


Abstract
The goals of this study were to develop compounds that were selective and highly efficacious agonists at alpha-7 receptors, while varying in antagonist activity; and to test the hypothesis that these compounds had memory-related and neuroprotective actions associated with both agonist and antagonist alpha-7 receptor activities. Three compounds were identified; E,E-3-(cinnamylidene)anabaseine (3-CA), E,E-3-(2-methoxycinnamylidene) anabaseine (2-MeOCA) and E,E-3-(4-methoxycinnamylidene) anabaseine (4-MeOCA) each displaced [125I]α-bungarotoxin binding from rat brain membranes and activated rat alpha-7 receptors in a Xenopus oocyte expression system fully efficaciously. The potency series for binding and receptor activation was 2-MeOCA > 4-MeOCA = 3-CA and 2-MeOCA = 3-CA > 4-MeOCA, respectively. No compound significantly activated oocyte-expressed alpha-4beta-2 receptors. Although each cinnamylidene-anabaseine caused a long-term inhibition ofalpha-7 receptors, as measured by ACh-application 5 min later, this inhibition ranged considerably, from less than 20% (3-CA) to 90% (2-MeOCA) at an identical concentration (10 μM). These compounds improved passive avoidance behavior in nucleus basalis lesioned rats, with 2-MeOCA most potent in this respect. In contrast, only 3-CA was neuroprotective against neurite loss during nerve growth factor deprivation in differentiated rat pheochromocytoma (PC12) cells. Choline, an efficacious alpha-7 agonist without antagonist activity, was also protective in this model. These results suggest that the neurite-protective action of alpha-7 receptor agonists may be more sensitive to potential long-term antagonist properties than acute behavioral actions are.
Molecular, biochemical and physiological studies demonstrate the presence of multiple nicotinic receptor subunits in brain and other tissues (Alkondon and Albuquerque, 1993; Deneris et al., 1991;Papke, 1993). One of the predominant nicotinic receptor subtypes in the brain contains the alpha-7 subunit.Alpha-7-containing nicotinic receptors are concentrated especially in telencephalic regions such as hippocampus and neocortex, based on high-affinity α-BTX binding (Clarke et al., 1985;Marks et al., 1986). These receptors function as homo-oligomers when expressed in oocytes, in which they demonstrate characteristic high-affinity binding to α-BTX, high calcium permeability and rapid desensitization (Couturier et al., 1990; Sequela et al., 1993; de Fiebre et al., 1995). Presynaptic nicotinic alpha-7 receptors in brain explants and primary cultures modulate calcium influx sufficiently to induce transmitter release and synaptic facilitation (McGehee et al., 1995; Gray et al., 1996). Ganglionic nicotinicalpha-7 receptors also appear to modulate intraneuronal calcium concentrations at perisynaptic sites (Zhang et al., 1996).
Recent studies with selective alpha-7 agonists suggest that these receptors are involved in memory-related behaviors and neuronal survival (Martin et al., 1994). The best studied selectivealpha-7 agonist is GTS-21, also referred to as DMXB in some studies, which recently began clinical trials for use in Alzheimer’s disease. GTS-21 has been found to enhance performance in several spatial and nonspatial memory-related paradigms in rats and rabbits (Meyer et al., 1994, 1997b; Woodruff-Pak et al., 1994). It also exerts neuroprotective activity against trophic factor deprivation in PC12 cells (Martin et al., 1994), N-methyl-d-aspartate receptor activation in primary neuronal cultures (Shimohama et al., in press,1998), fimbrial transections in vivo (Martin et al., 1994) and neocortical atrophy after nucleus basalis lesions (Meyeret al., in press, 1997a). Despite these behavioral and neuroprotective actions of GTS-21, however, it is only a modestly efficacious partial agonist at alpha-7 receptors, with about 20% of the activity of ACh itself. In addition, it is not effective behaviorally in all species, including mice (Briggs et al., 1997)
It would be important for understanding alpha-7 nicotinic receptor function to have available fully efficacious, selective agonists with little or no antagonist activity. Such agents could also be important therapeutically for conditions associated with memory-related dysfunction. However, the potential behavioral or neuroprotective effects of other selective, more efficaciousalpha-7 agonists, such as those described in this study, have not been investigated extensively. DMAC has been shown to activatealpha-7 nicotinic receptors selectively (de Fiebre et al., 1995), and one study reported that it had memory-enhancing activity (Meyer et al., 1994). DMAC was also the first compound found to exert the property of long-term inhibition ofalpha-7 receptors after their activation (de Fiebre et al., 1995), a property reported subsequently with ABT418 and epibatidine (Papke et al., 1997). Although the biological significance of this inhibitory activity is not known currently, it might be expected to interfere with long-term actions of the compound that depend on receptor activation such as neuroprotection.
This study investigated: 1) whether other anabaseine analogs could be developed that were highly potent and efficacious at alpha-7 receptors, while varying with respect to antagonist activity; and 2) whether these alpha-7 agonists had behavioral and neuroprotective actions hypothesized to result from selectivealpha-7 activation. We report for the first time that several 3-cinnamylidene anabaseine compounds, which vary with respect to substituents at the 2- or 4-position (fig.1), bind potently and efficaciously toalpha-7 receptors in rat brain tissue and activatealpha-7 receptors in a Xenopus oocyte expression system. These compounds, which differed significantly in their antagonist activities at alpha-7 receptors, were tested subsequently for their ability to improve passive avoidance behavior in nucleus basalis lesioned rats and to protect neurites from NGF deprivation in differentiated PC12 cells. Because our results indicated that the long-term neuroprotective actions of these compounds may be sensitive to their antagonist properties, choline was also investigated with respect to its neuroprotective activity as a prototypicalalpha-7-selective agonist with no antagonistic activity (Papke et al., 1996).

(Top) Structures of GTS-21 (DMXB), DMAC and related 3-cinnamylidene anabaseine compounds. CA refers to 3-cinnamylidene anabaseine moiety. (Bottom) Lowest energy conformation for structure of 3-CA as generated by a PM3 semiempirical computation with HyerChem version 4.5 (Hypercube, Inc., Gainesville, FL). The distance between the two nitrogen atoms is 4.41 A and the dihedral angle between the pyridine and reduced pyridine rings is 80 248 .
Materials and Methods
Preparation of E,E-3-(cinnamylidene)anabaseines
All chemicals were purchased from Aldrich Chemical Company, Inc (Milwaukee, WI). Cinnamylidene-anabaseines were synthesized and characterized as follows.
E,E-3-(Cinnamylidene)anabaseine dihydrochloride.
A solution containing 0.77 mmol anabaseine dihydrochloride (Zoltewicz and Cruskie, 1997), 1.9 mmol cinnamaldehyde, 20 ml ethanol and 8 drops of concentrated HCl was heated at reflux for 4 hr, then cooled to 0°C. Diethyl ether was added dropwise until no additional precipitate appeared (10–20 ml). The product was filtered to yield 200 mg of a fine yellow powder which was recrystallized by dissolving it in warm isopropyl alcohol and precipitating it with ether. The result was 180 mg (0.519 mmol) of a yellow solid (mp 210–213°C, 67% yield).1H NMR (dimethyl sulfoxide-d6):δ 8.93 (H6′, d, 5 Hz, 1H), 8.86 (H2′, s, 1H), 8.17 (H4′, d, 9 Hz, 1H), 7.77 (H5′, dd, 1H), 7.70 (H2, H6, m, 2H), 7.52 (Hβ, dd, 13 and 17 Hz, 1H), 7.40 (H3, H4, H5, m,3H), 7.29 (Hα, d, 17 Hz, 1H), 6.98 (Hγ, d, 12 Hz, 1H), 5.4 (N-H, b, 2H), 3.78 (CH2, m, 2H), 2.93 (CH2, m, 2H), 2.04 ppm (CH2, m, 2H). Anal. Calcd for C19H20N2Cl2: C, 65.70%; H, 5.80%; N, 8.07%. Found: C, 65.69%, H, 5.85%, N, 7.98%.
E,E-3-(2-Methoxycinnamylidene)anabaseine dihydrochloride.
A solution containing 0.86 mmol anabaseine dihydrochloride, 2.1 mmoltrans-2-methoxycinnamaldehyde, 20 ml ethanol and 8 drops concentrated HCl was heated at reflux for 4 hr while stirring. This was cooled to 0°C and the product was precipitated with diethyl ether (10–20 ml) to yield 220 mg of a yellow solid. This was recrystallized by dissolving it in warm isopropyl alcohol and precipitating it with diethyl ether to yield 200 mg (0.51 mmol) of an orange solid (mp 212–213°C, 59% yield). A portion was converted to the free base by use of saturated aqueous NaHCO3 and extracting the product into ethyl acetate; a yellow solid then was collected.1H NMR of the free base (CDCl3): δ 8.63 (H2′, d, 2 Hz, 1H), 8.57 (H6′, dd, 2 and 6 Hz, 1H), 7.70 (H4′, dt, 2, 2 and 9 Hz, 1H), 7.42 (H6, dd, 2 and 8 Hz, 1H), 7.27 (H5′, ddd, 2, 6 and 9 Hz, 1H), 7.17 (H4, dt, 2, 8 and 8 Hz, 1H), 7.05 (Hβ, dd, 12 and 17 Hz, 1H), 6.90 (Hα, d, 17 Hz, 1H), 6.85 (H5, t, 8 and 8 Hz, 1H), 6.78 (H3, d, 8 Hz, 1H), 6.32 (Hγ, d, 12 Hz, 1H), 3.80 (CH2, m, 2H), 3.78 (CH3O, s, 3H), 2.68 (CH2, m, 2H), 1.80 ppm (CH2, m, 2H). Anal. Calcd. for C20H22N2Cl2O.H2O: C, 60.76%; H, 6.12%, N, 7.08%. Found: C, 60.54%, H, 6.03%, N, 6.76%.
E,E-3-(4-Methoxycinnamylidene)anabaseine dihydrochloride.
A solution containing 1.7 mmol anabaseine dihydrochloride, 4.3 mmoltrans-4-methoxycinnamaldehyde, 20 ml of ethanol and 16 drops of concentrated HCl was heated at reflux for 4 hr while stirring. The reaction mixture was cooled to 0°C and the product was precipitated with diethyl ether (20–40 ml). The product was collected by filtration to give 410 mg of a yellow solid, which was recrystallized by dissolving it in warm isopropyl alcohol and precipitating it with diethyl ether to yield 350 mg (0.928 mmol) of an orange solid (mp 217–219°C, 54% yield). 1H NMR (dimethyl sulfoxide-d6): δ 8.98 (H6′, d, 6 Hz, 1H), 8.94 (H2′, s, 1H), 8.32 (H4′, d, 9 Hz, 1H), 8.78 (H5′, dd, 9 and 6 Hz), 7.66 (H2,6 d, 10 Hz, 2H), 7.38 (Hβ, dd, 17 and 12 Hz), 7.24 (Hα, d, 17 Hz, 1H), 6.95 (H3,5 and Hγ, m, 3H), 5.9 (NH, b, 2H), 3.76 (CH3, s, 3H), 3.74 (CH2, m, 2H), 2.89 (CH2, m, 2H), 2.04 ppm (CH2, m, 2H). Anal. Calcd. for C20H22N2OCl2: C, 63.67%; H, 5.88%; N, 7.43%. Found: C, 63.32%; H, 5.87%; N, 7.32%.
Animals
Male Sprague Dawley albino rats (250–350 g) were purchased from Charles River Laboratories (Boston, MA) and maintained in the University of Florida Health Center Vivarium according to National Institutes of Health guidelines, on a 12-hr light/dark cycle. They hadad libitum access to food (Purina rat chow) and water. Female Xenopus laevis (Nasco, Ft. Atkinson, WI) were housed in aquarium tanks maintained precisely at 18°C to reduce potential problems with seasonal variability in oocyte viability (Wu and Gerhart, 1991). Frogs were fed frog brittle (Nasco) and kept on a 12-hr light/dark cycle.
Oocyte Recordings
Oocytes were prepared and recordings made as described previously (de Fiebre et al., 1995). Frogs were anesthetized by submersion in 0.1% w/v (3)-aminobenzoic acid ethyl ester and several lobes of the ovary surgically removed through a small incision in the abdomen wall. Oocytes were freed from the follicle cells by treatment with collagenase (in calcium-free Barth’s solution: 88 mM NaCl, 1 mM KCl, 15 mM HEPES, pH 7.6, 0.33 mM MgSO4, 0.1 mg/ml gentamicin sulfate) for 2 hr at room temperature and stage 5 oocytes were isolated. Oocytes were rinsed and stored at 18°C in Barth’s saline (88 mM NaCl, 1 mM KCl, 15 mM HEPES, pH 7.6, 0.3 mM Ca(NO3)2, 0.41 mM CaCl2, 0.82 mM MgSO4, 0.1 mg/ml gentamicin sulfate) before and after microinjection with RNA (Wu and Gerhart, 1991). Microinjection was conducted with a Drummond Scientific “Nanoject Variable” automatic injector. Oocytes were injected with a 50-nl solution of specified nicotinic subunit rat-mRNA species (2–10 mg/ml; derived from the cDNA containing HIP 306 plasmid kindly provided by Dr. Jim Boulter, Salk Institute, San Diego, CA) and incubated 2 to 7 days at 18°C before electrophysiological recording.
For electrophysiological recordings, oocytes were perfused for several minutes in a Warner Instruments RC-8 recording chamber with a perfusion solution containing 115 mM NaCl, 2.5 mM KCl, 10 mM HEPES, pH 7.2, 1.8 mM CaCl2 and 1 μM atropine to block muscarinic responses. Perfusion was continuous at a rate of 10 ml/min. Drugs were diluted in perfusion solution and applied with a solenoid valve to switch from perfusion to drug solutions. Current responses to drug administration were studied under two electrode voltage clamp at a holding potential of −70 mV with a Dagan Corp (Minneapolis, MN) TEV-200 voltage clamp connected to a 386-SX IBM computer with a TL-1 DMA interface (Axon Instruments, Foster City, CA). Micropipettes were filled with 3 M KCl and had resistances of 0.5 to 2 megohms. Drug responses are analyzed with PClamp software (Axon Instruments, Foster City, CA). Oocytes with resting potentials of less than −30 mV were rejected.
High-Affinity [125I]α-BTX Binding
Rats were decapitated, and cerebral cortices were removed and homogenized in 10 vol of ice-cold KRH buffer (NaCl, 118 mM; KCl, 4.8 mM; MgSO4, 1.2 mM; CaCl2, 2.5 mM; and HEPES, 20 mM; pH adjusted to 7.5 with NaOH), and then assayed for high affinity [125I]α-BTX (New England Nuclear Co., Boston, MA) binding as described previously (de Fiebre et al., 1995). Binding assays were conducted at 4°C in KRH buffer. The final incubation contained 500 to 800 μg protein/250 μl with 2 nM [125I]α-BTX. Binding was terminated by diluting with 3 ml of ice-cold KRH buffer, followed immediately by filtration through glass fiber filters soaked in buffer containing 0.5% polyethylenimine. Nonspecific binding was determined with 100 μM unlabeled nicotine. IC50values were calculated and Ki values estimated by the equation of Cheng and Prusoff (1973).
Neuroprotection Assay
PC12 cell-culture methods were modified from those of Greene and Tischler (1976). Cells (American Type Culture Collection, Rockville, MD) were plated at 30 to 40% confluence and grown in Dulbecco’s Modified Eagle’s Medium containing 10% heat-inactivated horse serum, 5% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin and 0.5 mM l-glutamine. Chemicals used in the maintenance of cultures were obtained from Gibco/BRL (Grand Island, NY), except l-glutamine (Sigma Chemicals, St. Louis, MO). Cultures were maintained at 37°C, 94% O2/6% CO2 and 90 to 92% humidity in culture plates (Fisher Scientific, Orlando, FL) precoated with poly-l-lysine (10 g/l). Cultures were maintained for 7 days in serum-supplemented medium to which 100 ng/ml rat 2.5S NGF was added at day 1. On day 7, conditioned medium was replaced with serum- and antibiotic-free medium containing specified drugs. Four days later, the percent of cells expressing neurites was determined by dividing the number of neurite-expressing cells by total cell number. Cell number was estimated with use of the NIH Image program version 1.47 and the number of neurite-expressing cells counted manually in blind fashion (Martin et al., 1994). A Nikon inverted microscope (magnification, ×100) was attached to a Mac II computer viaa monochrome video camera (Cohu, Inc., San Diego, CA). Four random areas were counted per plate and there were four plates per treatment group, unless otherwise indicated. Each plate derived from a separate flask. Neurites were defined as processes at least two times the length of the perikaryon. To assess the reliability of the image analyses, a reticle was used for direct quantification of cells in the same view as the one analyzed in random samples. Differences among treatment groups were assessed by one-way ANOVA (Dixon and Massey, 1969).
Passive Avoidance Behavior
Passive avoidance behavior was measured in nucleus basalis-lesioned rats after intraperitoneal injections of drugs (base weight) or 0.9% saline diluent. For lesions, male 5-month-old Sprague Dawley rats were anesthetized with 30 mg/kg sodium pentobarbital (i.p.) and then infused bilaterally with 1 μl of 5 μg/μl ibotenic acid in phosphate-buffered saline, pH 7.4, into the nucleus basalis as described previously (Meyer et al., 1987). Infusion coordinates were anterior 7.0 mm, lateral 2.6 mm and vertical 6.5 mm according to Paxinos and Watson (1986). After surgery, animals were returned to their individual home cages, and for several days, were fed semisolid mash made from Purina Rat Chow. One month later, animals were trained in a two-chamber passive avoidance paradigm. Fifteen minutes after i.p. drug injection, they were placed in the lit compartment and allowed to enter the dark adjoining chamber. Animals that entered the second chamber received a mild foot shock (0.8 mA) for 1 sec. Rats were tested for latency to enter the dark chamber 24 hr later for up to 5 min; this test also began 15 min after injection. Statistical analyses used rank order nonparametric comparisons of latencies (Dixon and Massey, 1969).
Histological assessments of the ibotenic acid placements in the nucleus basalis were made after behavioral measurements by cholinesterase staining in formalin-fixed tissues. These injections typically reduced the number of cholinesterase-staining cells by more than 80% in the nucleus basalis, with some loss of staining in the globus pallidus and thalamus as well, as determined with the NIH Image 1.47 program.
Results
[125I]α-BTX bound to rat brain membranes in a nicotine-displaceable manner with Scatchard analyses indicating aKd of 1.9 nM (r = 0.98; single-site model). Each 3-cinnamylidene anabaseine blocked high-affinity, nicotine-displaceable [125I]α-BTX binding from rat brain membranes in a concentration-related manner (IC50 values: 3-CA, 80 nM; 4-MeOCA, 54 nM; 2-MeOCA, 8 nM) (fig.2).

Displacement of high-affinity α-BTX binding to rat brain membranes by 3-cinnamylidene anabaseines. Neocortices were removed and assayed for nicotine-displaceable, high-affinity [125I]α-BTX binding in the presence of specified drug concentrations, as described in the text. Each value is the mean ± S.E.M. of three preparations per group.
ACh displayed typical agonist activity at alpha-7 andalpha-4beta-2 nicotinic receptors expressed inXenopus oocytes (figs. 3 and4). The cinnamylidene-anabaseine compounds were also efficacious with respect to activating ratalpha-7 homo-oligomeric receptors, without activity at thealpha-4beta-2 combination. 3-CA and 2-MeOCA had similar alpha-7 EC50 potency values (3.2 ± 0.7 μM and 6.2 ± 2.2 μM, respectively), whereas 4-MeOCA was two to three times less potent (EC50= 15.9 ± 7.9 μM).

Responses of rat alpha-7 ACh receptor (AChR) to 3-CA, 2-MeOCA and 4-MeOCA. (A) Concentration-response relationships for the peak agonist-activated currents of oocytes injected with RNA coding for the ratalpha-7 subunit. All responses initially were measured relative to the individual oocyte response to 500 μM ACh, applied 5 min before drug application. Responses were normalized relative to the maximum response obtainable with ACh, so that a response of 1 represents full efficacy. The ratio of 500 μM ACh control response to ACh maximum was taken from Papke et al. (1997). (B) Residual inhibition of control (500 μM) ACh responses of ratalpha-7-injected oocytes after the application of 3-CA, 2-MeOCA or 4-MeOCA at specified concentrations. All responses are expressed relative to the oocyte-response to 500 μM ACh applied 5 min prior to the experimental agonist application.

Responses ofalpha-4beta-2 ACh receptor (AChR) to 3-CA, 2-MeOCA or 4-MeOCA. (A) Concentration-response relationships for the peak agonist-activated currents of oocytes injected with RNA coding for rat alpha-4beta-2 subunits. Responses are first expressed relative to the response to 10 μM ACh, applied 5 min before drug application, then normalized to the maximum response obtainable with ACh so that a response of 1 represents full efficacy. The ratio between 10 μM ACh and maximum ACh response was determined in separate experiments. (B) Residual inhibition of control (10 μM) ACh responses of ratalpha-4beta-2-injected oocytes after the application of 3-CA, 2-MeOCA or 4-MeOCA at the specified concentration. Responses are expressed relative to the individual oocyte response to 10 μM ACh, applied 5 min before experimental agonist application.
Antagonist properties subsequent to agonist activity were measured as a decrease in responsiveness to ACh applied 5 min after compound application, normalized to initial control applications (fig. 3). ACh administration had no long-term inhibitory activity, as noted previously. However, the cinnamylidene-anabaseines displayed concentration-dependent antagonist activity that varied in relative potency from what was observed with agonist activity. The IC50 values for 3-CA, 2-MeOCA and 4-MeOCA were 15 ± 6 μM, 2.3 ± 0.7 μM and 5.0 ± 2.3 μM, respectively. There was no clear relationship between agonist- and antagonist-potencies for alpha-7 receptors among these three compounds. None of the compounds elicited significant antagonism toalpha-4beta-2 receptors up to concentrations of 30 μM.
Bilaterally nucleus basalis-lesioned rats injected with saline diluent performed poorly in the passive avoidance paradigm compared with sham-operated controls (fig.5). The three cinnamylidene-anabaseines each improved passive avoidance behavior in the lesioned animals in an inverted U-shaped, dose-related manner when administered i.p. 2-MeOCA was effective at a dose of 0.1 mg/kg, whereas significant improvements in this behavior were observed at higher doses with the other two compounds. None of the agents altered performance during the training interval over the dose ranges in which test performance was improved, which suggests that there was no drug-induced change in locomotor function; higher doses did cause animals to be untrainable, with apparent loss of coordination without convulsions or unconsciousness (data not shown).

Effects of 3-cinnamylidene anabaseines on passive avoidance behavior in nucleus basalis lesioned rats. Adult Sprague Dawley albino rats were nucleus basalis-lesioned bilaterally and tested for passive avoidance behavior. Testing intervals were determined 15 min after i.p. injection of saline vehicle or specified drug, and described as mean ± S.E.M. of five to eight animals/group. NT, not tested; the animals could not be trained because of apparent motor deficits. *P < .05 compared with saline-injected group, one-way ANOVA.
Removal of NGF from differentiated PC12 cultures led to significant neurite loss 4 days later compared with plates in which NGF remained (fig. 6). 3-CA at 10 μM protected against NGF-deprivation neurite loss, whereas the other two compounds had no effect (fig. 6). Choline also protected against cell death in a 1 to 10 mM concentration range, which previously was found to activate these receptors selectively in oocytes. This effect was blocked with the nicotinic antagonist mecamylamine (fig.7).
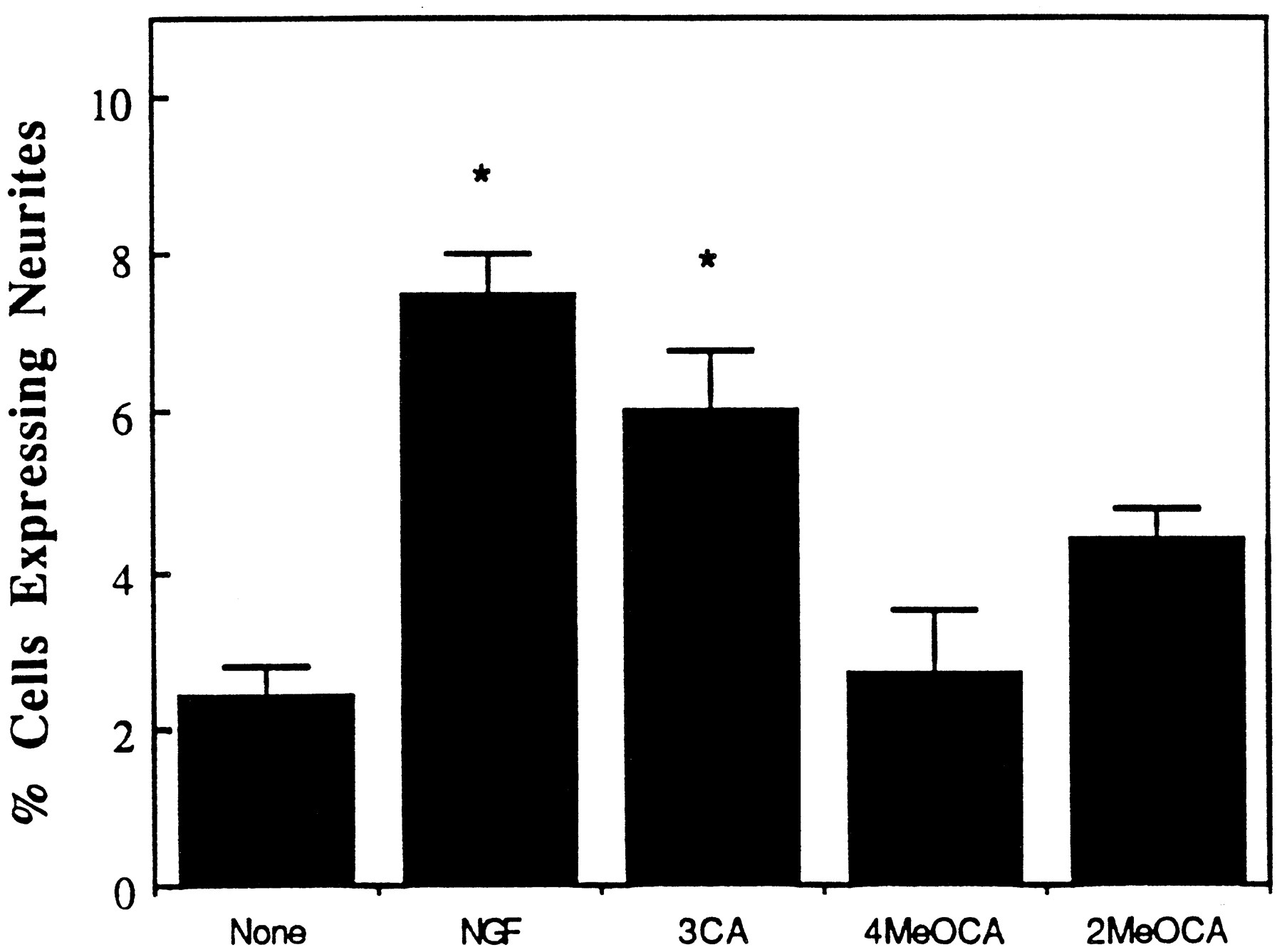
Effects of 3-cinnamylidene anabaseines on neurite viability in differentiated PC12 cells undergoing NGF deprivation. PC12 cells that had been differentiated for 1 week were exposed to the specified 3-cinnamylidene anabaseine (10 μM each) for 4 days, in the absence of NGF and serum. Some plates had 100 ng/ml NGF (minus serum) return for this 4-day interval. The percent of PC12 cells expressing neurites were quantified and expressed as the mean ± S.E.M. of three to four plates/group. *P < .05 compared with untreated cells (one-way ANOVA).
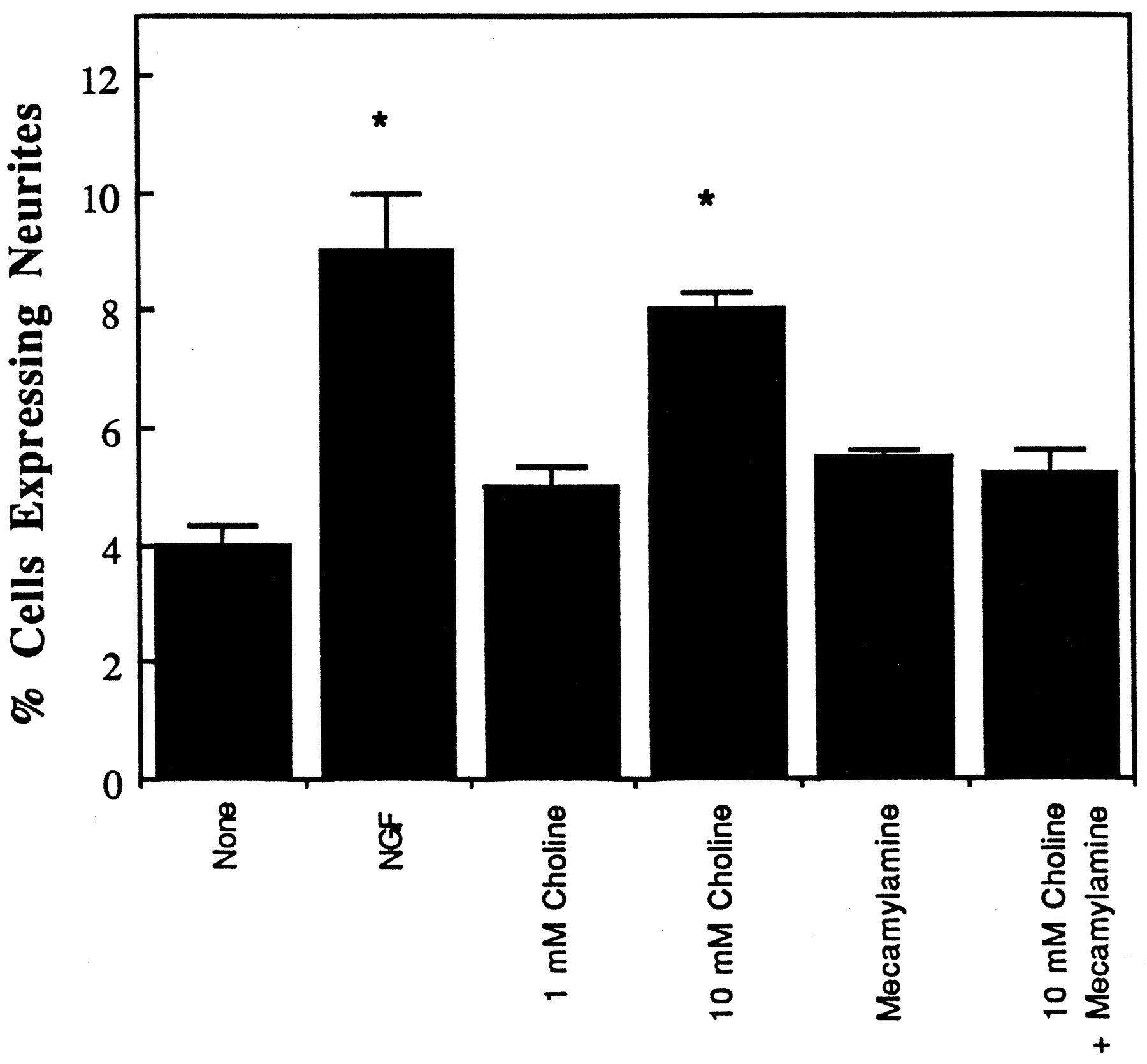
Effects of choline on neurite viability in differentiated PC12 cells under NGF deprivation. PC12 cells that had been differentiated for 1 week and then deprived of NGF + serum were exposed for 4 days to serum-free medium containing either nothing, 100 ng/ml NGF, the specified choline concentration, ± 10 μM mecamylamine. The fraction of PC12 cells expressing neurites at that time were quantified as described in the text and expressed as the mean ± S.E.M. of three to four plates/group. *P < .05 compared with untreated cells (one-way ANOVA).
Discussion
The mammalian brain possesses many nicotinic receptor subtypes, but the two predominant subsites apparently consist ofalpha-4beta-2 and alpha-7 subunits, based on binding and immunological studies (Clarke et al., 1985; Flores et al., 1992). Each of the 3-cinnamylidene anabaseines characterized to date (DMAC, 3-CA, 2-MeOCA, 4-MeOCA) selectively activates alpha-7 receptors overalpha-4beta-2 subtypes, which indicates at least some degree of nicotinic receptor selectivity. For DMAC, at least, this selectivity is seen over other nicotinic receptor subunit combinations as well (de Fiebre et al., 1995). The simplest 3-cinnamylamine anabaseine, 3-CA, was potent and efficacious, and also expressed relatively little antagonist activity at a neurite-protective concentration. Addition of the methoxy group to the 2-position resulted in the highest affinity for alpha-7 receptors of any compound yet reported, but also increased antagonist potency. The 4-substituted cinnamylidene anabaseine (4-MeOCA) had agonist and antagonist receptor properties similar to those reported for DMAC, which is also a 4-substituted cinnamylidene-anabaseine (de Fiebreet al., 1995): less affinity for the alpha-7 receptor than the unsubstituted 3-CA, but higher antagonist potency as well.
The presence of long-term antagonist activity, separate from desensitization, after acute administration of some cinnamylidene-anabaseines suggests that these antagonists remain bound to the antagonist site for an extended interval. However, there was no evidence for this phenomenon in binding-displacement studies. A very slow off-rate (several minutes) for cinnamylidene-anabaseines with significant antagonist activity (e.g., 4-MeOCA, 2-MeOCA) would be reflected as a many-fold increase in the potency of displacement compared with agents with much less antagonist activity (e.g., 3-CA). This was not observed. One potential explanation may be the presence of multiple binding sites for these compounds, with the antagonist site being separate from the α-BTX site and therefore not readily measured in this assay. Alternatively, the displacement of α-BTX binding in lysed membrane preparations may not reflect the antagonism that occurs in intact cells if this antagonism is voltage-dependent, as has been reported for ABT 418 (Papke et al., 1997).
Ibotenate-treated, nucleus basalis-lesioned rats offer a well studied model for memory-related dysfunction (Connor et al., 1991;Dekker et al., 1992; Meyer et al., 1987). The lowest effective doses of the cinnamylamine-anabaseine compounds reflect, at least in part, their relative potencies in this model. The rank ordering of these minimal doses for 3-CA, 2-MeOCA and 4-MeOCA are consistent with their relative receptor binding and activation concentrations in vitro. It also appears that compounds such as 2-MeOCA or DMAC (Meyer et al., 1994) that possess bothalpha-7 agonist and antagonist activity improve this behavior. In contrast, nicotinic receptor antagonists such as mecamylamine interfere with memory-related behaviors (Levin and Torry, 1996; Meyer et al., 1997b), which indicates that agonist activity may be essential for enhancement.
Nicotinic receptor agonists were found previously to exert neuroprotective or anti-apoptotic actions in a variety of model systems, including trophic factor deprivation in sympathetic ganglia (Koike et al., 1989), differentiated PC12 cells (Martinet al., 1994), lesioned brain dopamine neurons (Janson, 1988) and N-methyl-d-aspartate-induced toxicity in primary neocortical neurons (Akaike et al., 1994). In this study, choline and 3-CA each exhibited cytoprotective activity, which suggests a neuroprotective role for alpha-7 receptors. Choline activates rat alpha-7 receptors with a potency 10-fold lower than that of ACh, without effect at alpha-4,alpha-2, beta-2 or beta-4 containing subtypes (Papke et al., 1996). The potency of choline at oocyte-expressed alpha-7 receptors is similar to that observed for neuroprotection in the present assay, with activity seen in the 1 to 10 mM range in both systems.
In contrast, neither 2-MeOCA nor 4-MeOCA elicited neurite protection at the same concentration at which 3-CA was effective. This indicates either that alpha-7 receptor activation may be insufficient to elicit neuroprotection, or that some other property, such as antagonist activity, interferes with neuroprotection. The 10 μM concentration was selected based on the results of oocyte studies, which indicated that it would permit a comparison of the relative importance of agonist versus antagonist activities in this assay. Thus, 10 μM 3-CA and 2-MeOCA were hypothesized to activate a similar fraction of maximum peak current (about 75%), but differentially inhibit alpha-7 receptors: less than 20% inhibition for 3-CA and more than 90% inhibition for 2-MeOCA. The results of this study, along with those involving choline which has no apparent antagonist activity, are consistent with the hypothesis that neuroprotective activity in the PC12 cell model is attenuated byalpha-7 receptor antagonism when a similar level of receptor activation is present.
We hypothesize that activation of alpha-7 receptors is necessary for the neuroprotective and behavioral effects observed with 3-cinnamylidine- and 3-benzylidene-anabaseines, based in part on the ability of the antagonist mecamylamine to block their actions (Martinet al., 1994; Meyer et al., 1994, 1997b). These effects may be caused by the initial, transient increase in calcium influx, which then triggers intracellular transduction processes. An anti-apoptotic activity of nicotinic receptor activation was found to involve a protein kinase C-dependent pathway, at least in part (Wrightet al., 1993). We also have observed an increase in protein kinase C activity in PC12 cells treated with GTS-21 (data not shown), which suggests that alpha-7 nicotinic receptor subtypes may trigger this calcium-sensitive transduction pathway as well. Along this line, alpha-7 receptors have been found to be involved in synapse formation through a mechanism that appears to involve calcium homeostasis (Pugh and Berg, 1994).
However, because alpha-7 receptors desensitize in the presence of agonist, an important question about long-term actions of these compounds such as neuroprotection is whether these receptors remain in a state that can be activated after the initial application of agonist. Previous results indicate that the answer depends on the agonist as well as agonist concentration. Nicotine, for example, can desensitize receptors at concentrations below those necessary for activation in the Xenopus oocyte expression system (Briggset al., 1997). However, although high concentrations of the selective agonist GTS-21 cause rapid desensitization of virtually all the alpha-7 receptors in the same system, lower concentrations result in small but very protracted currents. Further, we found that because of a lower rate of desensitization, a 3 μM concentration of GTS-21 caused a total net charge of almost 50% that of 30 μM GTS-21 during a period of 100 sec, despite triggering a peak response of less than 10% of the higher concentration (data not shown). Therefore, low but activating concentrations of agonist may permit steady state levels of functional (i.e.,nondesensitized) receptor occupancy, at least in the absence of additional long-term antagonism. The neuroprotective action of 3-cinnamylidene-anabaseine was observed at a concentration somewhat less than that necessary for activating peak current.
Nicotinic agonists including ACh generally have a lower potency and affinity for the alpha-7 receptor than for other subtypes. This low affinity and potency may be important for activating (and thereby desensitizing) only a small fraction of thealpha-7 receptors at any given time, so that intracellular calcium accumulation is modulated carefully in a nontoxic manner. Mutant forms of alpha-7 receptors that do not desensitize rapidly have been found to be neurotoxic (Treinin and Chalfie, 1995) or reduce viability (Puchacz et al., 1995) after gene-transfer protocols.
The present observations are consistent with the hypothesis that choline is an endogenous agonist for alpha-7 receptors, at least under conditions in which it is being generated extracellularly at high concentrations near these receptors. This choline generation could occur through phospholipase-mediated metabolism of phosphatidylcholine, which is present predominantly on the extracellular surface of plasma membranes. This choline generation might be expected to have local trophic influences on nearby cells, protecting them from the original insult. As such, thealpha-7 receptor could play a critical role in maintaining differentiated cell survival, potentially even without ACh.
Acknowledgments
E.M.M. is a co-inventor of DMXB (GTS-21), which is licensed to Taiho Pharmaceuticals. C.M.dF. was supported in part by National Institutes of Health training grant AG00176 and NS32888 to RLP. The authors acknowledge Drs. M.P. Cruskie and C.D. Dill, who prepared the cinnamylidene-anabaseine compounds; Sherry Robinson, who conducted oocyte recordings; Guang-ling Huang, who conducted some behavioral studies; and Allison Dasta, who provided secretarial assistance.
Footnotes
-
Send reprint requests to: Edwin M. Meyer, Ph.D., Department of Pharmacology and Therapeutics, Box 100267, University of Florida College of Medicine, Gainesville, FL 32610.
-
↵1 Funded in part by NIH grant AG PO1 10481 and Taiho Pharmaceuticals.
- Abbreviations:
- DMXB
- GTS-21, E-3-(2,4-dimethoxybenzilidene)anabaseine
- ACh
- acetylcholine
- α-BTX
- α-bungarotoxin
- 3-CA
- E,E-3-(cinnamylidene)anabaseine
- 2-MeOCA
- E,E-3-(2-methoxycinnamylidene) anabaseine)
- 4-MeOCA
- E,E-3-(4-methoxycinnamylidene) anabaseine
- NGF
- nerve growth factor
- HEPES
- N-2-hydroxyethylpiperazine-N′-ethanesulfonic acid
- KRH
- Krebs-Ringer HEPES
- DMAC
- E,E-3-(4-dimethyl aminocinnamylidene) anabaseine
- ANOVA
- analysis of variance
- Received May 29, 1997.
- Accepted November 17, 1997.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}