


Abstract
Substance P (SP) participates in acute intestinal inflammation via binding to the G-protein-coupled neurokinin-1 receptor (NK-1R) and release of nuclear factor κ B (NF-κB)-driven proinflammatory cytokines from colonic epithelial cells. However, the signal transduction pathways by which SP-NK-1R interaction induces NF-κB activation and interleukin-8 (IL-8) production are not clear. Here, we examined participation of protein kinase C (PKC) in SP-induced IL-8 production in human nontransformed NCM460 colonocytes stably transfected with the human NK-1R (NCM460-NK-1R cells). SP (10-7 M) induced an early (1 min) phosphorylation of the PKC isoforms PKCδ, PKCθ, and PKCϵ, followed by I-κB kinase, IκBα, and p65 phosphorylation. Depletion of PKC by phorbol-12-myristate-13-acetate (10 μM) blocked SP-induced IκBα and p65 phosphorylation and IL-8 production. The PKCδ inhibitor rottlerin at a low concentration (1 μM), but not pseudosubstrate PKCθ and PKCϵ inhibitors (10 μM), significantly reduced IL-8 secretion. PKCδ silencing by RNA interference reduced PKCδ protein expression and SP-induced PKCδ phosphorylation that was associated with diminished IL-8 promoter and NF-κB luciferase activities in response to SP. Moreover, overexpression of wild-type PKCδ increased SP-induced IL-8 promoter- and NF-κB-driven luciferase activities that were rottlerin-sensitive. We conclude that PKCδ plays an important role in SP-induced proinflammatory signaling in human colonocytes.
Substance P (SP), an 11-amino acid neuropeptide member of the tachykinin family, is localized in the central nervous system (Mantyh, 2002), sensory neurons (Maggi, 1990), enteric nerves (Costa et al., 1982), and immune cells (Bost, 1995). Interaction of SP with its high-affinity neurokinin-1 receptor (NK-1R) mediates several important intestinal functions, such as motility (Holzer and Holzer-Petsche, 1997), mucosal permeability (Pothoulakis et al., 1994), chloride secretion (Riegler et al., 1999), and colonic epithelial cell proliferation (Castagliuolo et al., 2000; Koon et al., 2004). Several studies also confirm a major proinflammatory role for SP and the NK-1R in the pathogenesis and progress of intestinal inflammation of different etiologies (Pothoulakis et al., 1994; Mantyh et al., 1996; Stucchi et al., 2000). The proinflammatory responses to SP involve activation of the nuclear factor (NF)-κB (Lieb et al., 1997; Azzolina et al., 2003) and secretion of proinflammatory cytokines, including interleukin-6, interleukin-8 (IL-8), and tumor necrosis factor α (TNFα) (Laurenzi et al., 1990; Derocq et al., 1996; Lieb et al., 1996; Castagliuolo et al., 1997; Fiebich et al., 2000; Zhao et al., 2002). Interestingly, proinflammatory cytokines and NF-κB also regulate expression of NK-1R (Simeonidis et al., 2003).
Ligand binding to NK-1R stimulates phosphoinositide hydrolysis (Rollandy et al., 1989), mobilizes Ca2+ (Pradier et al., 1993), and activates mitogen-activated protein kinases (MAPKs) (Luo et al., 1996). SP stimulation also activates MAPKs and causes cell proliferation in human astrocytoma and colonic epithelial cells via pathways involving transactivation of epidermal growth factor receptor (EGFR) (Castagliuolo et al., 2000; Koon et al., 2004). Yang et al. (2002) reported that in human tracheal smooth muscle cells, SP stimulates MAPKs associated with cell proliferation through the protein kinase C (PKC) pathway. In spiral ganglion neurons, PKCs may also interact with Ca2+ mobilization to induce MAPK activation in response to SP (Lallemend et al., 2003). The PKC superfamily is classified into three subfamilies based on their domain structure and their ability to respond to Ca2+ and DAG (Newton and Johnson, 1998). Although the conventional PKC isoforms α, βI, βII, and γ are regulated by Ca2+ and DAG, the novel PKC isoforms δ, ϵ, η, and θ respond to DAG, but do not involve Ca2+. The third group of PKC isoforms includes the atypical PKC isoforms ζ, λ/ζ, and μ, which are regulated by Ca2+ or DAG. It was previously shown that, among these PKCs, SP could transiently induce tyrosine phosphorylation of PKCδ in rat parotid acinar cells (Soltoff and Toker, 1995) and Ser729 phosphorylation of PKCϵ in U373 MG human astrocytoma cells and primary rat astrocytes (Lieb et al., 2003). However, the physiological significance of activation of these PKCs is not known yet.
In addition to the MAPK pathway and protein kinase C activation, SP also activates the NF-κB pathway, which triggers release of IL-8 production in both U373 MG cells (Lieb et al., 1997) and human colonic epithelial cells transfected with NK-1R (Zhao et al., 2002). Although different isoforms of PKCs have been associated with the NF-κB pathway in response to various extracellular stimuli, whether PKCs are involved in SP's proinflammatory signaling is not known. In this study, we sought to investigate whether SP activates PKCs in nontransformed human colonic epithelial NCM-460 cells overexpressing NK-1R and examine their involvement in SP-stimulated NF-κB activation and IL-8 gene expression. We used colonocytes overexpressing NK-1R because previous studies demonstrated increased NK-1R mucosal expression in intestinal inflammation in both animal models (Castagliuolo et al., 1998; Pothoulakis et al., 1998; Stucchi et al., 2003; Weinstock et al., 2003) and humans with inflammatory bowel disease (Goode et al., 2000; Renzi et al., 2000). Our results present novel evidence that SP rapidly and persistently induces threonine phosphorylation of PKCδ, and this activation is critical for SP-induced NF-κB activation and IL-8 gene expression.
Materials and Methods
Cell Cultures. Nontransformed human colonic epithelial NCM460 cells overexpressing NK-1R (NCM460-NK-1R) were made as previously described by us (Zhao et al., 2002). NCM460-NK-1R cells were used in studies investigating both the proinflammatory (Zhao et al., 2002) and proliferative (Koon et al., 2004) responses to SP. Cells were cultured in M3D medium (INCELL Corporation, San Antonio, TX) containing 10% fetal calf serum (Invitrogen, Carlsbad, CA) and 1% penicillin/streptomycin (Invitrogen) solution.
Pharmacological Experiments. NCM460-NK-1R cells were seeded in 12-well plates (2 × 106 cells/well) overnight in M3D medium containing 10% fetal calf serum and 1% penicillin/streptomycin (Invitrogen). Cells were pretreated with rottlerin (0.1–1 μM mallotoxin), caffeic acid phenethyl ester (CAPE; 2–10 μg/ml), specific PKCθ pseudosubstrate peptide inhibitor (10 μM Myr-LHQRRGAIKQAKVHHVKC-NH2), and PKCϵ pseudosubstrate peptide inhibitors (10 μM EAVSLKPT) for 30 min or with phorbol-12-myristate-13-acetate (PMA; 10 μM) overnight (Calbiochem, San Diego, CA). Cells were then stimulated with SP or 0.1% trifluorocetic acid (vehicle control) for various time points in Western blot experiments and 4 h in ELISA and luciferase experiments. Trifluorocetic acid had no effect on IL-8 secretion or PKC-NF-κB signaling (data not shown).
IL-8 Measurements. IL-8 protein levels in conditioned media were determined by double-ligand ELISA using goat anti-(human IL-8) (R&D Systems, Minneapolis, MN) as previously described (Zhao et al., 2002). Results were expressed as mean ± S.E.M. (nanograms per milliliter).
Western Blot Analyses. SP-treated cells were lysed in 1× lysis buffer (62.5 mM Tris-HCl, 2% SDS, 10% glycerol, 0.01% bromophenol blue, and 1% 2-mercaptoethanol). Equal amounts of cell extracts were fractionated by 10% SDS-PAGE, and proteins were transferred onto nitrocellulose membranes (Bio-Rad, Hercules, CA) at 400 milliamps for 2 h at 4°C. Membranes were blocked in 5% nonfat milk in Tris-buffered saline/Tween 20 (50 mM Tris, pH 7.5, 0.15 M NaCl, and 0.05% Tween 20) and then incubated with antibodies against phospho-PKCϵ (Ser729) (Santa Cruz Biochemicals, Santa Cruz, CA), β-actin (Sigma-Aldrich, St. Louis, MO), phospho-PKCδ (Thr505), phospho-PKCθ (Thr538), phospho-IκB kinase (IKKα/β Ser180/Ser181), phospho-p65 (Ser536), and phospho-IκBα (Ser32/36) (Cell Signaling Technology Inc., Beverly, MA). Horseradish peroxidase-labeled antibodies were detected by enhanced chemiluminescence (Pierce Chemical, Rockford, IL). The image of the signal was exposed to X-ray film (Fujifilm, Tokyo, Japan).
PKCδ and p65 Knockdown by siRNA Transfection. Cells were seeded in 12-well plates (1 × 105 cells/well) overnight and transiently transfected with equal amounts of PKCδ siRNA, p65 siRNA, or control siRNA (Santa Cruz Biochemicals) using X-tremeGENE Transfection Reagent (Roche Applied Science, Indianapolis, IN) according to the manufacturer's instructions. Specific down-regulation of endogenous PKCδ/p65 protein expression was confirmed by Western blot analyses using a PKCδ/p65-specific antibody and the β-actin antibody.
Luciferase Assay. Cells were seeded in 12-well plates (2 × 105 cells/well) overnight and transiently cotransfected using Lipofectamine 2000 transfection reagent (Invitrogen) with siRNA of the PKCδ, p65, or control siRNA (Santa Cruz Biochemicals). In the same experiments, cells were also cotransfected with an IL-8 promoter luciferase construct, an NF-κB luciferase construct (a gift from Dr. Tom Mitchell and Dr. Bill Sugden, University of Wisconsin, Madison, WI), or with a control luciferase construct (pRL-TK; Promega, Madison, WI). For cotransfection experiments with a wild-type PKCδ-overexpressing plasmid (American Type Culture Collection, Manassas, VA) or its negative control, pCMV-β galactosidase plasmid (backbone of PKCδ-overexpressing construct) (BD Biosciences Clontech, Palo Alto, CA), the Effectene transfection reagent (QIAGEN, Valencia, CA) was used. Transfected cells were serum-starved for 24 h, followed by exposure to SP for 4 h. Firefly and Renilla luciferase activities in cell extracts were measured using a dual-luciferase reporter assay system (Promega). The relative luciferase activity was then calculated by normalizing IL-8 promoter/NF-κB driven firefly luciferase activity to control Renilla luciferase activity. Results are expressed as mean ± S.E.M. (n = 6).
Statistical Analyses. ELISA and luciferase assay results were analyzed using the Prism professional statistics software program (GraphPad Software Inc., San Diego, CA). Analyses of variance were used for intergroup comparisons.
Results
SP Induces Phosphorylation of PKCδ, PKCϵ, and PKCθ. Since SP can transiently stimulate PKCδ tyrosine (Soltoff and Toker, 1995) and PKCϵ serine phosphorylation (Lieb et al., 2003), we first examined whether SP activates these as well as other PKC members in human colonic epithelial cells by Western blot analyses using phospho-specific PKC antibodies. To do this, NCM460-NK-1R cells were stimulated with SP (10-7 M) for 1 to 60 min, and equal amounts of cell protein were used to determine phosphorylation of various PKCs. We found that SP potently induced PKCδ threonine phosphorylation by 1 min that remained activated through 60 min (Fig. 1A). PKCδ phosphorylation was dose-dependent, with detectable SP induction at 10-9 M (Fig. 1B). SP also induced transient PKCϵ phosphorylation within 1 to 5 min as well as PKCθ phosphorylation at 1 to 2 min and returned to basal levels after 2 min, whereas the β-actin signal indicates equal protein loading (Fig. 1A). However, SP did not induce phosphorylation of other PKCs including PKCα, PKCβ, PKCλ, PKCξ, and PKD/PKCμ (data not shown).

SP induces phosphorylation of PKCδ, PKCϵ, and PKCθ in human nontransformed colonocytes expressing NK-1R. A, serum-starved NCM460-NK-1R cells were exposed to SP (10-7 M) for the indicated time points. Cells were lysed, and equal amounts of protein were fractionated on 10% SDS-PAGE gels to determine the levels of phospho-PKCδ, phospho-PKCϵ, phospho-PKCθ, and β-actin. B, serum-starved NCM460-NK-1R cells were exposed to the indicated concentrations of SP for 2 min. Cells were lysed, and equal amounts of protein were fractionated on 10% SDS-PAGE to determine the levels of phospho-PKCδ and β-actin. Results are representative of three independent experiments.
SP Stimulates Phosphorylation of IKKs, IκBα, and p65 Subunit. To find out whether rapid activation of PKCδ, PKCϵ, and PKCθ is kinetically followed by activation of signaling molecules involved in the NF-κB pathway, the same time course samples were also subjected to Western blot analyses using antibodies against phosphorylated IKKs, IκBα, and NF-κB p65. The results indicate that SP (10-7 M) induced subsequent phosphorylation of IKKs, IκBα, and p65 at 5 to 10 min after SP stimulation (Fig. 2). These results suggest that activation of the NF-κB pathway is downstream of PKCδ, PKCϵ, and PKCθ phosphorylation in response to SP.
PKCδ Is Involved in SP-Induced NF-κB-Mediated IL-8 Gene Expression. Previous studies have shown that SP-induced IL-8 gene expression is NF-κB-dependent (Lieb et al., 1997; Zhao et al., 2002). To further confirm this, NCM460-NK-1R cells were pretreated with 2 to 10 μg/ml CAPE, a specific NF-κB pharmacologic inhibitor, and then treated with SP for 4 h. Our data showed that CAPE inhibited SP-induced IL-8 secretion in a dose-dependent manner (Fig. 3A).
To confirm whether SP-induced IL-8 transcription is NF-κB-dependent by a molecular approach, we used an NF-κB p65-specific siRNA to reduce expression of p65 protein. Compared with its control, transfection of the p65 siRNA significantly reduced SP-induced IL-8 promoter activity (Fig. 3BI) and NF-κB-dependent reporter gene expression (Fig. 3BII). To validate that the inhibitory effects of the p65 siRNA is mediated via specific inhibition of endogenous p65, lysates from transfected cells were used to determine the NF-κB p65 protein expression. Our results showed that the p65 siRNA dramatically reduced endogenous NF-κB protein expression, as well as SP-induced NF-κB phosphorylation, but had no influence to β-actin expression, confirming equal protein loading (Fig. 3C).
To determine whether SP-stimulated NF-κB-dependent IL-8 expression involves activation of PKCs, we first applied prolonged treatment with the PKC activator PMA that depletes protein expression of various PKCs, including PKCδ. Overnight PMA treatment led to high amounts of IL-8 in the media, even without SP stimulation (Fig. 3D), whereas addition of SP did not produce any further increase in IL-8 levels. When the PMA conditioned medium was replaced by fresh medium at 1 h before SP stimulation to remove residual IL-8, IL-8 levels returned to basal levels. Moreover, after the depletion of PKC, SP failed to significantly increase IL-8 levels, indicating that PKC depletion completely inhibited SP-induced IL-8 secretion (Fig. 3B).
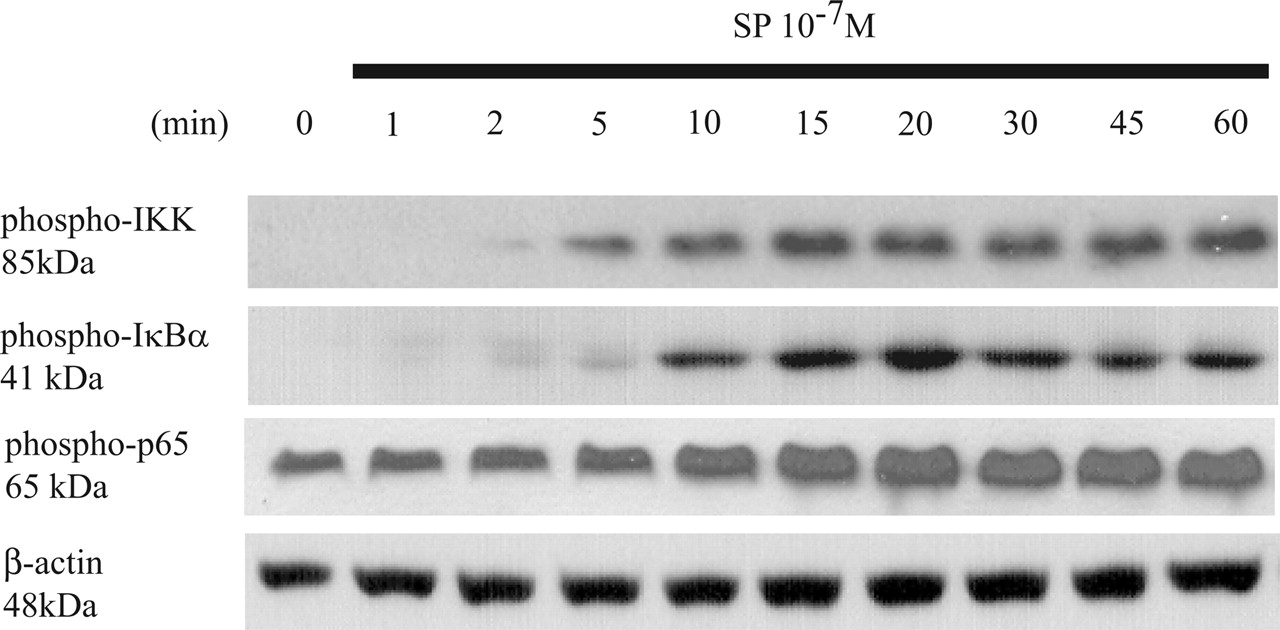
SP stimulates IKK, IκBα, and p65 subunit phosphorylation. Serum-starved NCM460-NK-1R cells were exposed to SP (10-7 M) for the indicated time points. Cells were lysed, and equal amounts of protein were fractionated on 10% SDS-PAGE to determine the levels of phospho-IKK, phospho-IκBα, phospho-p65, and β-actin. Results are representative of three independent experiments.

SP stimulates NF-κB-mediated IL-8 gene expression. A, NCM460-NK-1R cells were incubated in serum-free M3D media for 24 h and then pretreated with CAPE (2–10 μg/ml) for 30 min, followed by SP (10-7 M) stimulation for 4 h. B, NCM460-NK-1R cells were transiently transfected with siRNAs of NF-κB or negative control together with IL-8 promoter or NF-κB firefly reporter plasmids. All cells were also transfected with control pRL-TK plasmids for Renilla luciferase signal. The transfected cells were serum-starved overnight and treated with SP (10-7 M) for 4 h. Cell extracts were prepared to measure IL-8 promoter (BI) or NF-κB (BII) luciferase activity. Data are expressed as mean percentage relative firefly luciferase activity (normalized to control group without SP stimulation, which was normalized as 100%) ± S.E.M. and are representative of six independent samples. **, P < 0.01; ***, P < 0.001. C, p65 siRNA or control siRNA-transfected NCM460-NK-1R cells were stimulated with SP (10-7 M) for 15 min and then lysed. Equal amounts of protein were fractionated on 10% SDS-PAGE to determine the levels of phospho-p65, p65, and β-actin. Results are representative of three independent experiments. D, serum-starved NCM460-NK-1R cells were pretreated with PMA (10 μM). After 24 h, the medium was either left unchanged or replaced with fresh serum-free M3D media for 1 h, followed by SP (10-7 M) stimulation for 4 h. Conditioned media were collected and IL-8 levels were measured by ELISA. Data are expressed as means ± S.E.M. (n = 6). **, P < 0.01; ***, P < 0.001.
Next, we determined whether PKCθ or PKCϵ are involved in SP-stimulated NF-κB-dependent IL-8 synthesis by pretreating NCM460-NK-1R cells with PKCθ and PKCϵ pseudosubstrate peptide inhibitors (10 μM) for 30 min, followed by SP stimulation for 4 h. Our results showed that the PKCθ and PKCϵ pseudosubstrate peptide inhibitors had no significant effect in SP-induced IL-8 production (Fig. 4A). To validate the inhibitory effects of these pseudosubstrate inhibitors, Western blot analyses were used to measure SP-induced PKCθ and PKCϵ protein phosphorylation, compared with their nonphosphorylated proteins and β-actin. As shown in Fig. 4B, both these inhibitors significantly reduced SP-induced phosphorylation of PKCθ and PKCϵ, without affecting their protein expression levels. These results indicate that PKCθ and PKCϵ are not involved in SP-induced IL-8 synthesis.
To determine the role of PKCδ in SP-induced IL-8 production, we pretreated cells with a PKCδ-specific inhibitor, rottlerin, at relative low concentrations (0.1–1 μM). Previous results indicated that an inhibitory dose of rottlerin as low as 1 μM is PKCδ selective (Wang et al., 2003). Our results showed that SP-stimulated IL-8 secretion was significantly inhibited by rottlerin at 0.1 μM and substantially diminished at 1 μM (Fig. 5A). Next, we examined whether the effect of PKCδ inhibition on SP-induced NF-κB activation involves PKCδ. NCM460-NK-1R cells were pretreated with rottlerin (0.1 to 1 μM) and treated with SP for either 2 (to detect phosphorylation of PKCδ) or 15 (to detect phosphorylation of IKK and p65) min. The results show that rottlerin (1 μM) selectively inhibited SP-induced phosphorylations of PKCδ, IKK, and p65 in a dose-dependent fashion (Fig. 5B).
To further confirm the role of PKCδ in mediating SP-induced IL-8 expression, cells were transiently cotransfected with a PKCδ-specific siRNA or its control siRNA along with either IL-8 promoter or NF-κB luciferase constructs. Compared with its control siRNA, this PKCδ siRNA significantly reduced both basal and SP-induced IL-8 promoter activity (Fig. 6A) and NF-κB driven luciferase expression (Fig. 6B). To validate the specific inhibitory effects of PKCδ siRNA on the endogenous PKCδ expression, cells were also transiently transfected with this PKCδ siRNA or its control siRNA as described under Materials and Methods, and then equal amounts of cell protein were used to determine endogenous PKCδ protein expression. Our results showed that PKCδ silencing dramatically reduced the endogenous PKCδ protein expression as well as SP-induced PKCδ phosphorylation but had no effect on β-actin expression (Fig. 6C).

PKCδ, but not other PKCs, mediates SP-induced IL-8 secretion in human nontransformed colonocytes expressing NK-1R. A, serum-starved NCM460-NK-1R cells were pretreated with specific PKCθ and PKCϵ pseudosubstrate peptide inhibitors (10 μM) for 30 min, followed by SP (10-7 M) stimulation for 4 h. Conditioned media were collected for IL-8 ELISA measurements. Data are expressed as means ± S.E.M. and are representative of six independent samples. B, serum-starved NCM460-NK-1R cells were pretreated with specific PKCθ and PKCϵ pseudosubstrate peptide inhibitors (10 μM) for 30 min, followed by SP (10-7 M) stimulation for 1 to 5 min. Cells were lysed, and equal amounts of protein were fractionated on 10% SDS-PAGE to determine the levels of phosphorylated and nonphosphorylated PKCθ and PKCϵ proteins and β-actin. Results are representative of three independent experiments.
Overexpression of Wild-Type PKCδ Potentiates SP-Induced IL-8 Promoter and NF-κB Luciferase Activities. The role of PKCδ in SP-induced NF-κB activation was further characterized by the transfection of a PKCδ wild-type plasmid that overexpresses PKCδ. Without transfection of wild-type PKCδ, SP significantly increased IL-8 promoter activity by 4.9-fold (P < 0.001) (Fig. 7A) and NF-κB luciferase activity by 1.8-fold (P < 0.01) (Fig. 7B). However, in colonocytes transfected with PKCδ, SP-induced IL-8 promoter and NF-κB luciferase activities were increased by 4.5- and 3-fold, respectively (P < 0.001) (Fig. 7, A and B). The baseline activity of PKCδ wild-type transfected NCM460-NK-1R cells without SP stimulation had increased IL-8 promoter and NF-κB luciferase activities (by 11-fold, P < 0.001; 4-fold, P < 0.01, respectively). Consistent with our above results, rottlerin (1 μM) significantly blocked SP-induced IL-8 promoter (P < 0.001) and NF-κB luciferase (P < 0.01) activity with or without overexpression of wild-type PKCδ (Fig. 7, A and B). Taken together, our findings clearly demonstrate that PKCδ activation is an important step for SP-induced NF-κB activation and subsequent IL-8 gene expression.
Discussion
It is well documented that the level of the proinflammatory cytokine IL-8 is significantly increased in the patients with inflammatory bowel diseases including Crohn's disease and ulcerative colitis, which contributes to massive neutrophil infiltration into affected tissues (Murata et al., 1995). Previous reports have indicated that SP and NK-1R interaction plays an important role in the pathogenesis of small intestinal and colonic inflammation (Castagliuolo et al., 1997, 1998, 2000, 2002; Zhao et al., 2002). SP can directly stimulate IL-8 gene expression in human astrocytoma U373MG (Lieb et al., 1997) and in NK-1R stably transfected nontransformed human colonocytes (Zhao et al., 2002). Although PKCs (Palma et al., 1995), including PKCδ (Soltoff and Toker, 1995) and PKCϵ (Lieb et al., 2003), have been associated with SP/NK-1R signaling, their involvement in SP-induced colonocyte proinflammatory responses has never been explored. We report here for the first time that SP rapidly and persistently activates PKCδ, and this event is involved in SP-associated activation of the NF-κB pathway leading to IL-8 gene transcription.
We found that in colonocytes, SP causes PKCδ-threonine phosphorylation, a critical event involved in PKCδ activation. Results from several different experimental approaches support the importance of PKCδ in SP-mediated, NF-κB-driven IL-8 release. Thus, SP-mediated IL-8 production is selectively inhibited by low doses (<1 μM) of rottlerin, a specific pharmacological antagonist of PKCδ (Wang et al., 2003), as well as by down-regulation of PKC expression, including PKCδ. The selective roles of PKCδ in this SP response were further confirmed by inhibition of endogenous PKCδ expression by a specific PKCδ siRNA and by transfection of a wild-type PKCδ-expressing plasmid that enhances SP-induced NF-κB activation as well as IL-8 transcription. Our results also indicate that SP can rapidly but transiently activate PKCϵ and PKCθ but has no effect on PKCα, PKCβ, PKCλ, PKCξ, and PKD/PKCμ phosphorylation. However, our results with specific pseudosubstrate peptide inhibitors do not support a functional role for PKCϵ and PKCθ in SP-induced IL-8 secretion. Although SP had been shown to induce Ca2+ mobilization in U373MG cells (Renzi et al., 2000), SP-induced IL-8 synthesis was not affected by pretreatment of NCM-460-NK-1R cells with the intracellular Ca2+ calcium chelator 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetra(acetoxymethyl) ester (40 μM), the Ca2+ channel blocker 8-(N,N-diethylamino)-octyl-3,4,5-trimethoxybenzoate, HCl (100 μM), or the Ca2+-dependent PKC inhibitor Go6976 (40 μM) (data not shown). Taken together, our results clearly demonstrate that among several PKC isoforms, Ca2+-independent PKCδ plays a critical role in SP-mediated, NF-κB-dependent IL-8 gene expression. Consistent with our observations, the importance of PKCδ in regulating the NF-κB pathway has also been reported in several other systems. For example, lysophosphatidic acid-induced NF-κB and IL-8 expression is dependent on PKCδ activation in human bronchial epithelial cells (Cummings et al., 2004). PKCδ is also involved in TNFα-induced NF-κB-dependent gene expression in airway epithelial cells as inhibition of PKCδ either by rottlerin or by a dominant negative PKCδ mutant-attenuated TNFα-induced IL-8 promoter transcriptional activity (Page et al., 2003).

PKCδ is required for SP-induced activation of signaling molecules of the NF-κB pathway. A, serum-starved NCM460-NK-1R cells were pretreated with rottlerin (0.1–1 μM) or vehicle (DMSO) for 30 min, followed by SP (10-7 M) exposure for 4 h. Conditioned media were collected, and IL-8 levels were measured by ELISA. Data are expressed as means ± S.E.M. (n = 6). ** and ***, P < 0.01 and P < 0.001, respectively. B, serum-starved NCM460-NK-1R cells were pretreated with rottlerin (0.1–1 μM) or vehicle (DMSO) for 30 min, followed by SP (10-7 M) exposure for the indicated time points. Cells were lysed, and equal amounts of protein were fractionated on 10% SDS-PAGE to determine the levels of phospho-PKCδ (2 min), phospho-IKK (15 min), phospho-p65 (15 min), and β-actin. Results are representative of three independent experiments.
Our previous results indicated an important role for Rho GTPases in SP-induced NF-κB activation and IL-8 secretion in human NCM460-NK-1R colonocytes (Zhao et al., 2002). Recent reports also suggested that Rho A is upstream of PKC, including PKCδ in colonic epithelial cells (Massoumi et al., 2002), and intestinal smooth muscle cells (Harnett et al., 2005) Therefore, it is likely that SP-induced PKCδ activation and IL-8 secretion are also mediated via the Rho family of GTPases in NCM460 colonocytes. SP also induces MAPK via EGFR transactivation in human NCM460 and U373MG (Koon et al., 2004), but pharmacological inhibition of EGFR by AG1478 (2 μM) and ERK1/2 by PD98059 (25 μM) failed to affect SP-induced IL-8 secretion (data not shown), suggesting that SP-induced IL-8 secretion does not involve the EGFR/MAPK pathway.
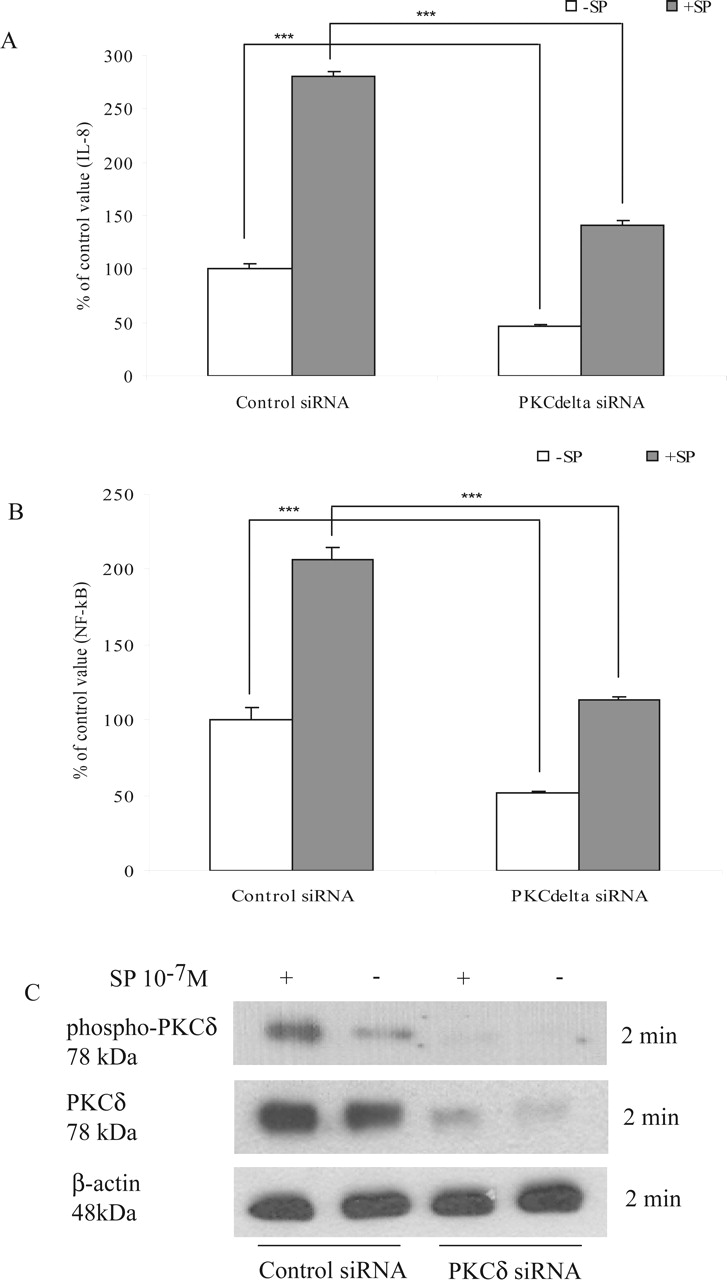
PKCδ siRNA significantly inhibited SP-induced IL-8 promoter and NF-κB luciferase activities. NCM460-NK-1R cells were transiently transfected with a PKCδ siRNAs or its control siRNA together with an IL-8 promoter or an NF-κB firefly reporter plasmid. All cells were also transfected with control pRL-TK plasmids for Renilla luciferase signal. The transfected cells were serum-starved overnight and treated with SP (10-7 M) for 4 h. Cell extracts were prepared to measure IL-8 promoter (A) or NF-κB (B) luciferase activity as described under Materials and Methods. Data are expressed as mean ± S.E.M. (n = 6). ***, P < 0.001. C, PKCδ siRNA or control siRNA-transfected NCM460-NK-1R cells were lysed, and equal amounts of protein were fractionated on 10% SDS-PAGE to determine the levels of phospho-PKCδ, PKCδ, and β-actin. Results are representative of three independent experiments.
Although NF-κB plays a pivotal role in intestinal inflammation (Jobin and Sartor, 2000), the role of PKCs, especially PKCδ, has not been systematically evaluated. PKC was found to be one of important mediators during 2,4,6-trinitrobenzenesulfonic acid-induced colitis in rats as the PKC inhibitors staurosporine and GF-109203X reduced 2,4,6-trinitrobenzenesulfonic acid-induced changes in mucosal PKC activity and the degree of mucosal damage (Brown et al., 1999). The selective PKCδ inhibitor rottlerin was found to inhibit proinflammatory cytokine production such as TNFα and IL-1β in human monocytes (Kontny et al., 2000). Also, PKCδ was activated in lung epithelial cells with asbestos-induced inflammation (Lounsbury et al., 2002). Our results provide strong evidence that the proinflammatory effect of SP in intestinal inflammation could be mediated via PKCδ. Thus, inhibition of PKCδ may represent a potential therapeutic approach for treatment of diseases associated with intestinal inflammation.

Overexpression of wild-type PKCδ potentiates SP-induced IL-8 promoter and NF-κB luciferase activities. NCM460-NK-1R cells were transiently transfected with a pCMV-β-galactosidase control or a wild-type PKCδ-overexpressing plasmid together with an IL-8 promoter or an NF-κB reporter construct plus an internal control pRL-TK plasmid. Transfected cells were serum-starved overnight and then pretreated with either rottlerin (1 μM) or vehicle (DMSO) for 30 min followed by SP (10-7 M) exposure for 4 h. Cell extracts were prepared to measure IL-8 promoter (A) or NF-κB (B) luciferase activity. Data are expressed as means ± S.E.M. (n = 6). ** and ***, P < 0.01 and P < 0.001, respectively.
Footnotes
-
This work was supported by National Institutes of Health Grant DK 47343 (to C.P.).
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.105.088013.
-
ABBREVIATIONS: SP, substance P; NK-1R, neurokinin-1 receptor; NF, nuclear factor; IL-8, interleukin-8; TNFα, tumor necrosis factor α; MAPK, mitogen-activated protein kinase; EGFR, epidermal growth factor receptor; PKC, protein kinase C; DAG, diacylglycerol; CAPE, caffeic acid phenethyl ester; PMA, phorbol-12-myristate-13-acetate; ELISA, enzyme-linked immunosorbent assay; PAGE, polyacrylamide gel electrophoresis; IKK, IκB kinase complex; siRNA, small interfering RNA; Go6976, 12-(2-cyanoethyl)-6,7,12,13-tetrahydro-13-methyl-5-oxo-5H-indolo(2,3-a)pyrrolo(3,4-c)-carbazole; AG1478, 4-(3-chloroanilino)-6,7-dimethoxyquinazoline; PD98059, 2′-amino-3′-methoxyflavone; DMSO, dimethyl sulfoxide.
- Received April 13, 2005.
- Accepted May 23, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}