parameters that define β2-adrenoceptor–mediated, CRE-dependent reporter activation in BEAS-2B cells by β2A, β2A-S, GS-5759, and GS-493163. Cells were treated with DCITC (100 nM) or vehicle for 60 minutes, washed in DCITC-free medium, and exposed to β2A (A), β2A-S (B), GS-5759 (C), or GS-493163 (D) at the concentrations indicated in the presence of GSK 256066 (10 nM). At 6 hours, cells were lysed, luciferase activity was determined, and E/[A] curves were constructed. The resulting pairs of curves were analyzed by operational model fitting from which estimates of KA, τ, n, Em, and p[A]50 (of the control curve) were derived (see Table 3). The horizontal dashed line in each panel represents baseline luciferase activity. (E) Linear and rank-order correlations between the pKA values of β2A, β2A-S, GS-5759, and GS-493163 determined from the experiments shown in (A–D) and the p[A]50 values of the same compounds (determined in the absence of GSK 256066) taken from Table 3. (F) The KA values of β2A and GS-5759 were used to calculate the relationship between fractional β2-adrenoceptor occupancy and CRE-dependent reporter activation. The diagonal dashed line is the line of identity where luciferase activity is a linear function of receptor occupancy. Data represent the mean ± S.E. mean of N independent determinations.
Pharmacodynamic parameters that define β2-adrenoceptor–mediated, CRE-dependent reporter activation in BEAS-2B cells by β2A, β2A-S, GS-5759, and GS-493163
Agonist E/[A] curves were constructed in cells treated with or without DCITC (100 nM for 60 minutes) and analyzed simultaneously by operational model fitting. p[A]50 and p[A′]50 refer to the concentration of compound that produced half maximum response in the absence and presence of DCITC, respectively. Parameters were derived from the data presented in Fig. 6, A–D.
Estimation of the Relative Efficacy of GS-5759, GS-493163, β2A, and β2A-S for β2-Adrenoceptor–Mediated, CRE-Dependent Reporter Activation.
Operational model fitting of the E/[A] curve data shown in Fig. 6, A–D was also used to determine if the spacer per se and/or the spacer linked to the pharmacophore mediating PDE4 inhibition affected the relative efficacy (τ) of β2A. As before, these experiments were necessarily performed in cells treated with GSK 256066 (10 nM) to ensure that any PDE4 inhibition produced by GS-5759 and GS-493163 did not complicate the estimation of relative efficacy values. As shown in Table 3, β2A, β2A-S, GS-5759, and GS-493163 were high-efficacy agonists with τ values of 27.5, 36.3, 27.5, and 25.1, respectively.
Using the KA values of GS-5759 and β2A estimated by fractional receptor inactivation (Table 3), β2-adrenoceptor occupancy expressed as a function of luciferase activity was described by a curvilinear relationship that deviated significantly from the line of identity (where response is a linear function of occupancy). It can be seen in Fig. 6F that the occupancy-response relationships of GS-5759 and β2A were almost superimposable, and that half maximal response required only 2–4% β2-adrenoceptor occupancy. Thus, for CRE-dependent transcription, a large β2-adrenoceptor reserve existed for these two agonists, which is consistent with their high operational efficacy values and KA/[A]50 ratios shown in Table 3. These data are significant because, although both pharmacophores of GS-5759 bound their cognate molecular targets over a similar concentration range (KAβ2/KIPDE4 = 0.66; Table 1), the β2-adrenoceptor reserve on 6×CRE BEAS-2B reporter cells rendered GS-5759 active at concentrations where PDE4 is unlikely to be inhibited ([A]50/KIPDE4 = 0.034; Table 1).
Effect of GS-5759 on Global Gene Expression.
To identify potential cAMP-induced genes (including RNA-coding genes) that may mediate the pharmacological actions of GS-5759, microarray-based gene expression profiling was performed in BEAS-2B cells. GS-5759 was used at a concentration (10 nM) 10-fold higher than its affinity for PDE4B1 and the β2-adrenoceptor (Table 1), which results in a robust activation of several putative, anti-inflammatory responses (Salmon et al., 2014; Tannheimer et al., 2014). Currently, it is unknown whether immediate and/or delayed targets of cAMP signaling contribute to the mechanism of action of GS-5759. Therefore, differentially expressed genes (DEGs) were identified at 1, 2, 6, and 18 hours after exposure to GS-5759. Of the 54,612 probe sets on each gene chip, 3039–5350 detected expression level changes that were statistically different from time-matched, vehicle-treated cells that depended on the time of exposure to GS-5759 and the number of probe sets per gene. Based on DAVID identifiers, this equates minimally to between 2091 and 3542 DEGs. The kinetics of global gene expression changes are shown in Fig. 7A at two arbitrarily set thresholds (>2-fold and >4-fold). In BEAS-2B cells exposed to GS-5759 for 1 hour, 188 DEGs were identified that were either upregulated (117) or downregulated (71) by a factor of >2-fold (Fig. 7A). After 2 hours of exposure, the number of DEGs with >2-fold expression changes increased to 210 (152 induced and 58 repressed), which declined to a value of 172 (134 induced and 38 repressed) by 6 hours (Fig. 7A). At the 18-hour time point, the number of genes statistically upregulated by GS-5759 waned further to 75, whereas the number of repressed genes increased to 67 (Fig. 7A). Similar kinetics were found if no fold threshold was set, although the number of DEGs was increased by ∼20-fold (data not shown). Figure 7B shows the expression level of all 54,612 probe sets at the 2-hour time point presented as a volcano plot, where red and green circles indicate >2-fold induction and repression, respectively. Volcano plots of data at 1, 6, and 18 hours are presented in Supplemental Fig. 2.

Changes in gene expression in BEAS-2B cells induced by GS-5759 and Ind/GSK. Total RNA was extracted from cells treated for 1, 2, 6, or 18 hours with GS-5759 (10 nM) (A), Ind/GSK (both 10 nM) (C), or vehicle and processed for gene expression changes by microarray. DEGs that were statistically different from vehicle-treated cells are shown at each time point at two arbitrarily set thresholds (>2-fold and >4-fold as indicated). The number of DEGs is indicated in parentheses next to each data point. (B) A volcano plot of all 47,400+ transcripts (by probe set) after treatment of cells with GS-5759 for 2 hours relative to time-matched, vehicle-treated cells (NS). Each probe set is represented by a circle colored gray (transcript changes ≤2-fold), red, or green (corresponding to transcripts that are induced and repressed by >2-fold, respectively). Yellow circles show GS-5759–induced genes with putative, therapeutic activities that were validated by RT-PCR (Fig. 9; Table 5). Values above the horizontal dotted line represent gene expression changes that were significantly different from vehicle [unadjusted P value <0.05, one-way analysis of variance (ANOVA)]. The vertical dotted line indicates baseline gene expression. (D) Significant (unadjusted P value <0.05) gene expression changes in BEAS-2B cells produced by the two treatments (9387 transcripts by probe set) subjected to rank-order and linear correlations. The values in the red and green quadrants correspond to the number of transcripts induced and repressed by the two treatments, respectively. The numbers in the gray quadrants indicate genes that were induced by GS-5759 but repressed by Ind/GSK, or vice versa. The solid and dashed diagonal lines represent linear regression and the line of identity, respectively. NS, not stimulated.
A primary objective of this study was to determine if the gene expression changes produced by GS-5759 replicated the effect of a β2-adrenoceptor agonist and a PDE4 inhibitor in combination. To that end, microarray-based gene expression profiling was determined in BEAS-2B cells exposed to Ind/GSK at concentrations (both 10 nM) that saturate their respective cognate targets. This was performed in parallel with the GS-5759 study to minimize differences in gene expression changes resulting from cell plasticity. Indacaterol and GSK 256066 (rather than β2A and GSK 256066a) were used for this analysis to provide information on a clinically relevant LABA and PDE4 inhibitor. As shown in Fig. 7C, the kinetics of statistically different gene expression changes in BEAS-2B cells produced by Ind/GSK were similar to those produced by GS-5759.
Relationship between GS-5759- and Ind/GSK-Induced Gene Expression Changes.
There was a significant linear and rank-order correlation between DEGs (by probe set) produced by GS-5759 and Ind/GSK at all time points (Fig. 7D; Supplemental Fig. 3). At 2 hours, 9387 probe sets indicated either induction (3056; 32.6%, red circles) or repression (5946; 63.3%, green circles) of gene expression produced by both treatments (Fig. 7D). The remainder (385; 4.1%, gray circles) were induced by GS-5759 and repressed by Ind/GSK, or vice versa. Similar data were obtained at the other time points (Supplemental Fig. 3). It is noteworthy that, although the lines of regression began close to the origin, they deviated significantly from unity in a time-dependent manner in favor of GS-5759 with slopes of 0.89, 0.88, 0.84, and 0.76 at 1, 2, 6, and 18 hours, respectively (Fig. 7D; Supplemental Fig. 3).
Ontology Analysis of GS-5759–Induced Genes.
The large number of gene expression changes effected by GS-5759 presumably reflects the fact that cAMP is a ubiquitous second messenger that regulates diverse physiologic processes. To gain information on genes that could be beneficial in COPD, GO analysis and functional annotation clustering were performed using DAVID at high stringency. Analyses of DEGs that were upregulated by >2-fold at each time point (Fig. 7A) identified 3, 13, 4, and 1 significant cluster(s) of functionally similar GO terms at 1, 2, 6, and 18 hours, respectively, at an enrichment score cutoff ≥1.2 (Huang et al., 2009). These functional clusters and the genes (with Affymetrix identifiers) used to generate them are provided in Supplemental Tables 2 and 3, respectively. Of relevance to respiratory therapeutics was that many functional clusters of GO terms relate to cytokines, inflammation and the immune system, apoptosis, gene transcription, T-lymphocyte/leukocyte function, and cell motility, although the degree of enrichment was dependent upon the time of exposure. An example of a functional cluster of overlapping GO terms with relevance to COPD relates to apoptosis and includes BAG1, BCL2L11, CASP4, CASP8, BCL6, and BCL7 (Table 4).
Gene ontology terms that relate to apoptosis are over-represented in all mRNA transcripts that are significantly upregulated by >2-fold in response to GS-5759 after 2-hour exposure
A large number of genes were repressed by GS-5759 and by Ind/GSK in BEAS-2B cells (Fig. 7; Supplemental Fig. 2). These are not discussed here but will form the subject of a separate report.
Validation of Microarray Data.
Figure 8 compares, as heat maps, the expression pattern of the top 75 GS-5759–induced genes by probe sets ranked highest to lowest at 1, 2, 6, and 18 hours. Nine induced genes (NR4A2, CDKN1C, RGS2, CRISPLD2, SOCS3, DUSP1, CD200, C5AR1, and FGFR2) identified on the array with potential relevance to COPD therapy (Table 5; Giembycz and Newton, 2014) were successfully validated by RT-PCR (Fig. 9). These genes are underlined in the heat maps shown in Fig. 8, and their expression level changes are given in each cell in log2 format. They are also shown as yellow circles on each volcano plot (Fig. 7B; Supplemental Fig. 2). Significant linear and rank-order correlations between the microarray and RT-PCR data were found after 2 hours of treatment using the probe set producing the greatest induction per gene (Supplemental Fig. 4, A and B). Similar results were obtained from cells treated with Ind/GSK (Supplemental Fig. 4, C and D). The same nine genes were also induced by indacaterol (10 nM) and GSK 256066 (10 nM; Fig. 9), suggesting that both pharmacophores in GS-5759 have the potential to regulate the same population of genes.

Heats maps illustrating GS-5759–induced gene induction determined by microarray in BEAS-2B cells. Total RNA was extracted from cells treated with GS-5759 (10 nM) or vehicle for 1, 2, 6, and 18 hours and processed for gene profiling by microarray. The top 75 induced genes by probe set that were statistically different from time-matched, vehicle-treated cells at each time point (unadjusted P value <0.05) were ranked highest to lowest and are presented as heat maps relative to changes of the same transcripts at the other three time points. The color scale is logarithmic (base 2) with the most intense red and green colors representing a 128-fold increase (log2 = 7) and an 8-fold decrease (log2 = 3) in gene expression, respectively. The right-hand side and left-hand side of every row in each heat map shows the gene name and corresponding Affymetrix identification number (Affy ID). Genes validated by RT-PCR are underlined and bolded in black, and their expression level changes are shown in each cell in log2 format.
Sensitivity of cAMP-inducible genes to GS-5759 in BEAS-2B cells and of the 6×CRE reporter

Validation of gene expression changes by RT-PCR. The expression of nine GS-5759- and Ind/GSK-induced genes was selected from the functional clusters of overlapping GO terms that relate broadly to inflammation and validated by RT-PCR (A–I). The left-hand side of each panel compares the kinetics of gene expression induced by GS-5759 and Ind/GSK determined with the cDNA used for the microarray. The middle panels show the effect of indacaterol (Ind, 10 nM) and GSK 256066 (GSK, 10 nM) on gene expression changes at the times indicated, and the right-hand panels show E/[A] curves for GS-5759–induced gene induction at 2 hours. These experiments used different mRNA. The dashed horizontal lines indicate baseline gene expression. Data represent the mean ± S.E. mean of N independent determinations. NS, Not stimulated.
GS-5759- and Ind/GSK-induced gene expression changes followed similar kinetics (Fig. 9). Indeed, a significant linear and rank-order correlation was found between these two treatments at 2 hours (Fig. 10), which is consistent with the global gene expression data shown in Fig. 7D. However, the potency of GS-5759 measured at 2 hours varied in a gene-dependent manner, with NR4A2 ([A]50 = 1.9 nM) and FGFR2 ([A]50 = 25 pM) differing in sensitivity by a factor of 75-fold (Fig. 9; Table 5). With the exception of FGFR2, these genes were less sensitive to GS-5759 than was the 6×CRE reporter (Table 5).
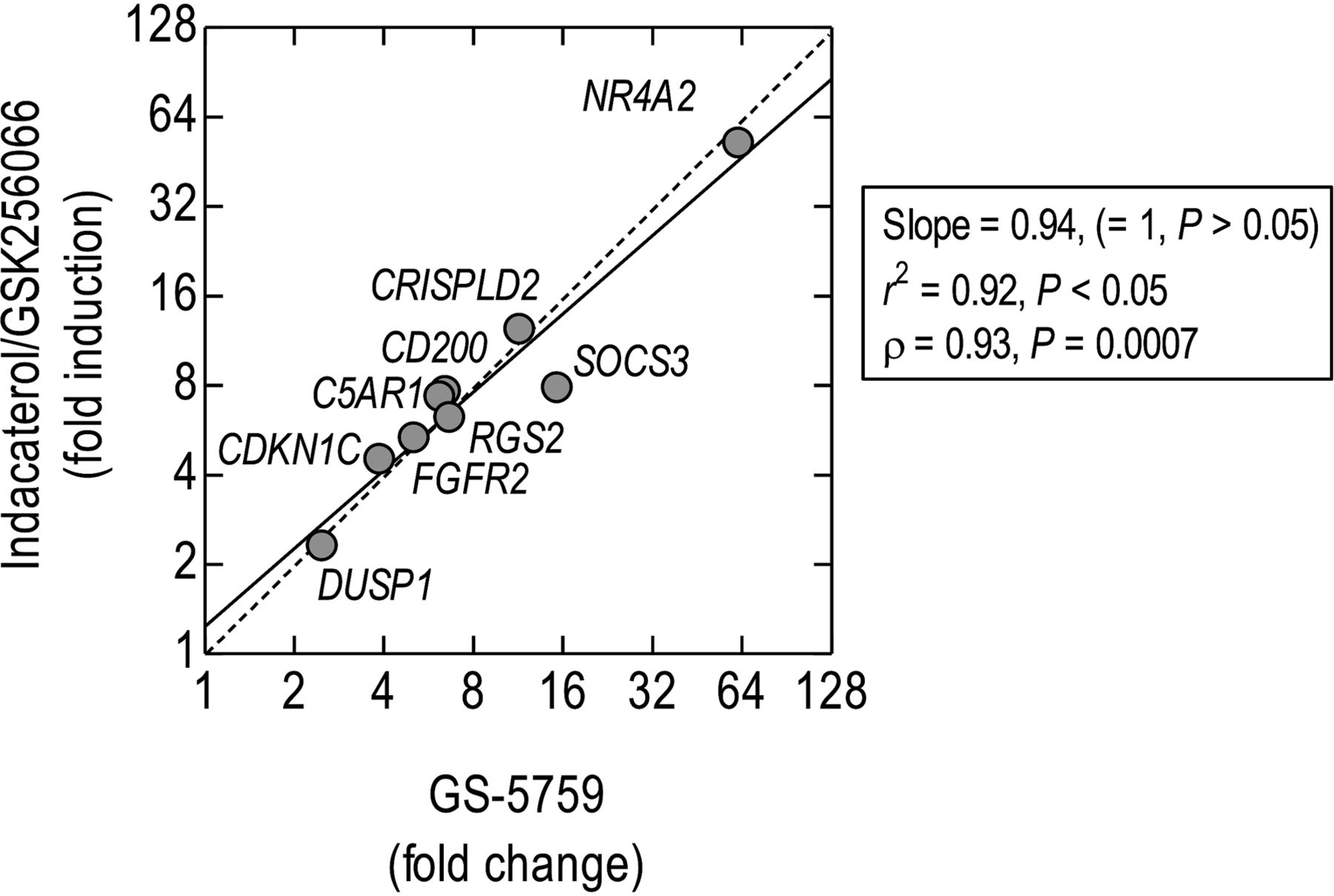
Relationship between gene expression changes induced in BEAS-2B cells by GS-5759 and Ind/GSK. Total RNA was extracted from cells treated for 2h with GS-5759 (10nM), or Ind/GSK (both 10nM) and processed for gene expression changes by RT-PCR relative to time-matched, vehicle-treated cells. The solid and dashed diagonal lines represent linear regression and the line of identity respectively.
Discussion
Herein, we describe the pharmacodynamics of a novel, bifunctional ligand, GS-5759, in the BEAS-2B human airway epithelial cell line using changes in gene expression as a therapeutically relevant output. GS-5759 was selected as a representative compound exhibiting β2-adrenoceptor agonism and PDE4 inhibition, where both functional groups have the potential to interact simultaneously and, potentially, synergize on target tissues by increasing cAMP synthesis and retarding cAMP degradation, respectively. These mutually cooperative properties are relevant in COPD, where maintaining airway caliber and suppressing inflammation are primary therapeutic objectives.
Bifunctional ligands for COPD that interact with two different 7-TMRs have been reported previously (Hughes et al., 2011; Hughes and Jones, 2011; Norman, 2013), with batefenterol, a muscarinic receptor antagonist and β2-adrenoceptor agonist (MABA; Hughes et al., 2015), being the most advanced development candidate having successfully completed several phase II trials (Bateman et al., 2013; Wielders et al., 2013). However, the pharmacology of ligands such as GS-5759, which targets a 7-TMR and an intracellular enzyme (Salmon et al., 2014; Tannheimer et al., 2014), is not well defined and formed the basis of the current investigation.
The dual pharmacology of GS-5759 is attributable to two structural elements linked covalently by a pent-1-yn-1-ylbenzene spacer: 1) a quinolinone-based orthostere, which is a well recognized pharmacophore conferring β2-adrenoceptor agonism (Yoshizaki et al., 1976; Milecki et al., 1987) and is found in batefenterol (Hughes et al., 2015) and related MABAs, including biphenyl-2-yl-carbamic acid 1-{9-[(R)-2-hydroxy-2-(8-hydroxy-2-oxo-1,2-dihydro-quinolin-5-yl)-ethylamino]-nonyl}-piperidin-4-yl ester (THRX 198321) (Steinfeld et al., 2011) and biphenyl-2-ylcarbamic acid 1-{3-[4-(4-{2-[(R)-2-hydroxy-2-(8-hydroxy-2-oxo-1,2-dihydroquinolin-5-yl)ethylamino]ethyl}phenoxy)phenyl]propyl piperidin-4-yl ester (THRX 200495) (McNamara et al., 2012); and 2) a quinoline 3-carboxamide present in GSK 256066a (Ochiana et al., 2015), which has low picomolar affinity for PDE4 (Table 1).
GS-5759 as a PDE4 Inhibitor.
Covalently linking GSK 256066a to β2A by a spacer to form GS-5759 was associated with a 35-fold loss in affinity for PDE4B1. Effects of this magnitude are commonly produced by attaching a spacer to a pharmacophore (Shonberg et al., 2011), indicating that the original, unconjugated parent compound often needs to exhibit high affinity for its cognate target to be suitable for incorporation into a bifunctional ligand. Nevertheless, GS-5759 still displayed an IC50 value against the isolated enzyme that was similar to roflumilast (Hatzelmann et al., 2010), which is the only PDE4 inhibitor currently approved for treatment of COPD (Giembycz and Field, 2010). Moreover, the KI of GS-5759 for PDE4B1 was comparable to its KA at the β2-adrenoceptor, indicating that both pharmacophores have potential to synergize in target tissues and contribute to the overall response of interest (vide infra).
Nature of the Binding of GS-5759 to the β2-Adrenoceptor.
On 6×CRE BEAS-2B reporter cells under conditions of PDE4 inhibition, GS-5759 and β2A interacted with ICI 118551 and propranolol in a surmountable, competitive manner, indicating that the quinolinone-containing moiety binds to the orthosteric site of the β2-adrenoceptor. However, GS-5759 was more potent than β2A, which was not due to concurrent PDE4 inhibition (discussed later). Similar, albeit more modest, effects were obtained with β2A-S and GS-493163, a structural analog of GS-5759. Three explanations could account for the higher potency of GS-5759 (and related molecules): GS-5759 has higher intrinsic efficacy, GS-5759 has higher affinity, or GS-5759 has higher intrinsic efficacy and higher affinity. To address these possibilities, β2A, β2A-S, GS-5759, and GS-493163 E/[A] curve data were analyzed by operational model fitting before and after fractional, irreversible β2-adrenoceptor inactivation with DCITC. To ensure that operational parameter estimates could be compared, cells were treated with GSK 256066 (10 nM) to eliminate any contribution that PDE4 inhibition could make to the position or shape of GS-5759 E/[A] curves. Although this intervention will increase the operational efficacy of test compounds, it does not invalidate the assessment of relative activities. Using this approach, the four compounds had similar τ values, KA/[A]50 ratios, and, therefore, occupancy-response relationships. In contrast, GS-5759, GS-493163, and β2A-S bound to the β2-adrenoceptor with 35-, 7-, and 7-fold higher affinity, respectively, than did β2A (Table 3). A plot of p[A]50 versus pKA of the four ligands was linear, with a rank-order correlation and slope of unity suggesting that the higher potencies of GS-5759, GS-493163, and β2A-S were due to an increase in affinity.
The mechanism underlying the enhanced strength of binding merits discussion. It is possible that these bifunctional ligands and the β2A-S fragment interact with the orthosteric site of the β2-adrenoceptor via their quinolinone-containing moiety and simply have higher affinities than β2A. However, recent evidence suggests this interpretation may be incorrect. Studies examining the dual pharmacology of the MABA, THRX 198321, led Steinfeld et al. (2011) to propose that the muscarinic receptor antagonist (MA) pharmacophore binds an allosteric site within the β2-adrenoceptor, resulting in an increase in affinity of the quinolinone-containing orthostere. Although allosterism could account for the THRX 198321 data and the decrease in the KA values of GS-5759 and related molecules described here, it is not a necessary requirement. Indeed, our results can be more simply explained by “forced proximity” binding (Vauquelin and Charlton, 2013). Theoretically, both pharmacophores of a bifunctional ligand can form nonallosteric interactions with a given molecular target (so-called bivalent binding; Hughes et al., 2012) that synergize, resulting in an increase in affinity over the monofunctional and monovalent parent compounds (Valant et al., 2012; Vauquelin and Charlton, 2013). Thermodynamically, this gain in affinity is explained by a reduction in the total energy necessary for the second pharmacophore to bind (Mammen et al., 1998), which is less than the overall energy required for the binding of each monofunctional parent compound. Thus, the interaction of the first pharmacophore “forces” its tethered companion to adopt a position in close proximity to its binding site (Vauquelin and Charlton, 2013). Consequently, the local concentration of the dissociated pharmacophore is elevated for as long as its covalently linked partner remains bound, thereby increasing the probability of it reassociating with its cognate target (Mammen et al., 1998; Valant et al., 2012).
Evidence that GS-5759 bound to the β2-adrenoceptor in a bivalent manner was the agonist-dependent discrepancy in pKB values of ICI 118551 (9.53 and 9.12 with β2A and GS-5759, respectively). Although the difference was modest (KBβ2A/KBGS-5759 = ∼2.6), it was statistically significant and obvious from inspection of the E/[A] curves shown in Fig. 2 (red arrows). Moreover, these results were replicated with propranolol, which would be predicted for an antagonist-independent phenomenon. According to the “forced proximity” model (Vauquelin and Charlton, 2013), ICI 118551 and propranolol compete with the quinolinone-containing moiety of GS-5759 at the orthosteric site of the receptor in the same way they compete with the monofunctional parent compound, β2A. However, because the quinolinone-containing orthostere is covalently linked to its companion PDE4 inhibitor, it remains in close apposition to its target site for longer and achieves a higher local concentration than does β2A, which escapes into the bulk of the solution relatively unhindered. Thus, although ICI 118551 and propranolol behave as competitive antagonists, a given concentration will be less able to prevent the rebinding of the tethered, quinolinone-containing orthostere of GS-5759 compared to β2A (Vauquelin and Charlton, 2013). “Forced proximity” also predicts an increase in target residence time of a bivalent ligand (Vauquelin and Charlton, 2013), which could explain the long duration of action of GS-5759 at β2-adrenoceptors in guinea pig trachea (Tannheimer et al., 2014).
Balancing the Activity of Both Pharmacophores.
A necessary requirement in the development of a bifunctional ligand is that both pharmacophores bind their cognate targets over a similar concentration range. This is important for GS-5759 because the two pharmacophores are designed to interact, and potentially synergize, in the same tissues by increasing cAMP synthesis and inhibiting cAMP degradation. GS-5759 is a balanced molecule in the sense that its affinity for the β2-adrenoceptor and PDE4 are similar (KAβ2/KIPDE4 = 0.66). However, the potency of GS-5759 will vary in a tissue- and function-dependent manner because of differences in β2-adrenoceptor density and efficiency of coupling to adenylyl cyclase. Consequently, establishing the ideal activity ratio of the two pharmacophores is a development challenge. The finding that GS-5759 was 29-fold more potent in activating the luciferase reporter than at inhibiting PDE4B1 illustrates this point. Our results indicate that PDE4 activity was unaffected over the active concentration range of the compound given that GSK 256066 produced ∼2-fold, sinistral displacements of GS-5759 and β2A E/[A] curves. The high potency of GS-5759 on BEAS-2B reporter cells may reflect the artificial nature of the plasmid, which contains six CREs derived from the regulatory sequences of four different genes (Himmler et al., 1993). Indeed, many genes shown in Table 5 were less sensitive to GS-5759, implying that PDE4 inhibition may play a more important role in their expression. Thus, the importance of identifying compounds that maximize coincident PDE4 inhibition and β2-adrenoceptor agonism in a given target cell or tissue on the relevant functions of interest is clear. Ensuring an appropriately balanced pharmacology of the two pharmacophores is essential given that the clinical dose of a bifunctional β2-adrenoceptor agonist and PDE4 inhibitor will likely be determined by its bronchodilator activity (Wang et al., 2012).
Role of the Spacer.
The KA of the β2A-S fragment for the β2-adrenoceptor was 7-fold higher than the β2A parent compound, implying that the spacer may have contributed to the gain in affinity of GS-5759. Similar effects at muscarinic receptors have been reported for MA orthosteres containing polymethylene chains [denoted as MA linked to nonane (MA-L) by Steinfeld et al. (2011)]. Nevertheless, in those studies, the contribution of the spacer to the gain in affinity was questioned. Steinfeld et al. (2011) argued that the hydrophobicity of the nonamethylene moiety in the MA-L fragment of the MABA, THRX 198321, may facilitate its binding to a microdomain within the receptor that would not occur with the intact molecule because of the polar nature of its tethered, companion β2A orthostere. Our calculated estimates of distribution coefficients (MedChem Designer; Simulations Plus Inc., Lancaster, CA) predict a 7800-fold increase in the lipophilicity of MA-L over the unconjugated MA orthostere and a reduction in total polar surface area (tPSA), which are consistent with this suggestion (Supplemental Fig. 5). However, the contribution of the spacer to the overall gain in affinity of GS-5759 is less clear given that the β2A-S fragment contains the amine functionality (absent in MA-L) used to link GSK 256066a (Supplemental Fig. 5). Accordingly, the tPSA is higher in β2A-S than in β2A, and the increment in lipophilicity is relatively modest (149-fold), resulting in a molecule that is 7.35 × 105 times more polar than MA-L. Moreover, the affinity of GS-5759 was reduced to the level of the β2A-S fragment upon methylsulphonylation of the 4-amine of the quinoline to form GS-493163.
It is noteworthy that the higher affinity of β2A-S over β2A was not found by Steinfeld et al. (2011) for the same β2A orthostere with a nonamethylene chain (denoted as BA-L [β2-agonist linked to nonane] in that study). Notwithstanding methodological differences between agonist affinity determinations, this discrepancy may be explained by physicochemical properties of the two spacers. Thus, in addition to differences in tPSA and logD values, nonane and 4-(pent-1-yn-1-yl)aniline have six and two rotatable bonds, respectively, which could differentially affect the molecular flexibility and receptor binding characteristics of the β2A-S and BA-L fragments (Supplemental Fig. 5).
Gene Expression Profiling.
An important property of a bifunctional ligand is that the therapeutic activity of each pharmacophore in combination is retained. This is particularly important if the bifunctional ligand displays a unique mechanism of action as we propose here. To establish if this was the case, DEGs resulting from treatment of BEAS-2B cells with GS-5759 and Ind/GSK were compared. At each time point, there was a highly significant linear and rank-order correlation between the two treatments that began close to the origin. This finding was confirmed by RT-PCR validation of nine genes selected from the array, indicating that the magnitude of most gene expression changes produced by Ind/GSK was replicated, quantitatively, by GS-5759. However, for reasons that are currently unclear, the lines of regression deviated slightly from unity slopes favoring gene expression changes produced by GS-5759.
We reported previously that LABAs and PDE4 inhibitors induce a variety of genes in BEAS-2B cells (Kaur et al., 2008a; Moodley et al., 2013; BinMahfouz et al., 2015). This was confirmed again here with indacaterol and GSK 256066, implying that both pharmacophores of GS-5759 can behave similarly and potentially interact in a synergistic manner. However, as discussed earlier, the contribution of PDE4 inhibition to the mechanism of action of GS-5759 depends on the sensitivity of responses of interest to β2-adrenoceptor agonism. This will apply within and across tissue types and will have to be determined empirically.
Some probe sets designed to detect the same gene did not give a positive signal. For example, three of 11 probe sets that reportedly detect FGFR2 mRNA transcripts were inactive, and an additional four probe sets suggested gene repression (Supplemental Fig. 6). Although these findings may be genuine, perhaps due to alternative mRNA splicing, they expose a problem. Many transcripts (e.g., RGS2, C5AR1) are detected on Affymetrix GeneChips by a single probe set, indicating that the size of the cAMP-regulated transcriptome is probably underestimated.
On gene expression, the potency of GS-5759 varied by a factor of 75-fold, with FGFR2 and NR4A2 being the most and least sensitive, respectively. Similar results have been reported for glucocorticoid-inducible genes (Reddy et al., 2009; Uings et al., 2013; Joshi et al., 2015a), indicating that a given transcription factor (e.g., CRE-binding protein in this study) does not activate the promoter of cognate target genes equally. This may be explained, minimally, by differences in the number and sequence of the response element(s) to which the transcription factor can bind and the three-dimensional architecture of the promoters.
Conclusions.
GS-5759 is a novel, bifunctional β2-adrenoceptor agonist and PDE4 inhibitor with a unique mechanism of action. Evidence is presented that GS-5759 interacts with the native β2-adrenoceptor on BEAS-2B cells in a nonallosteric, bivalent manner. Our data suggest that the pharmacophore that mediates PDE4 inhibition may also bind a concavity within the receptor that increases the affinity, local concentration, and residence time of its companion β2-adrenoceptor agonist for the orthosteric site that is explained by a “forced proximity” interaction (Vauquelin and Charlton, 2013). Consequently, the duration of action of GS-5759 is prolonged.
Therapeutically, the polypharmacology of GS-5759 may be clinically superior to either of the component pharmacophores alone through its ability to increase airway caliber and suppress inflammation (Salmon et al., 2014). Synergy between the two pharmacophores at target cells and tissues with an attendant improvement in clinical outcomes is also predicted. As discussed earlier, this interaction should predominate in cells where β2-adrenoceptor density is limiting and/or coupling efficiency to adenylyl cyclase is weak (Giembycz and Newton, 2014). If clinical superiority of bifunctional ligands, such as GS-5759, is established over LABA monotherapy, they could become a logical, first-line treatment option for patients with COPD.
Acknowledgments
The authors acknowledge Sylvia Hill for generating the samples used for gene expression profiling.
Authorship Contributions
Participated in research design: Giembycz, Hamed, Joshi, Newton, Phillips, Salmon, Tannheimer, Wright, Yan.
Conducted experiments: Hamed, Joshi, Newton, Yan.
Contributed new reagents or analytic tools: Kim, Phillips.
Performed data analysis: Giembycz, Hamed, Joshi, Newton, Yan.
Wrote or contributed to the writing of the manuscript: Giembycz, Hamed, Joshi, Newton, Phillips, Salmon, Tannheimer, Wright, Yan.
Footnotes
- Received September 19, 2016.
- Accepted December 5, 2016.
↵1 Current affiliation: University of Pittsburgh, Center for Vaccine Research, Pittsburgh, Pennsylvania.
↵2 Current affiliation: RespirPharm Solutions, Sammamish, Washington.
↵3 Current affiliation: Merck Research Laboratories, Boston, Massachusetts.
↵4 T.J., D.Y., and O.H. contributed equally to this work.
This study was supported by the Canadian Institutes for Health Research [Grant MOP 93742], the Alberta Lung Association, and an unrestricted educational research grant from Gilead Sciences Inc., Seattle, WA. M.A.G. holds a Tier 1 Canada Research Chair in Pulmonary Pharmacology. R.N. is an Alberta Innovates–Health Solutions (AI-HS) Senior Scholar. T.J. and D.Y. hold Alberta Lung Association and AI-HS studentship awards, respectively.
The authors state no conflict of interest.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- β2A
- β2-adrenoceptor agonist orthostere
- β2A-S
- β2A linked to 4-(pent-1-yn-1-yl)aniline
- C5AR1
- complement component 5a receptor 1
- CGP 20712A
- 2-hydroxy-5-[2-[[2-hydroxy-3-[4-[1-methyl-4-(trifluoromethyl)imidazol-2-yl]phenoxy]propyl]amino]ethoxy]benzamide
- COPD
- chronic obstructive pulmonary disease
- CRE
- cAMP-response element
- DAVID
- Database for Visualization and Integrated Discovery
- DCITC
- 5(2-(((1′-(4′-isothiocyanatephenylamino)thiocarbonyl)amino)-2-methylpropyl)amino-2-hydroxypropoxy)-3,4-dihydrocarbostyril
- DEG
- differentially expressed gene
- FGFR2
- fibroblast growth factor receptor 2
- GO
- gene ontology
- GS-493163
- (R)-6-[(3-{[4-(5-{[2-hydroxy-2-(8-hydroxy-2-oxo-1,2-dihydroquinolin-5-yl)ethyl]amino}pent-1-yn-1-yl)phenyl] carbamoyl}phenyl)sulphonyl]-4-[(3-methoxyphenyl)(methylsulphonyl)amino]-8-methylquinoline-3-carboxamide trifluoroacetic acid
- GS-548834
- 5-[(1R)-2-{[5-(4-aminophenyl)pent-4-yn-1-yl]amino}-1-hydroxyethyl]-8-hydroxyquinolin-2(1H)-one bis-trifluoroacetic acid
- GS-5759
- (R)-6-[(3-{[4-(5-{[2-hydroxy-2-(8-hydroxy-2-oxo-1,2-dihydroquinolin-5-yl)ethyl]amino}pent-1-yn-1-yl)phenyl] carbamoyl}phenyl)sulphonyl]-4-[(3-methoxyphenyl)amino]-8-methylquinoline-3-carboxamide trifluoroacetic acid
- GS-709344
- 8-hydroxy-5-((R)-1-hydroxy-2-methylaminoethyl)-1H-quinolin-2-one
- GSK 256066
- 6-[3-(dimethylcarbamoyl)benzenesulphonyl]-4-[(3-methoxyphenyl)amino]-8-methylquinoline-3-carboxamide
- GSK 256066a
- 4-(3-methoxyphenylamino)-8-methyl-6-(3-(methyl carbamoyl)phenylsulphonyl)quinoline-3-carboxamide
- ICI 118551
- (2R,3R)-1-[(2,3-dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl) amino]-2-butanol
- Ind/GSK
- indacaterol/GSK 256066 in combination
- LABA
- long-acting β2-adrenoceptor agonist
- MA
- muscarinic receptor antagonist
- MABA
- muscarinic receptor antagonist/β2-adrenoceptor agonist
- MA-L
- MA linked to nonane
- NR4A2
- nuclear receptor subfamily 4, group A, member 2
- ONO-AE1-259
- (Z)-7-[(1R,2R,3R,5R)-5-chloro-3-hydroxy-2-[(E,4S)-4-hydroxy-4-(1-prop-2-enylcyclobutyl)but-1-enyl]cyclopentyl]hept-5-enoic acid
- PDE
- phosphodiesterase
- RGS2
- regulator of G-protein signaling 2
- RT-PCR
- reverse-transcription polymerase chain reaction
- SFM
- serum-free medium
- THRX 198321
- biphenyl-2-yl-carbamic acid 1-{9-[(R)-2-hydroxy-2-(8-hydroxy-2-oxo-1,2-dihydro-quinolin-5-yl)-ethylamino]-nonyl}-piperidin-4-yl ester
- THRX 200495
- biphenyl-2-ylcarbamic acid 1-{3-[4-(4-{2-[(R)-2-hydroxy-2-(8-hydroxy-2-oxo-1,2-dihydroquinolin-5-yl)ethylamino]ethyl}phenoxy)phenyl]propyl piperidin-4-yl ester
- 7-TMR
- 7-transmembrane-spanning receptor
- tPSA
- total polar surface area
- Copyright © 2017 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}