


Abstract
The benzopyranopyrrole S33084 displayed pronounced affinity (pKi = 9.6) for cloned human hD3-receptors, and >100-fold lower affinity for hD2 and all other receptors (>30) examined. S33084 concentration dependently, potently, and competitively (pA2 = 9.7) antagonized dopamine (DA)-induced [35S]guanosine-5′-O-(3-thio)triphosphate (GTPγS) binding at hD3-receptors. It also concentration dependently abolished stimulation by DA of hD3-receptor-coupled mitogen-activated protein kinase. Administered alone, S33084 did not modify dialysate levels of DA in the frontal cortex, nucleus accumbens, or striatum of freely moving rats, nor the firing rate of ventrotegmental dopaminergic cell bodies. Furthermore, it had minimal effect on DA turnover in mesocortical, mesolimbic, and nigrostriatal projection regions. However, S33084 dose dependently blocked the suppressive influence of the preferential D3-agonist PD128,907 on frontocortical release of DA. Furthermore, it likewise antagonized the inhibitory influence of PD128,907 on the electrical activity of ventrotegmental dopaminergic neurons. Although less potent than S33084, GR218,231 likewise behaved as a selective hD3- versus hD2-receptor antagonist and its neurochemical and electrophysiological profiles were similar. In contrast, L741,626 was a preferential antagonist at hD2 versus hD3sites. In vivo, on administration alone, L741,626 increased frontocortical, mesolimbic, and (more potently) striatal DA release, enhanced the firing rate of dopaminergic perikarya, and accelerated cerebral DA synthesis. It also blocked the actions of PD128,907. In conclusion, S33084 is a novel, potent, selective, and competitive antagonist at hD3-receptors. Although GR218,231 behaves similarly, L741,626 is a preferential D2-receptor antagonist. DA D2- but not D3-(auto) receptors tonically inhibit ascending dopaminergic pathways, although the latter may contribute to phasic suppression of DA release in frontal cortex.
Dopamine (DA) D3-receptors are closely related to their D2 counterparts in their primary structure, coupling to intracellular transduction mechanisms, and recognition patterns for chemically diverse ligands (Sokoloff and Schwartz, 1995;Levant, 1997; Missale et al., 1998). These observations raise the question of their respective functional roles, pathophysiological significance, and therapeutic interest as targets for the treatment of schizophrenia, depression, Parkinson's disease, drug abuse, and other disorders in which a dysfunction of dopaminergic systems and “D2”-receptors has been implicated (Sokoloff and Schwartz, 1995; Levant, 1997). In an attempt to elucidate the functional significance of D3- versus D2-receptors, several complementary approaches have been adopted: 1) transgenic mice lacking D2- and/or D3-receptors (Koeltzow et al., 1998; Jung et al., 1999; Xu et al., 1999); 2), specific antisense probes directed against D2- or D3-receptors (Tepper et al., 1997; Ekman et al., 1998); 3) correlation analyses of agonist potency in eliciting actions in vivo relative to affinities (and efficacies) at D2- and D3-receptors in vitro (Gobert et al., 1995;Millan et al., 1995; Gainetdinov et al., 1996; Levant, 1997); and 4) ligands interacting preferentially with D2- or D3-receptors (Levant, 1997; Wustrow and Wise, 1997; Audinot et al., 1998).
With regard to the last strategy, the agonist bromocriptine shows a modest preference for D2-receptors, activation of which contributes to its antiparkinsonian properties (Newman-Tancredi et al., 1997; Perachon et al., 1999). DA D2-receptor blockade is implicated in extrapyramidal side effects of neuroleptics, such as haloperidol, which displays a modest preference for D2-receptors. Correspondingly, little effort has been devoted to identification of selective D2 antagonists. Nevertheless, the arylpiperazine derivative L741,626 [4-(4-chlorophenyl)-1-(1H-indol-3-ylmethyl)piperidin-4-ol], was serendipitously found to possess substantial selectivity for hD2- versus hD3-receptors (Bowery et al., 1996; Kulagowski et al., 1996; Pillai et al., 1998). Interestingly, most of the better-known agonists possess higher affinity for cloned hD3- versus hD2-receptors (Millan et al., 1995; Sokoloff and Schwartz, 1995; Levant, 1997). Furthermore, the aminotetralin derivative 7-hydroxy-2-dipropylaminotetralin (7-OH-DPAT) and the chromano-oxazine PD128,907 are widely employed as “selective” agonists for the characterization of D3 sites (Pugsley et al., 1995; Sokoloff and Schwartz, 1995; Levant, 1997;Wustrow and Wise, 1997). Indeed, studies of G-protein coupling and intracellular transduction mechanisms indicate that both PD128,907 and 7-OH-DPAT preferentially activate hD3 versus hD2 sites (Sokoloff and Schwartz, 1995; Levant, 1997; Missale et al., 1998; Cussac et al., 1999; Newman-Tancredi et al., 1999; Vanhauwe et al., 1999). However, the degree of selectivity for D3 over D2sites remains under discussion, and it is likely that the separation found in vitro is not matched in vivo. Indeed, it is now clear that both PD128,907 and 7-OH-DPAT can elicit actions in vivo via D2-receptors (Levant, 1997), so caution is required in attributing their effects to activation of D2- and/or D3-receptors.
All of the above-mentioned findings underline the key importance of selective D3-receptor antagonists, but their identification has proven challenging. After cloning of hD3-receptors, several antagonists were observed to possess a mild (<5-fold) preference for D3- over D2-sites, namely, the substituted aminotetralins AJ76 and UH232, although the latter may be a partial agonist (Griffon et al., 1995; Sokoloff and Schwartz, 1995; Levant, 1997; Wustrow and Wise, 1997). Subsequently, the aminoindane U99194 and the substituted benzamides nafadotride and GR103,691 were described as “selective” D3 antagonists (Murray et al., 1995; Sautel et al., 1995; Haadsma-Svensson and Svensson, 1998). However, U99194 displays low affinity (100–200 nM) and only a mild preference (∼10-fold) for hD3-receptors (Audinot et al., 1998). The preference of nafadotride for hD3- versus hD2-sites is likewise modest (<10-fold; Levant, 1997; Audinot et al., 1998). Moreover, although GR103,691 is a selective (60-fold) D3- versus D2-receptor antagonist, it possesses significant affinity for 5-hydroxytryptamine (serotonin; 5-HT)1A and α1-adrenergic receptors, and shows poor bioavailability (Murray et al., 1995; Audinot et al., 1998). Compared with the above-mentioned characteristics, the aminotetralin antagonist S14297 displays substantial potency and selectivity for hD3- versus hD2-receptors, as well as satisfactory bioavailability (Gobert et al., 1995; Millan et al., 1995; Audinot et al., 1998). However, S14297 showed partial agonist activity in stimulating hD3-receptor-coupled mitogen-activated protein (MAP) kinase (Cussac et al., 1999). Furthermore, the only modest preference of S14297 for D3- versus muscarinic receptors compromises its use as an experimental tool (Millan et al., 1995).
Clearly, there remains a need for improved, selective antagonists at dopamine D3-receptors. Characterization of structure-activity relationships in a series of benzopyrano[3,4-c]pyrroles identified the cyano-substituted, diphenyl derivative S33084 as a potent D3-receptor ligand (Dubuffet et al., 1999; Cussac et al., 2000a,b; Fig. 1). A principle objective of this study was, thus, with various cellular paradigms, to characterize the interaction of S33084 at dopamine D3- compared with D2-receptors. In this respect, its actions were compared with those of the D2 antagonist L741,626 (vide supra) and GR218,231 (Murray et al., 1996), a novel hD3-receptor antagonist (Newman-Tancredi et al., 1999). A complementary aim of this study was to exploit S33084, GR218,231, L741,626, and a combined neurochemical and electrophysiological approach for a characterization of the potential role of D3- compared with D2-receptors in the modulation of cerebral dopaminergic transmission.

Chemical structure of S33084.
Materials and Methods
Animals.
In vivo studies used, in line with our extensive previous studies of the modulation of dopaminergic transmission (Gobert et al., 1995, 1996; Lejeune and Millan, 1995; Millan et al., 1995), male Wistar rats weighing 250 to 325 g (Iffa-Credo, L'Arbresle, France). They were maintained in sawdust-lined cages with unrestricted access to food and water. The laboratory temperature was held at 21 ± 1°C and humidity was controlled at 60 ± 5%. There was a 12-h light/dark cycle, with lights on from 7:30 AM to 7:30 PM. Before experimentation, all animals were adapted for at least 1 week to laboratory conditions.
Determination of Drug Affinities.
Procedures used for the determination of drug affinities at multiple native and cloned human dopaminergic, serotonergic, and adrenergic receptors, and at other binding sites, have been described in detail in Millan et al. (1998). They are summarized in Table 1. Isotherms were subjected to nonlinear regression analysis with PRISM (GraphPad, San Diego, CA) to yield IC50 values. These values were subsequently transformed intoKi values according to the Cheng-Prusoff equation: Ki = IC50/(1 + L/Kd), where L corresponds to the radioligand concentration andKd to its dissociation constant (Cheng and Prusoff, 1973).
Determination of the affinity of S33084, GR218,231, and L741,626 at multiple monoaminergic receptors
Antagonist Properties at hD2- and hD3-Receptors: [35S]Guanosine-5′-O-(3-thio)triphosphate (GTPγS) Binding.
The protocol used to quantify the binding of [35S]GTPγS (1000 Ci/mmol; NEN, Les Ulis, France) at hD2- and hD3-receptors has been described in detail inNewman-Tancredi et al. (1999). The buffer composition was as follows: HEPES (20 mM), NaCl (100 mM), GDP (3 μM), and MgSO4 (3 mM). Incubations were performed at a 22°C and pH 7.4 for 60 min. Drug actions were evaluated both alone and in the presence of a fixed concentration of DA (3 and 1 μM for hD2- and hD3-sites, respectively). Agonist efficacy (alone) was expressed relative to that of a maximally effective concentration of DA (10 μM, defined as 100%). For antagonist studies, concentration-response curves of the blocking properties of drugs versus DA were analyzed as described inNewman-Tancredi et al. (1999) to yield pKb values. In addition, the concentration-response relationship for activation by DA of [35S]GTPγS binding at hD3-receptors was performed in the presence of incremental concentrations of S33084, and pA2values were derived by Schild analysis.
Activation of MAP kinase at hD3-Receptors.
As described in Cussac et al. (1999), Chinese hamster ovary (CHO) cells transfected with hD3-receptors were cultivated in six-well plates until 90% confluent, washed, and incubated overnight in serum-free medium. Drugs were diluted in this medium and added to cells to yield the desired, final concentration. After a 5-min preincubation with the test drug, cells were exposed to DA (1 μM) for 5 min. Subsequently, 0.25 ml/well of Laemmi sample buffer (containing 200 mM dithiotreitol) was added. Whole-cell lysates were boiled at 95°C for 3 min. Thereafter, 14 μl of the cell extract was loaded onto 15-well, 10% polyacrylamide gels and “fully” activated MAP kinase was detected by use of a monoclonal antibody specifically directed against phosphorylated [extracellular signal receptor-activated kinase 2 (ERK2) pp42MAPK and pp44MAPK (ERK1)] forms on both threonine and tyrosine residues (NanoTools, Denzlingen, Germany). This was followed by enhanced chemiluminescence detection with horseradish peroxidase as a secondary antibody (Amersham, Les Ulis, France).
Modulation of Electrical Activity of Dopaminergic Neurons in Anesthetized Rats.
The procedure used for evaluation of drug actions on the electrical activity of dopaminergic perikarya localized in the ventrotegmental area (VTA) has been described in detail inLejeune and Millan (1995). Rats were anesthetized with chloral hydrate (400 mg/kg i.p.) and, after placement in a stereotaxic apparatus, a tungsten microelectrode was lowered into the VTA: coordinates AP, 5.5 from bregma; L, 0.7; and H, −7/8.5 from dura. Dopaminergic neurons were identified according to their waveform (Lejeune and Millan, 1995) and baseline activity was monitored for 5 min. The influence of S33084, GR218,231, and L741,626 alone on firing rate was evaluated on their administration in cumulative doses at intervals of 3 to 5 min and drug effects were evaluated for 60 s at their time of peak action. For examination of their antagonist actions, a single dose of the antagonist was administered 1 min after PD128,907 (0.005 mg/kg i.v.) and drug effects were evaluated 2 to 3 min after antagonist injection. Spike 2 software (CED, Cambridge, England) was used to accomplish data acquisition. The data are expressed as percentage of change from preinjection, basal values (defined as 0%). They were analyzed by ANOVA followed by Newman-Keuls test.
Modulation of Cerebral Synthesis of DA, Noradrenaline (NA), and 5-HT.
The modulation of cerebral synthesis of DA, NA, and 5-HT was evaluated as described in Millan et al. (1998). DA and 5-HT synthesis was determined in the striatum (rich in DA but not NA) and NA and 5-HT synthesis was evaluated in the hippocampus (rich in NA but not DA). Drug actions were measured 60 min after administration and 30 min after injection of the decarboxylase inhibitor NSD1015 (100 mg/kg s.c.). HPLC analysis followed by electrochemical detection was used for determination of tissue levels of l-dopa and 5-hydroxytryptophan (5-HTP) as described in Millan et al. (1998). Levels of l-dopa and 5-HTP were expressed relative to those of vehicle values (defined as 0%). Data were analyzed by ANOVA followed by Dunnett's test.
Modulation of Cerebral DA Turnover.
As described in Millan et al. (1998), the ratio of levels of the DA metabolite dihydroxyphenylalaninecarboxylic acid (DOPAC) to those of DA were characterized in projection targets of the mesocortical pathway (frontal cortex; FCX), the mesolimbic pathway (nucleus accumbens and olfactory tubercles), and the nigrostriatal pathway (striatum) 30 min after administration of drugs. HPLC and electrochemical detection were used for determination of levels of DOPAC and DA as described in Millan et al. (1998). DOPAC/DA ratios were expressed relative to vehicle values (defined as 0%). Data were analyzed by ANOVA followed by Dunnett's test.
Modulation of Dialysate Levels of DA, NA, and 5-HT in FCX, Nucleus Accumbens, and Striatum of Freely Moving Rats.
The techniques used herein for characterization of drug influence on DA, NA, and 5-HT levels simultaneously determined in single dialysate samples of the FCX or the nucleus accumbens (shell region) and contralateral striatum of freely moving rats have been documented in detail (Gobert et al., 1996; Millan et al., 1998). Coordinates for FCX were AP, +2.2; L, ±0.6; DV, −0.2); for nucleus accumbens AP, +1.7; L, +1.1; DV, −4.1), and for striatum AP, +0.5; L, −2.8; DV, −3.0. All studies were undertaken 5 days after placement of guide cannulas. Samples were taken every 20 min. After three basal samples (defined as 0%), S33084 GR218,231, L741,626, or vehicle were injected and sampling pursued for an additional 3 h. For interaction studies with PD128,907 (0.16 mg/kg s.c.), this agonist was administered 20 min after the antagonist. Assay sensitivity was 0.1 to 0.2 pg/sample DA, NA, and 5-HT. Data were analyzed by ANOVA with sampling time as the repeated within-subject factor.
Drug Doses, Solution, Salts, and Sources.
For the in vivo procedures, full dose-response relationships were evaluated for S33084, GR218,231, and L741,626 in each case. The maximal dose evaluated for L741,626 was 40.0 mg/kg s.c., and as discussed in Millan et al. (2000)and Results, this dose allows for the full expression of its antagonist actions at D2-receptors. For S33084 and GR218,231, a maximal dose of 10 mg/kg was defined. This dose limit was determined by their maximal solubility. Furthermore, relative to their very high affinities at D3-receptors, and in vivo actions, this maximal dose limit is more than sufficient to permit the full expression of their potential antagonist properties at D3-receptors (Results; Millan et al. 2000). All drug doses are in terms of the base. PD128,907 was dissolved in sterile water. For S33084, GR218,231, and L741,626, a few drops of lactic acid were added and the pH adjusted to as close to neutrality as possible (>5.0). Drugs were injected in an injection volume of 1 ml/kg s.c. or 0.5 ml/kg i.v. (electrophysiology). PD128,907 was obtained from Research Biochemicals (Natick, MA) and L741,626 from Tocris Cookson (Bristol, UK). S33084 and GR218,231 were synthesized by G. Lavielle and J.-L. Peglion (Institut de Recherches Servier), respectively. S33084 and GR218,231 were synthesized as described in Dubuffet et al. (1999)and Murray et al. (1996), respectively. For both S33084 and GR218,231, microanalysis (carbon, hydrogen, and nitrogen atomic composition) yielded values within 0.4% of theoretical values for the formula given, and HPLC analysis also demonstrated >99% purity.
Results
Binding Profile of S33084 at Dopaminergic Receptors (Tables 1 and2; Fig.2).
At cloned hD3-receptors, S33084 displaced [125I]iodosulpride with very high affinity (Table 2; Fig. 2). S33084 likewise competed with [125I]iodosulpride at cloned hD2-receptors, but its affinity was considerably (120-fold) lower at these sites (Table 2). A similar pattern of data was acquired with [3H]spiperone; the D3-to-D2 selectivity of S33084 also was marked (125-fold) (Table 1). The affinity of S33084 for native, rat striatal D2-receptors labeled by [3H]raclopride was similar to that for cloned hD2-receptors, i.e., >100-fold lower than for cloned hD3-receptors (Table 1). The affinity of S33084 for cloned hD4-, hD1-, and hD5-receptors was weak: in each case, >1000-fold lower than for hD3-receptors (Table 1). S33084 also displayed >1000-fold lower affinity for native, rat DA uptake sites compared with its affinity for hD3-receptors (data not shown).
Interaction of S33084, GR218,231, and L741,626 at cloned hD3- versus hD2-receptors

Interaction of S33084, GR218,231, and L741,626 at hD3- compared with hD2- and hD4-receptors expressed in CHO cells. Isotherms were obtained in competition experiments with [3H]spiperone as described in Materials and Methods. Data are mean ± S.E. of at least three independent experiments performed in triplicate.
Binding Profile of GR218,231 and L741,626 at Dopaminergic Receptors (Tables 1 and 2; Fig. 2).
In analogy to S33084, GR218,231 exhibited a pronounced preference for hD3- versus hD2-sites labeled by either [125I]iodosulpride (60-fold selectivity) or [3H]spiperone (100-fold) (Table 2; Fig. 2). Its absolute affinity at hD3 sites was some 5-fold lower than that of S33084. The affinity of GR218,231 for native, rat striatal D2-receptors was 80-fold inferior to its affinity at hD3-receptors (Table 1). Furthermore, GR218,231 displayed weak (1000-fold lower) affinity for hD4-, hD1-, and hD5- versus hD3-receptors (Table 1). L741,626 presented an opposite pattern of interaction at hD3- and hD2-receptors compared with S33084 and GR218,231. Its affinity at hD3 sites labeled by [125I]iodosulpride and [3H]spiperone was, thus, modest (Table 2; Fig.2). However, it showed more pronounced (∼15-fold in each case) affinity for hD2- versus hD3-receptors labeled by [125I]iodosulpride and [3H]spiperone (Table 2). L741,626 also manifested marked affinity for native, rat striatal D2 sites labeled by [3H]raclopride (Table 1). The affinity of L741,626 for hD4-receptors was modest, ∼70-fold lower than for hD2-receptors. Furthermore, L741,626 showed modest affinity for hD1- and hD5- versus hD2-receptors (Table 1).
Binding Profile of S33084, GR218,231, and L741,626 to Nondopaminergic Receptors (Table 1).
At diverse, native and cloned 5-HT receptor subtypes indicated in Table 1, the affinity of S33084 was at least 500-fold lower than at hD3-receptors. It also displayed >1000-fold lower affinity at 5-HT reuptake sites compared with hD3-receptors (data not shown). GR218,231 also showed marked (>100-fold) selectivity for hD3 versus various 5-HT receptor types (Table 1). Furthermore, the affinity of L741,626 for the various 5-HT receptor types indicated in Table 1 was > 200-fold lower than for hD2-receptors. S33084 and GR218,231 also displayed considerably (>500-fold) lower affinity for α1- and α2A-adrenergic receptors (Table 1) and NA reuptake sites (data not shown) compared with hD3 sites. Similarly, the affinity of L741,626 for α2A-adrenergic receptors (Table 1) and NA uptake sites (data not shown) was low, whereas it displayed 60-fold lower affinity for α1-adrenergic receptors compared with hD2-receptors (Table 1). Compared with hD3-receptors, S33084 showed >1000-fold lower affinity for cloned human, muscarinic (M1) receptors and ς1-sites (Table 1), as well as histamine (H1) γ-aminobutyric acid, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, andN-methyl-d-asparate receptors and several other sites indicated in the legend to Table 1.
Antagonist Properties at hD3- versus hD2-Receptors: [35S]GTPγS Binding (Fig.3).
At cloned hD3- and hD2-receptors transfected into CHO cells, DA concentration dependently and markedly (1.5- and 2.2-fold, respectively) stimulated [35S]GTPγS binding with pEC50 values of 8.16 ± 0.10 and 6.45 ± 0.05, respectively (data not shown; Newman-Tancredi et al., 1999). In contrast, S33084, GR218,231, and L741,626 all failed to modify [35S]GTPγS binding when applied alone. S33084 potently and concentration dependently suppressed stimulation of [35S]GTPγS binding at hD3-receptors with a pKb of 9.61 ± 0.11. It was, however, considerably less potent in blocking the action of DA at hD2 sites displaying a pKb of 7.75 ± 0.05. In confirmation of our previous study (Newman-Tancredi et al., 1999), a similar pattern of data was acquired for GR218,231, which displayed pKb values of 9.02 ± 0.05 and 7.27 ± 0.15 at hD3- and hD2-sites, respectively. In contrast, L741,626 more potently blocked the action of DA at hD2- than at hD3-receptors with pKb values of 8.74 ± 0.01 and 7.38 ± 0.08, respectively. In the presence of incremental concentrations of S33084, the concentration-response curve for stimulation by DA of hD3-receptors was displaced in parallel to the right without any loss of maximal stimulation, indicative of competitive antagonist activity. Furthermore, these data generated a linear Schild plot with a slope of 1.04 ± 0.09 (r = 0.96), yielding a pA2 value of 9.69. This value is very similar to the pKi of S33084 at hD3-receptors (9.6; Table 1).

Antagonist actions of S33084, GR218,231, and L741,626 at cloned hD3- compared with hD2-receptors, as determined by inhibition of DA-stimulated [35S]GTPγS binding. A and B, concentration-dependent inhibition of DA-stimulated [35S]GTPγS binding at hD2- and hD3-receptors, respectively, by S33084, GR218,231, and L741,626. C, concentration-response curves for stimulation of [35S]GTPγS binding at hD3-receptors by DA in the presence of incremental concentrations of S33084. D, Schild transformation of data in C. Data are mean ± S.E. of at least three independent experiments performed in triplicate.
Antagonist Properties at hD3-Receptors: MAP Kinase Activation (Fig. 4).
In CHO cells transfected with hD3-receptors, DA activated (phosphorylated) both ERK1 and ERK2 species of MAP kinase (Cussac et al., 1999). In contrast to DA, S33084 failed to induce either ERK2 (Fig. 4) or ERK1 (data not shown) forms of MAP kinase. Furthermore, after pretreatment of cells for 5 min with S33084, the induction of both forms of MAP kinase by DA (1 μM) was concentration dependently abolished (Fig. 4; data not shown). This action was expressed specifically inasmuch as the induction of MAP kinase by fibroblast growth factor (20 ng/ml) was not modified by S33084 (10 μM; data not shown). At a single concentration, GR218,231 (1 μM) likewise abolished the action of DA without itself inducing MAP kinase (data not shown). In view of its low affinity for hD3-receptors, L741,626 was not evaluated in this protocol.

Antagonist actions of S33084 at cloned hD3-receptors as determined by inhibition of DA-stimulated MAP kinase activity. CHO-hD3 cells were incubated with S33084 for 5 min, followed by addition of DA (1 μM). Active forms of MAP kinase were detected as described inMaterials and Methods. Immunoblots shown are from a representative experiment repeated at least three times with comparable results.
Influence on Basal Levels of DA, 5-HT, and NA in Dialysates of Freely Moving Rats (Figs. 5 and6).
Over a broad range of doses, S33084 failed to modify basal levels of DA, NA, or 5-HT simultaneously determined in single dialysate samples of the FCX of freely moving rats (Fig. 5). At a dose of 2.5 mg/kg s.c., S33084 did not modify basal levels of DA in the nucleus accumbens or striatum (Fig. 6). Levels of 5-HT were likewise unaffected (data not shown). Similarly, GR218,231 did not influence levels of these monoamines in any structure examined (Figs. 5 and 6). In contrast to S33084 and GR218,231, L741,626 dose dependently elevated levels of DA in the FCX (Fig. 5). It also dose dependently (0.16–10.0 mg/kg s.c.) elevated levels of DA in both the accumbens and, more potently, the striatum (Fig. 6). These actions were specific inasmuch as levels of 5-HT and/or NA were not significantly altered in the same dialysis samples (Fig. 5; data not shown).
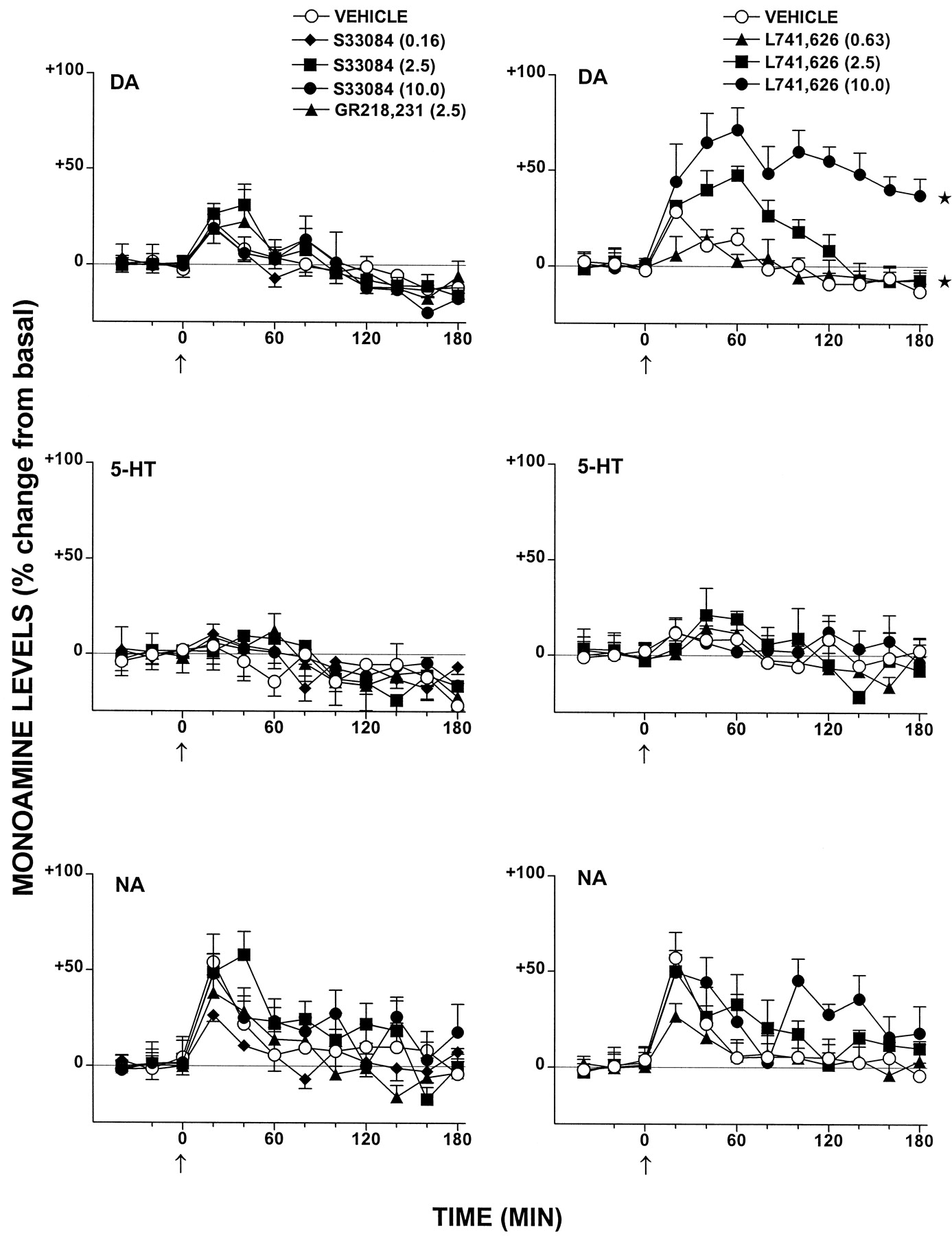
Influence of S33084, GR218,231, and L741,626 on extracellular levels of DA compared with 5-HT and NA in the FCX of freely moving rats. Dialysate levels are expressed as a percentage of basal, preinjection values that were defined as 0%. These values were 1.2 ± 0.1, 0.8 ± 0.1, and 1.4 ± 0.2 pg/20 μl of dialysate for DA, 5-HT, and NA, respectively. Data are mean ± S.E. (n ≥ 5 per value). ANOVA with dose as between-factor and with time as within-factor was performed over 20 to 180 min. DA: influence of S33084, F3,23= 1.0, P > .05; influence of GR218,231,F1,13 = 0.1, P > .05; and influence of L741,626, F3,22 = 20.8, P < .01. 5-HT: influence of S33084,F3,23 = 0.4, P > .05; influence of GR218,231, F1,14 = 0.3, P > .05; and influence of L741,626,F3,22 = 0.2, P > .05. NA: influence of S33084, F3,22 = 0.8, P > .05; influence of GR218,231,F1,12 = 0.1, P > .05; and influence of L741,626, F3,21 = 1.1, P > .05. Asterisks indicate significance (*P < .05) of differences to respective vehicle values in Dunnett's test.

Influence of S33084, GR218,231, and L741,626 on extracellular levels of DA in the nucleus accumbens and striatum of freely moving rats. Dialysate levels are expressed as a percentage of basal, preinjection values that were defined as 0%. These values were 3.5 ± 0.3 and 9.3 ± 0.5 pg/20 μl of dialysate for the nucleus accumbens and striatum, respectively. Data are mean ± S.E. (n ≥ 5 per value). ANOVA with dose as between-factor and with time as within-factor was performed over 20 to 180 min. Striatum: influence of S33084,F1,19 = 0.3, P > .05; influence of GR218,231, F1,19 = 2.5, P > .05; and influence of L741,626,F4,34 = 103.4, P < .01. Accumbens: influence of S33084,F1,19 = 0.5, P > .05; influence of GR218,231, F1,19 = 0.8, P > .05; and influence of L741,626,F4,31 = 16.7, P < .01. Asterisks indicate significance (*P < .05) of differences to respective vehicle values in Dunnett's test.
Effect of Coadministration of S33084 and L741,626 (Fig.7).
It is possible that D2- and D3-receptors might fulfill a complementary, redundant role in the tonic control of DA release. In this case, release of DA via D3-receptor blockade might immediately lead to engagement of colocalized D3-autoreceptors, thereby masking its actions. Therefore, the influence of their concomitant blockade was examined by coadministration of S33084 and L741,626. However, as shown in Fig. 7, even in the presence of L741,626 to block D2-receptors, S33084 failed to elevate FCX levels of DA. Similarly, the facilitatory influence of L741,626 on DA release was not significantly enhanced after pretreatment with S33084. These data do not, thus, reveal any complementary role of D3- with D2-autoreceptors in the tonic control of frontocortical DA release.

Influence of sequential administration of S33084 and L741,626 on DA release in FCX. Dialysate levels are expressed as a percentage of basal, preinjection values that were defined as 0%. Data are mean ± S.E. (n ≥ 5 per value). ANOVA with dose as between-factor and with time as within-factor was performed over 40 to 200 min. A, L741,626 followed by S33084 versus L741,626 and vehicle, F1,14 = 1.1,P > .05. B, S33084 followed by L741,626 versus vehicle and L741,626, F1,8 = 0.1,P > .05.
Influence on Suppression by PD128,907 of Dialysate Levels of DA in FCX (Fig. 8).
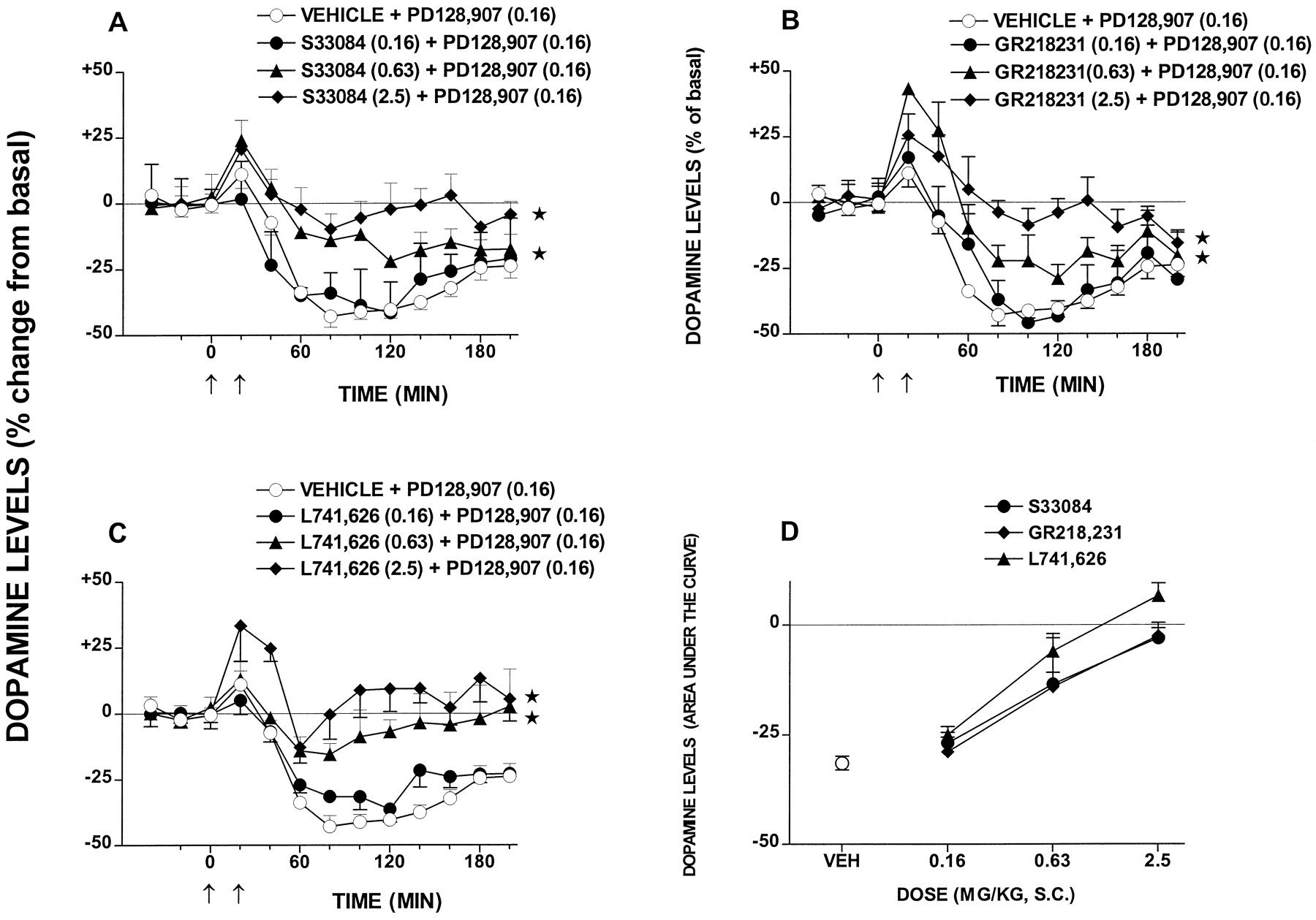
PD128,907 markedly reduced dialysate levels of DA in FCX (Fig. 8) without significantly modifying those of 5-HT or NA (data not shown). This action of PD128,907 was dose dependently attenuated by both S33084 and GR218,231. Similarly, L741,626 dose dependently blocked the action of PD128,907. The ID50 values (95% CL) for blocking the action of PD128,907 were 0.97 (0.43–2.2), 0.95 (0.44–2.07), and 0.53 (0.29–0.95) mg/kg s.c. for S33084, GR218,231, and L741,626, respectively.

Influence of S33084, GR218,231, and L741,626 on the inhibition by PD128,907 of DA release in FCX. A through C, effect of various doses of S33084, GR218,231, and L741,626 on the PD128,907-induced decrease in DA levels. D, dose-response relationships for S33084, GR218,231, and L741,626 (area under the curve analysis) over the whole period of sampling. Dialysate levels are expressed as a percentage of basal, preinjection values that were defined as 0%. Data are mean ± S.E. (n ≥ 5 per value). ANOVA with dose as between factor and with time as within factor was performed over 40 to 200 min. A, F3,25= 24.7, P < .01; B,F3,23 = 21.9, P < .01; and C, F3,22 = 17.8,P < .01. Asterisks indicate significance (*P < .05) of differences to respective vehicle values in Dunnett's test. In D, ANOVA as follows:F2,40 = 2.9, P > .05.
Influence on Inhibition of Dopaminergic Neuron Firing by PD128,907 (Figs. 9 and10).
The electrical activity of VTA-localized dopaminergic neurons was markedly inhibited by PD128,907 at a dose of 0.005 mg/kg i.v. This action of PD128,907 was dose dependently and significantly reduced by S33084 (Fig. 9), but S33084 did not itself modify firing rate (data not shown). Likewise, administered alone, GR218,231 did not modify the activity of dopaminergic neurons (data not shown) but, in its presence, the inhibitory influence of GR218,231 was dose dependently abrogated (Fig.9). L741,626 elicited a dose-dependent and marked acceleration in the firing rate of dopaminergic neurons on administration alone (Fig. 10). The action of PD128,907 was dose dependently reversed by L741,626. The ID50 values (95% CL) for blocking the action of PD128,907 were 0.57 (0.19–1.71), 0.36 (0.13–0.81), and 0.27 (0.09–0.80) mg/kg i.v. for S33084, GR218,231, and L741,626, respectively.

Influence of S33084 and GR218,231 on the inhibition by PD128,907 of electrical activity of dopaminergic neurons in the VTA. Data are mean ± S.E. (n ≥ 5 per value). Top and left, blockade by S33084 of the actions of PD128,907 (0.005 mg/kg i.v.). ANOVA, F3,24 = 7.0,P < .01. Top and right, representative recording of the influence of S33084 (0.25 mg/kg i.v.) on the inhibitory action of PD128,907 (0.005 mg/kg i.v.). Bottom and left, blockade by GR218,231 of the actions of PD128,907 (0.005 mg/kg i.v.). ANOVA,F4,26 = 10.7, P < .01. Bottom and right, representative recording of the influence of GR218,231 (0.25 mg/kg i.v.) on the inhibitory action of PD128,907 (0.005 mg/kg i.v.). Asterisks indicate significance of difference to respective vehicle values. *P < .05.

Influence of L741,626 on the electrical activity of dopaminergic neurons in the VTA. Data are mean ± S.E. (n ≥ 5 per value). Top and left, enhancement of the spontaneous activity of dopaminergic neurons in the VTA. ANOVA,F6,24 = 32.3, P < .01. Top and right, representative dose-response influence of L741,626 on the electrical activity of dopaminergic cell bodies. Bottom and left, blockade by L741,626 of the action of PD128,907 (0.005 mg/kg i.v.). ANOVA, F3,22 = 22.0,P < .01. Bottom and right, representative recording of the influence of L741,626 (0.25 mg/kg i.v.) on the inhibitory action of PD128,907 (0.005 mg/kg i.v.). Asterisks indicate significance of difference to respective vehicle values. *P < .05.
Influence on Cerebral Turnover and Synthesis of DA, NA, and 5-HT (Figs. 11 and12).
S33084 and GR218,231 exerted no significant influence on DOPAC:DA ratios in projection regions of mesocortical pathways (FCX), mesolimbic pathways (accumbens and olfactory tubercle), and the nigrostriatal pathway (striatum). In contrast, L741,626 provoked a dose-dependent and significant elevation in each structure examined, which was most pronounced in the striatum and least marked in the FCX. S33084 also did not significantly modify striatal DA synthesis as determined by accumulation ofl-dopa after inhibition of decarboxylase. Likewise, GR218,231 exerted little influence on striatal DA synthesis. In contrast, striatal DA synthesis was dose dependently increased by L741,626. S33084, GR218,231, and L741,626 exerted no significant influence on striatal and hippocampal 5-HT synthesis and on hippocampal synthesis of NA.

Influence of S33084, GR218,231, and L741,626 on cerebral DA turnover. Induction of DA turnover is expressed as a percentage of basal, preinjection DOPAC/DA ratios that were defined as 0%. Basal levels of DOPAC and DA in nanograms per milligram protein were as follows: accumbens, DA = 94.0 ± 4.0, DOPAC = 40.4 ± 0.5; FCX, DA = 1.1 ± 0.1, DOPAC = 0.8 ± 0.1; olfactory tubercles, DA = 67.2 ± 4.8, DOPAC = 7.7 ± 0.6; and striatum, DA = 128.4 ± 9.2, DOPAC = 12.9 ± 0.8. Data are mean ± S.E. (n≥ 5 per value). ANOVA values are as follows. Frontal cortex: GR218,231, F5,18 = 1.8,P > .05; L741,626,F5,15 = 9.0, P < .01; and S33084, F6,48 = 0.5,P > .05; olfactory tubercles: GR218,231,F5,19 = 1.2, P > .05; L741,626, F5,19 = 20.0,P < .01; and S33084,F6,18 = 0.3, P > .05; nucleus accumbens: GR218,231,F5,19 = 0.7, P > .05; L741,626, F5,19 = 23.2,P < .01; and S33084,F6,18 = 0.5, P > .05; and striatum: GR218,231, F5,25 = 0.1, P > .05; L741,626,F5,19 = 12.2, P < .01; and S33084, F6,55 = 0.1,P > .05. Asterisks indicate significance of difference to respective vehicle values. *P < .05.

Influence of S33084, GR218,231, and L741,626 on cerebral turnover of DA, 5-HT, and NA. Induction of DA, 5-HT, and NA synthesis is expressed as a percentage of basal, preinjection levels of the precursors, l-dopa (DA and NA) and 5-HT (5-HTP). Basal levels of l-dopa in nanograms per milligram protein were 22.2 ± 1.4 and 1.4 ± 0.4 in striatum and hippocampus, respectively, and basal levels of 5-HTP were 2.3 ± 0.1 and 2.7 ± 0.2 in striatum and hippocampus, respectively. Data are mean ± S.E. (n ≥ 5 per value). ANOVA values are as follows. Striatum, l-dopa: S33084,F5,20 = 0.9, P > .05; GR218,231, F5,33 = 6.9,P < .01; and L741,626,F6,38 = 36.0, P < .01. Striatum, 5-HTP: S33084, F5,20 = 2.2, P > .05; GR218,231,F5,33 = 1.1, P > .05; and L741,626, F6,39 = 2.1,P > .05. Hippocampus, l-dopa: S33084,F5,20 = 0.7, P > .05; GR218,231, F5,34 = 0.7,P > .05; and L741,626,F6,39 = 0.4, P > .05. Hippocampus, 5-HTP: S33084, F5,20= 0.2, P > .05; GR218,231,F5,33 = 0.7, P > .05; and L741,626, F6,39 = 1.7,P > .05. Asterisks indicate significance of difference to respective vehicle values. *P < .05.
Discussion
Receptor Profile.
The pronounced affinity and selectivity of S33084 at hD3- versus D2-receptors is underpinned by studies of [3H]S33084 that binds with high affinity (Kd = 9.7) to hD3-receptors (Cussac et al., 2000a). S33084 likewise displays high affinity and (>100-fold) selectivity for cloned and native, rat D3- versus D2-receptors (Cussac et al., 2000b). These observations, and the marked (>100-fold) selectivity of S33084 versus all (>30) receptors examined, underline its utility for exploration of the functional role of D3-receptors. Indeed, S33084 is substantially more selective than other D3 antagonists characterized in vivo (Wustrow and Wise, 1997; Audinot et al., 1998). Furthermore, although several selective antagonists at hD3- versus hD2-receptors were recently documented, their functional actions in vivo remain to be examined (Boyfield et al., 1997; Yuan et al., 1998; Austin et al., 1999). GR218,231 was originally characterized with two different cell lines (Murray et al., 1996) and, with a common CHO expression system, the present investigation confirms its marked selectivity for hD3- versus hD2-receptors. Similarly, we extend previous studies demonstrating the preference of L741,626 for hD2-receptors (Kulagowski et al., 1996).
Antagonist Properties at hD3- versus hD2-Receptors.
Activation of hD3-receptors enhances [35S]GTPγS binding to G-proteins (Missale et al., 1998; Newman-Tancredi et al., 1999; Vanhauwe et al., 1999), and this response was potently suppressed by S33084, demonstrating antagonist properties at hD3 sites. GR218,231 behaved similarly. Confirming their selectivity, only markedly higher concentrations blocked [35S]GTPγS binding at hD2-receptors. Furthermore, S33084 displaced the DA stimulation-response curve for induction of [35S]GTPγS binding at hD3-receptors without compromising its maximal effect, yielding a pA2 (9.7) close to its pKi (9.6), and demonstrating competitive interaction with hD3 sites. In contrast to S33084, L741,626 more potently suppressed hD2- versus hD3-receptor-mediated [35S]GTPγS binding, revealing its opposite preference for hD2 sites. Downstream of G-protein coupling, hD3-receptors activate MAP kinase (Cussac et al., 1999) and S33084 and GR218,231 both abolished DA-stimulated MAP kinase. Inasmuch as this parameter is highly sensitive, this finding underpins “pure” antagonist properties of S33084 and GR218,231 at hD3-receptors, an interpretation supported by in vivo studies (Results; Millan et al., 2000).
In Vivo Actions at D3- versus D2-Receptors.
Although the above-mentioned observations demonstrate the striking selectivity of S33084 (and GR218,231) for hD3- versus hD2-receptors in vitro, and a marked preference of L741,626 for hD2- versus hD3-receptors, the question arises concerning their actions in vivo. Drug selectivity may best be established in vivo by determination of active dose ranges in well-defined models reflecting activity at specific receptors. However, although functional models of activity at D2 sites are available (see below; Millan et al., 2000), no functional response in vivo has, as yet, been unambiguously attributed to D3-receptors. This difficulty, common to all studies of D3-receptor function, encourages caution in the interpretation of actions of even highly selective ligands, such as S33084. Nevertheless, it is reasonable to make the following inferences. First, in accordance with their 100-fold higher affinities versus L741,626 at D3-receptors, S33084 and GR218,231 should be ∼100-fold more potent than L741,626 in models exclusively mediated by D3-receptors. Second, as discussed in Bristow et al. (1997) and Millan et al. (1998)for selective D4-receptor antagonists, one may exploit residual (“surrogate”) actions of S33084 and GR218,231 at D2-receptors for an estimation of doses at which they should selectively block D2-receptors. Thus, based on weak actions of S33084 and GR218,231 in (certain) models involving D2-receptors (Millan et al., 2000), any effects in models reflecting only D3-receptor activation should be seen at ∼100-fold lower doses or ∼0.1 mg/kg s.c. These estimations provide a framework for cautious interpretation of the actions of S33084.
Autoreceptor Modulation of Dopaminergic Transmission.
Dopaminergic neurons are tonically inhibited by dendritic and terminal autoreceptors, operating in interaction with DA transporters (Gobert et al., 1995; Koeltzow et al., 1998; L'hirondel et al., 1998; Dickinson et al., 1999). Dopaminergic neurons display a high density of D2-receptors, whereas D3-(auto)receptors are present in only low concentrations (Levant, 1997; Tepper et al., 1997; Suzuki et al., 1998). Nevertheless, transduction mechanisms potentially permitting an inhibitory role of D3 sites have been identified (Werner et al., 1996; Kuzhikandathil and Oxford, 1999; Liu et al., 1999).
Tonic Control of Cerebral DA Synthesis.
The observation that S33084 and GR218,231 do not modify cerebral DA synthesis supports studies of D3-receptor-deficient mice (Koeltzow et al., 1998; Jung et al., 1999) in suggesting that D3-receptors do not play a major role in the modulation of DA turnover. Nevertheless, antisense probes neutralizing D3-receptors increased nucleus accumbens DA synthesis (Nissbrandt et al., 1995) and, in knockout mice lacking D2- and D3-receptors,Jung et al. (1999) observed a more pronounced increase in striatal DA turnover than in D2-receptor-deficient counterparts. These observations suggest that D3-autoreceptors might play a minor role, complementary to that of D2-autoreceptors, in modulation of DA synthesis (Gobert et al., 1995). Correlation analyses with (nonselective) dopaminergic antagonists suggested a role of D2 sites in the tonic control of cerebral DA synthesis (Gobert et al., 1995; Gainetdinov et al., 1996), and a major, tonic, inhibitory influence of D2-autoreceptors was revealed herein with L741,626. This finding is important because D2-receptor-deficient mice display no consistent alterations in levels of DA or tyrosine hydroxylase, presumably due to compensatory mechanisms, including enhanced translation of D3-receptor mRNA and alterations in DA clearance via the DA transporter (Kelly et al., 1998; Dickinson et al., 1999;Jung et al., 1999).
Tonic Control of Mesolimbic and Striatal DA Release.
Neither S33084 nor GR218,231 elevated resting extracellular levels of DA in nucleus accumbens or striatum, suggesting that D3-autoreceptors do not play a prominent role in tonic control of DA release herein. Furthermore, although transgenic mice lacking D3-receptors, and rats treated with D3-receptor antisense, showed enhanced spontaneous DA release in the accumbens (Ekman et al., 1998), Koeltzow et al. (1998) proposed that this effect reflects a short-loop, feedback control of mesolimbic DA release via postsynaptic D3 sites. In contrast to S33084 and GR218,231, L741,626 markedly enhanced dialysis levels of DA, demonstrating that D2-autoreceptors tonically regulate limbic and striatal DA release. Previous studies of the interaction of L741,626 with dopaminergic agonists (Bowery et al., 1996) likewise suggested a role of D2-autoreceptors in phasic control of striatal DA release. Interestingly, no alterations in basal, striatal DA release were observed in D2-receptor-deficient mice, presumably due to compensatory mechanisms (L'hirondel et al., 1998; Dickinson et al., 1999). Thus, the present facilitatory influence of L741,626 on DA release and synthesis underlines the continuing importance of pharmacological tools for elucidation of potential roles of D2- (and D3-) receptors.
Tonic and Phasic Control of Mesocortical DA Release.
The lack of influence of S33084 and GR218,231 versus L741,6216 on resting frontocortical levels of DA suggest that D3-receptors do not, in contrast to their D2 counterparts, tonically regulate DA release in FCX. It is unlikely that this inactivity of S33084 and GR218,231 reflects release of DA onto colocalized D2-autoreceptors because, in the presence of L741,626, S33084 still failed to increase frontocortical DA levels. However, in analogy to S14297 (Gobert et al., 1995), S33084 and GR218,231 dose dependently blocked the inhibitory influence of PD128,907 on DA levels. There are several possible interpretations. First, there may be a selective implication of D3-receptors in the phasic control of frontocortical DA release. This is, however, unlikely because active doses of S33084 and GR218,231 were superior to those estimated as D3-receptor selective (vide supra). Furthermore, L741,626 was active at doses similar to those of S33084 and GR218,231. Nevertheless, D3-autoreceptor isoforms (splice variants or post-translationally modified) may differ from postsynaptic populations (Jung et al., 1999), and affinities of S33084 and GR218,231 versus L741,626 for (terminal) D3-(auto)receptors controlling DA release in rat FCX may differ from hD3-receptors characterized in CHO cells. Second, actions of S33084 and GR218,231 might reflect blockade of D2-receptors. However, this can be largely ruled out because 1) doses of S33084 and GR218,231 inhibiting PD128,907 were not significantly greater than those of L741,626; 2) in contrast to L741,626, basal DA release in FCX was not affected by S33084 or GR218,231; and 3) over this dose range, S33084 and GR218,231 do not block other D2-receptor-mediated responses (Millan et al., 2000). A third possibility is that actions of S33084 and GR218,231 reflect physical and/or functional interactions between D3- and D2-autoreceptors either at the recognition site or intracellular level (Missale et al., 1998). And last, regarding a possible role of both D2- and D3-autoreceptors, previous studies provided evidence for two distinct classes of dopaminergic autoreceptor modulating cerebral DA release (Pizzi et al., 1993; Patel et al., 1994). Thus, the present data with L741,626 demonstrate a role of D2-autoreceptors in the phasic control of DA release in FCX, but further study is required to characterize the effect of S33084 and the potential implication of D3-receptors.
Electrical Activity of VTA Dopaminergic Perikarya.
The lack of modification by S33084 and GR218,231 of the electrical activity of VTA dopaminergic perikarya concurs with a lack of change in D3-receptor-deficient mice and rats treated with D3 antisense probes (Tepper et al., 1997;Koeltzow et al., 1998) in indicating that D3-receptors do not tonically control the electrical activity of dopaminergic perikarya. In contrast, firing rate was accelerated by L741,626, revealing the tonic inhibitory role of D2-autoreceptors. For phasic actions, in analogy to S14297 (Lejeune and Millan, 1995), S33084 and GR218,231 attenuated the inhibitory influence of PD128,907 on VTA neurons, raising the possibility of a phasic role of D3 sites. The high doses of S33084 and GR218,231 required suggest, however, caution in the interpretation of these actions (see above). Nevertheless, antisense probes to D2- and D3-receptors additively attenuated the inhibitory influence of apomorphine on the electrical activity of substantia nigra dopaminergic neurons (Tepper et al., 1997), suggesting a cojoint role of D3 and D2 sites. For the phasic role of D2-autoreceptors, L741,626 blocked the inhibitory influence of PD128,907 on VTA-localized dopaminergic neurons herein and it prevented the inhibitory influence of PD128,907 at VTA- and substantia nigra-localized dopaminergic neurons in vitro (Bowery et al., 1996). Furthermore, the inhibitory influence of DA on dopaminergic perikarya is absent in D2-receptor-deficient mice (Mercuri et al., 1997), whereas PD128,907 is equipotent in inhibiting VTA- and substantia nigra-localized dopaminergic neurons in wild-type versus D3-receptor knockout mice (Koeltzow et al., 1998).
Potential Inverse Agonist Actions.
One possible explanation for a lack of intrinsic influence of S33084 and GR218,231 on dopaminergic transmission might be that they behave as “neutral antagonists” rather than “inverse agonists” at D3-(auto)receptors. However, evidence for constitutive activity at hD3 sites is contradictory (Griffon et al., 1997; Malmberg et al., 1998; Cussac et al., 1999; Newman-Tancredi et al., 1999; Vanhauwe et al., 1999) so this seems unlikely. For the “tonic” role of D2-receptors, if “inverse agonist” properties (Nilsson et al., 1996; Hall and Strange, 1997) were required to enhance dopaminergic transmission, this could explain 1) the lack of influence of D2 antisense probes and null mutations for D2-receptors on dopaminergic pathways (vide supra), and 2) increases in dopaminergic transmission of contrasting magnitude (proportional to “negative efficacy”) by various “antagonists” (Gobert et al., 1995). However, inverse agonist properties of L741,626 have not, to date, been demonstrated and unambiguous proof of the significance of inverse agonist actions at D2 sites in vivo is needed (Millan et al., 1999). The present interpretation that L741,626 enhances dopaminergic transmission by interrupting tonic activity at D2-autoreceptors seems, then, more parsimonious.
Serotonergic and Adrenergic Transmission.
Although fragmentary data suggest that dopaminergic mechanisms modulate adrenergic and serotonergic transmission (Rossetti et al., 1989; Suzuki et al., 1998;Adell and Artigas, 1999), S33084, GR218,231, L741,626, and PD128,907 failed to modify release and turnover of 5-HT and NA herein. Alterations in 5-HT and NA turnover have likewise not been reported in D3- or D2-receptor-deficient mice (Koeltzow et al., 1998; Dickinson et al., 1999). D2- and D3-receptors do not, thus, exert a pronounced influence on serotonergic or adrenergic pathways.
Conclusions
In conclusion, S33084 is a potent, selective, and competitive antagonist at D3-receptors which, together with GR218,231 and L741,626, should be of considerable use for exploration of their pathophysiological significance. Based on actions of these ligands, D2- but not D3-autoreceptors fulfill a major role in the tonic inhibition of dopaminergic transmission. Nevertheless, the influence of S33084 on dopaminergic pathways, and the possibility that D3-(auto)receptors contribute to the phasic modulation of DA release, justify further examination. In this regard, although this study focused on the FCX, it would be of interest to examine a potential role of D3-receptors in structures such as the Isles of Calleja or subregions of the nucleus accumbens that are enriched in D3-receptors (Levant, 1997).
Acknowledgments
We thank C. Langaney for secretarial assistance and C. Chaput, L. Cistarelli, C. Melon, V. Pasteau, and M. Touzard for technical assistance.
Footnotes
-
Send reprint requests to: Dr. Mark J. Millan, Institut de Recherches Servier, Centre de Recherches de Croissy, Psychopharmacology Department, 125 Chemin de Ronde, 78290 - Croissy-sur-Seine, France.
- Abbreviations:
- DA
- dopamine
- L741,626
- 4-(4-chlorophenyl)-1-(1H-indol-3-ylmethyl)piperidin-4-ol
- 7-OH-DPAT
- 7-hydroxy-2-dipropylaminotetralin
- 5-HT
- 5-hydroxytryptamine (serotonin)
- MAP
- mitogen-activated protein
- GR218,231
- 2(R,S)-(di-n-propylamino)-6-(4-methoxyphenylsulfonyl methyl)-1,2,3,4-tetrahydronaphthalene
- S33084
- (3aR, 9bS)-N-[4-(8-cyano-1,3a,4,9b-tetrahydro-3H-benzopyrano[3,4-c]pyrrole-2-yl)-butyl]-(4-phenyl) benzamide
- GTPγS
- guanosine-5′-O-(3-thio)triphosphate
- CHO
- Chinese hamster ovary
- ERK
- extracellular signal receptor-activated kinase
- VTA
- ventrotegmental area
- NA
- noradrenaline
- 5-HTP
- 5-hydroxytryptophan
- DOPAC
- dihydroxyphenylalaninecarboxylic acid
- FCX
- frontal cortex
- PD128,907
- (+)-(4aR,10bR)-3,4,4a,10b-tetrahydro-4-propyl-2H,5H-[1]benzopyrano-[4,3-b]-1,4-oxazin-9-ol
- U99194
- (5,6-dimethoxy-indan-2-yl)dipropylamine
- GR103,691
- 4′-acetyl-N-{4-[(2-methoxy-phenyl)-piperazin-1-yl]-butyl}-biphenyl-4-carboxamide
- S14297
- (+)-[7-(N-N-dipropylamino)-5,6,7,8-tetrahydro-naphto(2,3b)dihydro-2,3-furane]
- NSD1015
- m-hydroxybenzylhydrazine
- Received October 25, 1999.
- Accepted February 21, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}