


Abstract
Antagonist affinity measurements have traditionally been considered important in defining the receptor or receptor subtypes present within cells or tissues. Any change in this value has normally been taken as evidence for the presence of a second receptor. However, highly efficacious ligands induce a time and phosphorylation-dependent change in the β2-adrenoceptor resulting in 10-fold lower affinity for antagonists. Also the β1-adrenoceptor is now considered to exist in two different active conformations which are distinguished by their pharmacological properties. In this study, the site of action of a range of β-agonists and β-antagonists was determined using the human β1-adrenoceptor stably expressed in Chinese hamster ovary cells with cyclic AMP response element reporter genes. Adrenaline and noradrenaline were confirmed as having agonist actions via the catecholamine site, whereas all antagonists had higher affinity for the catecholamine rather than secondary site. However, the rank order of affinity for the two sites was different suggesting that they are indeed separate entities. The measurements of antagonist affinity at the catecholamine site, however, were found to depend upon the agonist present. For example, xamoterol, cimaterol, terbutaline, and formoterol agonist responses were more readily antagonized by CGP 20712A[2-hydroxy-5-(2-[{hydroxy-3-(4-[1-methyl-4-trifluoromethyl-2-imidazolyl]phenoxy)propyl}amino]ethoxy)benzamide] than the catecholamine responses themselves. This, however, was not related to agonist efficacy as has previously been reported for the human β2-adrenoceptor. Therefore, it may be that some agonists (e.g., cimaterol) purely activate the catecholamine site and others purely activate the secondary site (e.g., CGP 12177 [(–)-4-(3-tert-butylamino-2-hydroxypropoxy)-benzimidazol-2-one]), whereas the others (e.g., catecholamines) activate both sites to differing degrees.
Antagonist affinity measurements have traditionally been considered important in defining the receptor or receptor subtypes present within cells or tissues. Antagonist affinity measurements (KD values) were therefore considered to be constant for a given receptor-antagonist interaction regardless of which agonists were present or which downstream responses were measured (Kenakin et al., 1995). As a consequence, any change in this value has been taken as evidence for the presence of a second receptor population (Arunlakshana and Schild, 1959; Black et al., 1965, 1972). However, previous studies with the β2-adrenoceptor have suggested that this is not always the case (Baker et al., 2003a). Here, short-term cAMP accumulation studies gave similar values for antagonist affinity regardless of which competing agonist was present. However, in longer-term gene transcription assays, isoprenaline and adrenaline required 10-fold higher antagonist concentrations to inhibit the responses of that when salbutamol or terbutaline were used. It transpired that this depended upon the efficacy of the competing agonist. Highly efficacious ligands (isoprenaline and adrenaline) induced a time-dependent, phosphorylation-dependent change in the β2-adrenoceptor resulting in 10-fold lower affinity for antagonists (Baker et al., 2003a).
This affinity assumption also does not hold for the β1-adrenoceptor because it is now considered to exist in two different active conformations (Granneman, 2001; Molenaar, 2003; Arch, 2004). Although the molecular nature of these conformations is unknown, they are distinguishable by their pharmacological properties. Agonist responses occurring via the classical “catecholamine” site of the β1-adrenoceptor are readily inhibited by classical β-antagonists, whereas agonist responses occurring via the “secondary” (low-affinity) site are relatively resistant to antagonism (Granneman, 2001). This was first demonstrated with (–)-4-(3-tert-butylamino-2-hydroxypropoxy)-benzimidazol-2-one (CGP 12177) (originally considered as a β-antagonist; Staehelin et al., 1983). CGP 12177 inhibits isoprenaline-induced β1-adrenoceptor responses as a high affinity neutral antagonist; however, at higher concentrations (i.e., with lower affinity) it produces an agonist response which is relatively resistant to antagonism by classical β-antagonists (Pak and Fishman, 1996; Konkar et al., 2000a,b; Lowe et al., 2002; Baker et al., 2003b). Further studies from knockout animals and transfected cell system receptors showed that both pharmacological profiles were dependent upon the presence of only the β1-adrenoceptor and not a further subtype of β-receptor, hence the current two-state receptor model (Pak and Fishman, 1996; Kaumann and Molenaar, 1997; Kaumann et al., 1998; Cohen et al., 2000; Konkar et al., 2000a,b; Granneman, 2001; Kaumann et al., 2001; Baker et al., 2003b). This is very different from the human β2-adrenoceptor, where CGP 12177 is a typical partial agonist (i.e., similar KD values) from binding (0.14 nM), partial antagonism (0.09–0.17 nM), and agonism (EC50 0.22 nM; Baker et al., 2002).
The secondary low-affinity site of the β1-adrenoceptor may be important in its own right as certain physiological events appear specifically linked to β1-low-affinity state, rather than catecholamine site, actions (e.g., relaxation in blood vessels, Kozlowska et al., 2003; Mallem et al., 2004). Furthermore, the plasma concentration of carvedilol (100 ng/ml = 300 nM) used in human cardiovascular diseases is sufficient to activate this secondary site (Sawangkoon et al., 2000; Baker et al., 2003b). However, whereas the β1-low-affinity state appears to signal via Gs-proteins in recombinant cell systems (Pak and Fishman, 1996; Konkar et al., 2000b; Baker et al., 2003b), this conformation may also couple to Gi-proteins in rat heart and blood vessels (Kompa and Summers, 1999; Mallem et al., 2004).
Since the discovery of the two β1-adrenoceptor sites, several studies have examined the site of action of different ligands. Most classical antagonists and adrenaline, noradrenaline, isoprenaline, and dobutamine were found to preferentially act via the catecholamine site, whereas LY 362884 agonist actions occurred via the secondary site (Konkar et al., 2000b; Lowe et al., 2002; Joseph et al., 2004). Several “β-antagonists” also have intrinsic agonist actions of their own (Lowe et al., 2002; Baker et al., 2003b). Some of these agonist responses were potently antagonized by neutral β1-antagonists (suggesting catecholamine site agonism, e.g., acebutolol), others were relatively resistant to antagonism (suggesting secondary site activation, e.g., carvedilol) although other compounds had agonist activity at both sites (alprenolol, pindolol; Baker et al., 2003b). Now, when evaluating the overall pharmacology of any particular β-adrenoceptor ligand at the β1-adrenoceptor, the site of action and efficacy at each site must be determined.
The aim of this study was therefore to determine the site of action of several β-agonists and β-antagonists at the human β1-adrenoceptor using stably transfected recombinant cell systems, then explore whether the antagonist affinity measurements for these antagonists varied at the two sites of the β1-receptor in an efficacy-dependent manner analogous to that seen at the human β2-adrenoceptor.
Materials and Methods
Materials. Cell culture reagents were from Sigma Chemical (Poole, Dorset, UK) except fetal calf serum which was from PAA Laboratories (Teddington, Middlesex, UK). [3H]adenine and [14C]cAMP were obtained from Amersham Biosciences UK, Ltd. (Little Chalfont, Buckinghamshire, UK). The Luclite Plus Assay System was from PerkinElmer (Groningen, Netherlands). CGP 12177, betaxolol, pronethalol, bisoprolol, ICI 118551, sotalol, salbutamol, timolol, and xamoterol were from Tocris Cookson Inc. (Bristol, UK). Carvedilol was a gift from GlaxoSmithKline (Uxbridge, Middlesex, UK), and bupranolol was a gift from Prof. Sian Harding (Imperial College, London). Sigma Chemical supplied all other reagents.
Cell Culture. CHO cells stably expressing both the human β1-adrenoceptor (at 79 fmol/mg protein) and a six cyclic AMP response element (CRE)-luciferase reporter gene (six CRE upstream of a luciferase response gene) were used for the luciferase experiments (CHO-β1-luciferase cells; Baker et al., 2003b). CHO cells stably expressing the human β1-adrenoceptor (at 1147 fmol/mg protein) and a six CRE-secreted placental alkaline phosphate (SPAP) reporter gene (six CRE upstream of a SPAP reporter gene) were used for the SPAP and cAMP experiments (CHO β1-SPAP cells; Baker et al., 2003b). Both cells lines were grown in Dulbecco's modified Eagle's medium/nutrient mix F-12 (DMEM/F-12) containing 10% fetal calf serum and 2 mM l-glutamine in a humidified 5% CO2/95% air atmosphere at 37°C.
CRE-Luciferase Production. CHO-β1-luciferase cells were grown to confluence in white-sided 96-well view plates in 200 μl of DMEM/F-12 containing 10% fetal calf serum and 2 mM l-glutamine. This was removed and replaced with 200 μl of serum-free media (i.e., DMEM/F-12 containing 2 mM l-glutamine only) or 200 μl of serum-free media containing an antagonist at the final required concentration and the cells incubated for 1 h at 37°C (5% CO2). Twenty microliters of agonist (diluted in serum-free media) were then added to each well and the plate incubated at 37°C (5% CO2) for 5 h. The media and drugs were removed, a white base was added to the plate, and luciferase activity was detected as described previously (Baker et al., 2003b). For the experiments seen in Fig. 3, serum-free media or a fixed concentration of agonist in serum-free media were added to the wells and immediately followed by the addition of 20 μl of CGP 12177 and the plates incubated at 37°C for 5 h.

CRE-luciferase activity in CHO-β1-luciferase cells response to isoprenaline (isop) (a), adrenaline (adren) (b), noradrenaline (norad) (c), cimaterol (cimat) (d), and CGP 12177 (e) in the absence and presence of 100 nM (a–d) or 10 μM (e) betaxolol (betax). Bars show basal luciferase activity in response to 10 μM isoprenaline and in response to 100 nM (a–d) or 10 μM (e) betaxolol alone. Data points are mean ± S.E.M. from a single experiment in each case. These individual experiments are representative of 4 (a), 7 (b), 5 (c), 6 (d), and 5 (e) separate experiments.
CRE-SPAP Production. CHO-β1-SPAP cells were grown to confluence in 24-well plates in 1 ml of DMEM/F-12 containing 10% fetal calf serum and 2 mM l-glutamine. The media were removed from each well, replaced with 1 ml of serum-free media, and incubated for 24 h (37°C, 5% CO2). On the morning of experimentation, the serum-free media were removed from each well and replaced with either a further 1 ml of serum-free media or 1 ml of serum-free media containing CGP 20712A at the final required concentration, and the cells were incubated for 1 h at 37°C (5% CO2). Ten microliters of agonist were then added to each well, and the plates were incubated for 5 h (37°C, 5% CO2). SPAP secretion was then determined as described previously (McDonnell et al., 1998).
[3H]cAMP Accumulation. Cells were grown to confluence in 24-well plates in 1 ml of DMEM/F-12 containing 10% fetal calf serum and 2 mM l-glutamine. The media were removed and the cells prelabeled with [3H]adenine by incubation with 2 μCi/ml [3H]adenine in serum-free media for 2 h at 37°C (5% CO2). The [3H]adenine was removed and each well washed by the addition and removal of 1 ml of serum-free media. One milliliter of serum-free media containing 1 mM 3-isobutyl-1-methylxanthine with or without the final required concentration of CGP 20712A was added to each well and the cells incubated for 1 h at 37°C (5% CO2). Ten microliters of agonist were added to each well, and the plates were incubated for 10 min at 37°C before the reaction was terminated by the addition of 50 μl of concentrated HCl per well. The plates were then frozen, and [3H]cAMP was separated from other 3H nucleotides by Dowex and alumina column chromatography, with each column being corrected for efficiency by comparison with [14C]cAMP recovery as previously described (Donaldson et al., 1988).
Data Analysis. Sigmoidal agonist concentration-response curves were fitted using the following equation through computer-assisted nonlinear regression using the program GraphPad Prism 2:  where Emax is the maximal response, [A] is the agonist concentration, and EC50 is the concentration of agonist that produces 50% of the maximal response.
where Emax is the maximal response, [A] is the agonist concentration, and EC50 is the concentration of agonist that produces 50% of the maximal response.
Antagonist KD values were then calculated from the shift of the agonist concentration responses in the presence of a fixed concentration of antagonist using the following equation:  where DR (dose ratio) is the ratio of the agonist concentration required to stimulate an identical response in the presence and absence of a fixed concentration of antagonist [B].
where DR (dose ratio) is the ratio of the agonist concentration required to stimulate an identical response in the presence and absence of a fixed concentration of antagonist [B].
In experiments where three different fixed concentrations of antagonist were used, Schild plots were constructed using the following equation:  These points were then fitted to a straight line. A slope of 1 then indicates competitive antagonism (Arunlakshana and Schild, 1959).
These points were then fitted to a straight line. A slope of 1 then indicates competitive antagonism (Arunlakshana and Schild, 1959).
A two-site analysis was used for the experiments shown in Fig. 3 using the following equation:  where basal is the response in the absence of agonist or antagonist, Ag is the response to a fixed concentration of agonist, [C] is the concentration of CGP 12177, IC50 is the concentration of CGP 12177 that inhibits 50% of the response of the fixed agonist, CGPstim is the maximum stimulation by CGP 12177, and EC50 is the concentration of CGP 12177 that stimulated a half-maximal CGP 12177 response.
where basal is the response in the absence of agonist or antagonist, Ag is the response to a fixed concentration of agonist, [C] is the concentration of CGP 12177, IC50 is the concentration of CGP 12177 that inhibits 50% of the response of the fixed agonist, CGPstim is the maximum stimulation by CGP 12177, and EC50 is the concentration of CGP 12177 that stimulated a half-maximal CGP 12177 response.

CRE-luciferase activity in CHO-β1-luciferase cells in response to isoprenaline (isop) (a), adrenaline (adren) (b), noradrenaline (norad) (c), cimaterol (cimat) (d), and CGP 12177 (e) in the absence and presence of various concentrations of carvedilol (carv). Bars show basal luciferase activity in response to 10 μM isoprenaline alone. The response to each carvedilol concentration alone was assessed in each experiment (0.3 nM to 10 μM) and was never significantly different from basal and for simplicity is not shown. The Schild slopes are 0.91 ± 0.08 (n = 4) (a), 1.01 ± 0.03 (n = 4) (b), 0.97 ± 0.06 (n = 3) (c), 1.03 ± 0.08 (n = 3) (d), and 1.01 ± 0.08 (n = 4) (e). Data points are mean ± S.E.M. from a single experiment in each case. These individual experiments are representative of 4 (a), 4 (b), 3 (c), 3 (d), and 4 (e) separate experiments.
A 10 μM (maximal) isoprenaline concentration was included in each plate for each separate experiment for CRE-luciferase, CRE-SPAP, and [3H]cAMP accumulation (with the exception of Fig. 3) to allow agonist responses to be expressed as a percentage of the isoprenaline maximum for each experiment. All data are presented as mean ± S.E.M. of triplicate determinations, and n in the text refers to the number of separate experiments.
Results
CRE-Luciferase Production. Isoprenaline stimulated an increase in luciferase activity in CHO-β1-luciferase cells that was 6.2- ± 0.3-fold over basal (log EC50 –8.02 ± 0.06, n = 19). Adrenaline (log EC50 –6.85 ± 0.05, 102.8 ± 1.1% isoprenaline maximum, n = 21), noradrenaline (log EC50 –7.30 ± 0.05, 103.2 ± 1.2% isoprenaline maximum, n = 20), and cimaterol (log EC50 –7.65 ± 0.02, 93.0 ± 1.4% isoprenaline maximum, n = 20) stimulated similar responses. CGP 12177 (log EC50 –7.43 ± 0.02, n = 17) stimulated a response of 37.8 ± 1.6% of isoprenaline and was a partial agonist in this cell system.
The β1-selective antagonist CGP 20712A inhibited the isoprenaline, adrenaline, and noradrenaline responses to yield very similar log KD values of –9.19 ± 0.12, n = 9, –9.12 ± 0.09, n = 14, and –9.15 ± 0.09, n = 12, respectively. These values are markedly different from that obtained from antagonism of the CGP 12177 response (log KD –7.09 ± 0.09, n = 7, p < 0.0001 analysis of variance Newman-Keuls post hoc). Similar results were obtained with the other β-antagonists studied (see Table 1 and Figs. 1 and 2) suggesting that adrenaline and noradrenaline were stimulating responses via the same site as isoprenaline, i.e., the catecholamine site of the β1-adrenoceptor. When cimaterol was the agonist, all of the antagonist KD values obtained were significantly different from those obtained with isoprenaline as agonist (see Table 1). However, all of the receptor-ligand interactions appeared to be competitive because the Schild slopes achieved were not significantly different from unity (e.g., Fig. 2). This is similar to previous studies (Konkar et al., 2000a,b; Lowe et al., 2002).
Log KD values of 12 antagonists in the presence of five different agonists from CRE-luciferase activity experiments using CHO-β1-luciferase cells Values are mean ± S.E.M. from n separate determinations.
CGP 12177 was then added to fixed concentrations of the agonists (adrenaline, noradrenaline, and cimaterol). At low concentrations, CGP 12177 inhibited the agonist stimulation. However, at higher concentrations of CGP 12177, its stimulatory effects were clearly seen (Fig. 3). In each case, the concentration of CGP 12177 required to inhibit the agonist was about two orders of magnitude less than that required to cause the stimulation.
CRE-SPAP Production. Isoprenaline stimulated an increase in CRE-SPAP production (log EC50 –8.46 ± 0.12, 2.4- ± 0.1-fold over basal, n = 13) that was inhibited by CGP 20712A to yield a log KD value of –9.24 ± 0.08 (n = 13). Adrenaline and noradrenaline responses were similarly inhibited by CGP 20712A (see Table 2). A range of other β-agonists were then examined, and the responses to each were antagonized by CGP 20712A. The results obtained demonstrate a range of KD values for CGP 20712A with xamoterol and cimaterol being the most potently antagonized and SR 59230A and CGP 12177 being the least (see Fig. 4 and Table 2).
Log EC50 values and percentage isoprenaline maximum response of CRE-SPAP production from CHO-β1-SPAP cells to a range of β-agonists
Log KD values obtained from CGP 20712A antagonism of these agonists is also shown. Values are mean ± S.E.M. of n determinations.

CRE-luciferase production in CHO-β1-luciferase cells in response to CGP 12177 in the absence and presence of fixed concentrations of 1 μM adrenaline (adren) (a), 300 nM and 1 μM noradrenaline (norad) (b), and 10, 30, and 100 nM cimaterol (cimat) (c). Bars represent basal CRE-luciferase production in response to each of the agonists as detailed in the individual legends. Data points are triplicate determinations from a single experiment which are representative of 3 (a), 3 (b), and 4 (c) separate experiments.
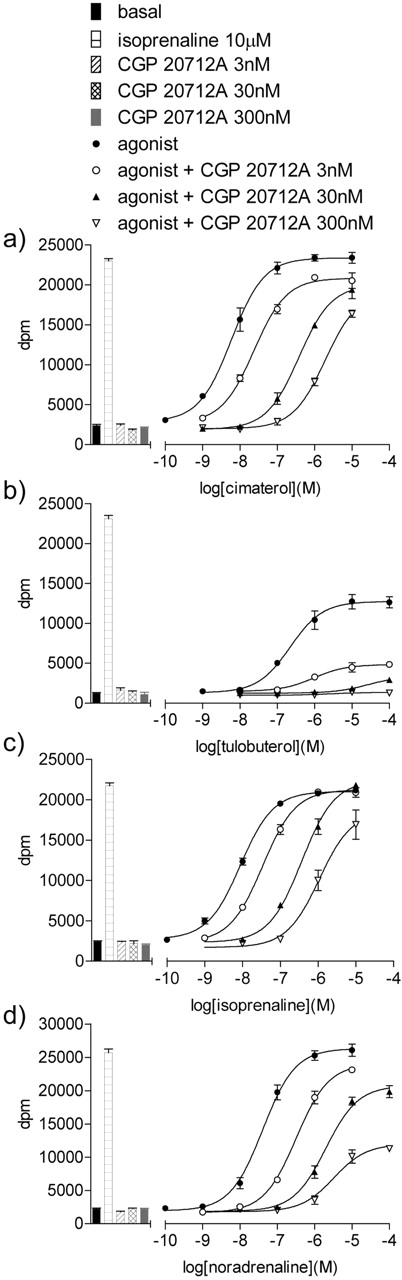
[3H]cAMP Accumulation. Isoprenaline stimulated an increase in [3H]cAMP accumulation (10-min agonist incubation) that was 10.1- ± 1.9-fold over basal (log EC50 –8.27 ± 0.16, n = 4). This response was antagonized by increasing concentrations of CGP 20712A to yield a log KD value for CGP 20712A of –9.25 ± 0.07, n = 12. This was a competitive inhibition as the Schild slope obtained was 0.95 ± 0.05, n = 4 (Fig. 5). Cimaterol stimulated a response (log EC50 –8.19 ± 0.07, 97.9 ± 1.7% isoprenaline maximum, n = 4) that was antagonized by CGP 20712A to yield a log KD value of –9.48 ± 0.06 (n = 10; Schild slope 0.98 ± 0.07, n = 3, Fig. 5). Thus, although this too appeared competitive, the log KD value for CGP 20712A was significantly different (p = 0.02, unpaired t test) from that obtained when isoprenaline was the agonist. The response to noradrenaline was antagonized by CGP 20712A to yield a log KD value similar to that obtained when isoprenaline was the agonist. However, the increasing concentrations of CGP 20712A caused a reduction in the maximum response to noradrenaline unmasking the lower efficacy of this ligand compared with isoprenaline and cimaterol (Fig. 5, Table 3). A log KD value was not obtained for CGP 20712A when tulobuterol was the agonist because even in the presence of the lowest concentration of CGP 20712A, the maximum tulobuterol response was significantly reduced showing a low efficacy for this ligand at the human β1-adrenoceptor (Fig. 5, Table 3).
Log EC50 values, percentage isoprenaline maximum response and log KD values for CGP 20712A at the level of [3H]cAMP accumulation from CHO-β1-SPAP cells
Values are mean ± S.E.M. of n determinations.
Discussion
It is now accepted that the β1-adrenoceptor has two separate sites or conformations to which ligands can bind (Pak and Fishman, 1996; Konkar et al., 2000a,b; Granneman, 2001; Joseph et al., 2004). Agonists stimulating responses through the catecholamine site are readily inhibited by antagonists in a manner similar to that of isoprenaline. Agonist responses occurring via the secondary site are relatively resistant to inhibition with antagonists in a similar manner to those of CGP 12177. Thus, in a system with only the β1-adrenoceptor present, the site of action of a new agonist can be determined by comparing inhibition of its response with inhibition of isoprenaline and CGP 12177 responses (Konkar et al., 2000a,b; Lowe et al., 2002; Baker et al., 2003b; Joseph et al., 2004). In this study, the agonist actions of adrenaline and noradrenaline were inhibited in a very similar manner to that of isoprenaline and are therefore acting through the catecholamine site of the human β1-adrenoceptor. This is in agreement with previous findings in the ferret (Lowe et al., 2002).
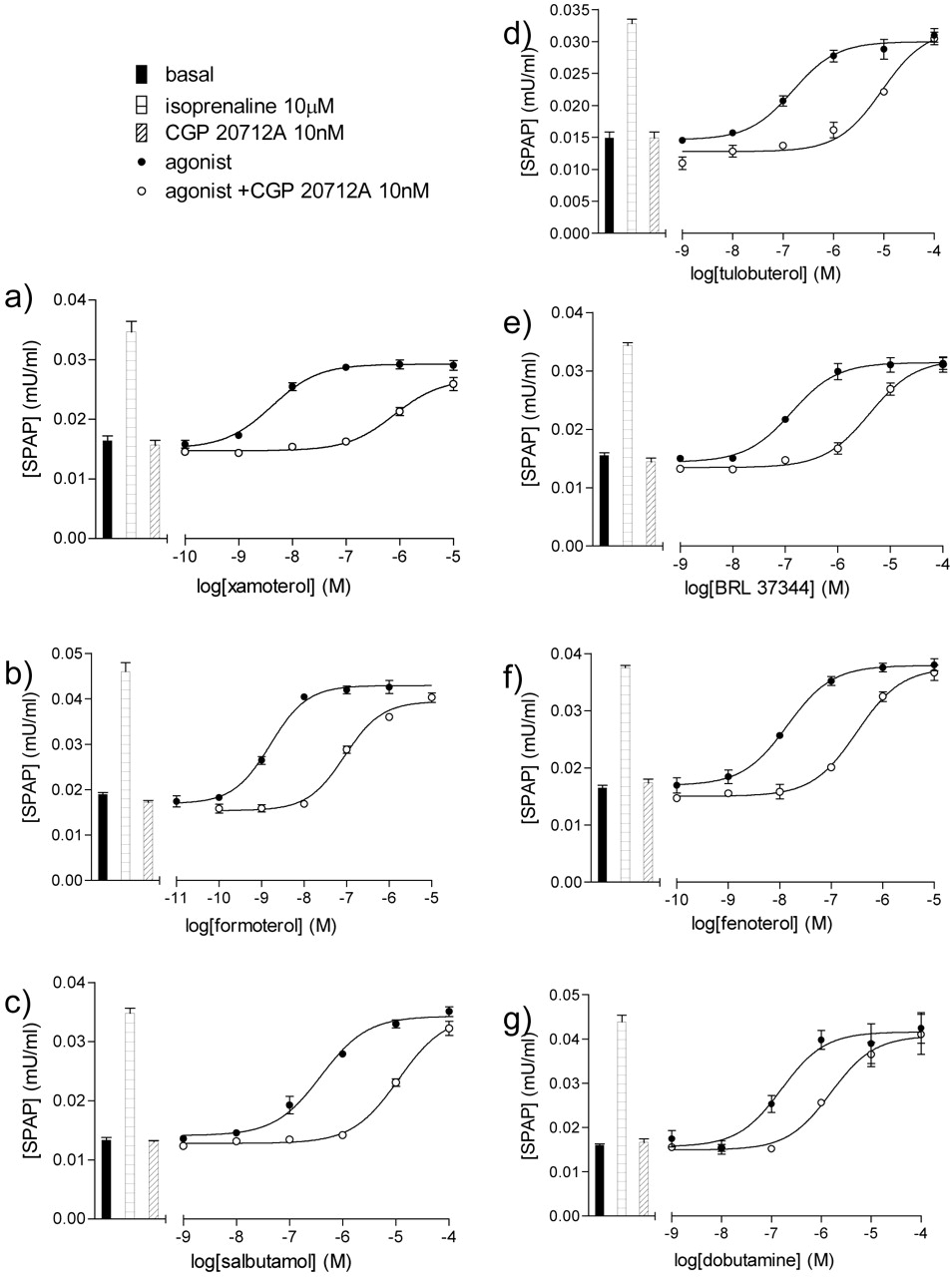
CRE-SPAP production in CHO-β1-SPAP cells in response to xamoterol (a), formoterol (b), salbutamol (c), tulobuterol (d), BRL 37344 (e), fenoterol (f), and dobutamine (g) in the presence and absence of 10 nM CGP 20712A. Bars represent basal CRE-SPAP production in response to 10 μM isoprenaline and 10 nM CGP 20712A. Data points are mean ± S.E.M. of triplicate determinations from single experiments, each representative of 11 (a), 12 (b), 8 (c), 11 (d), 11 (e), 12 (f), and 12 (g) separate experiments.

[3H]cAMP accumulation in CHO-β1-SPAP cells in response to cimaterol (a), tulobuterol (b), isoprenaline (c), and noradrenaline (d) in the absence and presence of 3, 30, or 300 nM CGP 20712A. Bars represent basal [3H]cAMP accumulation in response to 10 μM isoprenaline, 3 nM CGP 20712A, 30 nM CGP 20712A, or 300 nM CGP 20712A alone. The Schild slope is 0.98 ± 0.07, n = 3 (a) and 0.95 ± 0.05, n = 4 (c). Calculation of Schild slopes is not possible for (b) and (d) due to noncompetitive antagonism. Data points are mean ± S.E.M. of triplicate determinations. Each of these single experiments is representative of 3 (a), 4 (b), 4 (c), and 4 (d) separate experiments.
All of the 12 antagonists used in this study have higher affinity (KD value) for the catecholamine site than the secondary site (Table 1). This is also true for CGP 12177 itself and carvedilol (which are neutral antagonists of the catecholamine site and agonists of the secondary site) and alprenolol and pindolol (which are agonists of both sites; Pak and Fishman, 1996; Konkar et al., 2000b; Lowe et al., 2002; Baker et al., 2003b). Thus, regardless of the efficacy of the ligand, no ligand has yet been described that is able to bind to the secondary site with greater affinity. However, the rank order of antagonist affinities at the catecholamine site (which is identical with isoprenaline, adrenaline, and noradrenaline) is markedly different from the rank order of affinities at the secondary site (Table 1 and Fig. 6 where the poor correlation is clearly demonstrated). ICI 118551, bupranolol, and nadolol all have higher than expected affinity, and atenolol has a lower than expected affinity for the secondary site than if it was just a low-affinity mirror of the catecholamine site. This is similar to findings recently reported by Joseph et al. (2004) also at the human β1-adrenoceptor, but there appear to be some differences between species (cf. Lowe et al., 2002 in the ferret heart, this report and Joseph et al., 2004 using the human β1-adrenoceptor). Thus, as well as the differences in ligand efficacy at the two sites, the different ligand affinities also suggest that the secondary site is indeed a separate pharmacological entity.
Cimaterol stimulated responses that were inhibited by all antagonists to yield much higher affinity values than those achieved with CGP 12177, suggesting that cimaterol is not acting via the secondary site. However, all of the KD values obtained showed a consistently higher affinity than when the catecholamines were present. This raises the possibility that cimaterol is acting through a third, higher-affinity conformation of the β1-adrenoceptor and that many ligand-dependent states of the β1-adrenoceptor may exist (Molenaar, 2003; Arch 2004; Fig. 6). The rank order of antagonist affinity (Table 1; Fig. 6), however, is the same for cimaterol as for the catecholamines, suggesting that cimaterol is likely to stimulate the β1-adrenoceptor via the catecholamine site. Cimaterol also behaved exactly the same as the catecholamines in the presence of CGP 12177 (Fig. 3). Here, CGP 12177 inhibited all of the responses (as it is a high affinity neutral antagonist of the catecholamine site) at concentrations substantially below that required for CGP 12177 stimulation via the secondary site (Pak and Fishman, 1996; Konkar et al., 2000b; Baker et al., 2003b). Again, cimaterol appears to be acting via the catecholamine site.

Correlation between antagonist log KD values with isoprenaline as agonist (x-axis) versus those obtained with adrenaline, noradrenaline, cimaterol, or CGP 12177 as agonist (y-axis). The log Kd values are those described in Table 1. The correlation coefficients and slopes are as follows: 0.98 and 1.06 for isoprenaline versus adrenaline, 0.99 and 1.04 for isoprenaline versus noradrenaline, 0.99 and 1.04 for isoprenaline versus cimaterol, and 0.76 and 0.98 for isoprenaline versus CGP 12177, respectively.
Interestingly, in all three cases presented in Fig. 3 and that previously reported in Baker et al. (2003b), the maximum stimulation achieved by CGP 12177 was always higher in the presence of the agonist than in its absence, even though the higher CGP 12177 concentrations should be completely inhibiting the agonist-catecholamine site stimulation. This, however, was not seen when examining the pharmacology of the rat β1-adrenoceptor expressed in CHO cells in membrane cAMP assays (Konkar et al., 2000b). The reason for this is unknown but may yet be explained when better understanding of the molecular structure of the secondary site and its interaction with signaling complexes is known.
To further explore the discrepancy in antagonist affinity in the presence of cimaterol (rather than isoprenaline, adrenaline, or noradrenaline), experiments were undertaken with CHO-β1-SPAP cells. This not only validated this finding in a completely separate cell line at a different receptor expression level and with a different reporter gene, but the higher receptor expression allowed easier examination of other agonist responses. By comparing the affinity of CGP 20712A as an antagonist of the agonist responses with the affinity of it as an antagonist of the isoprenaline responses (to denote the catecholamine site) and CGP 12177 responses (to denote the secondary site), it can be seen that most of the β-agonists were stimulating responses through the catecholamine site. There was, however, a full range of antagonist affinity values obtained (see Table 2). This is at odds with the accepted dogma that antagonist affinity is an innate property of that receptor-ligand interaction and is immune to influences from the competing agonist (Kenakin et al., 1992).
A change in antagonist affinity (KD) measurement within the same assay that depended purely upon which competing agonist was present was reported at the human β2-adrenoceptor (Baker et al., 2003a). At the β2-adrenoceptor, highly efficacious ligands induced a time-dependent, phosphorylation-dependent change in the β2-adrenoceptor that resulted in a 10-fold lower affinity for antagonists (Baker et al., 2003a). To determine whether the apparent changes in antagonist affinity at the catecholamine site of the β1-adrenoceptor in this study (Table 2) were also dependent upon the efficacy of the competing ligand, short-term cAMP accumulation assays were performed on four agonists, cimaterol from the top of Table 2, tulobuterol from the middle, and isoprenaline and noradrenaline from the bottom.
First, study of the EC50 values of the agonists suggest that receptor desensitization was not the cause. In the β2-adrenoceptor study (Baker et al., 2003a), the concentration response curves of less efficacious agonists became left-shifted over time (i.e., EC50 in long-term assays was to the left of the EC50 obtained in the same cells in short-term assays). This is likely due to the magnification in the signaling cascade from cAMP to CRE gene transcription that occurs at the level of protein kinase A (Yuan et al., 1994; January et al., 1998). At the β2-adrenoceptor, concentration response curves of the more efficacious agonists, however, became right-shifted over time suggesting receptor desensitization. In this β1-adrenoceptor study, the concentration responses of all four agonists became left-shifted over time. Second, the log KD values for CGP 20712A at the β1-adrenoceptor were different even in the short-term cAMP assay suggesting that the time of agonist incubation was therefore not a factor (Table 3). Third, there was no correlation between agonist efficacy and antagonist affinity. Isoprenaline and cimaterol appeared the most efficacious of the ligands (parallel shifts were seen with increasing concentrations of CGP 20712A) despite being at opposite extremes of Table 2. Noradrenaline appeared less efficacious than isoprenaline and tulobuterol (from the middle of the table) a partial agonist whose low efficacy and lack of receptor reserve was made clearly evident by the collapsing concentration response curves with increasing CGP 20712A concentrations (similar to that seen in Hopkinson et al., 2000, and Baker et al., 2003a). The agonist efficacy (isoprenaline = cimaterol > noradrenaline > tulobuterol) and time of incubation therefore was not responsible for the change in antagonist affinity ranking (cimaterol > tulobuterol > isoprenaline = noradrenaline).
In conclusion, there is increasing evidence that the secondary site of the human β1-adrenoceptor is indeed a separate entity from the catecholamine site. There is a different rank order in which antagonists bind to the two sites (Table 1). There are different efficacies of ligands at the two sites (Baker et al., 2003b), and CGP 12177 in the presence of a catecholamine-site agonist seems to give a greater response than alone. However, no ligand, regardless of efficacy, has yet been described to bind to the secondary site with higher affinity than the catecholamine site. Furthermore, the measurement of antagonist affinity at the catecholamine site depends upon the agonist present (Table 2). The reason for this is not known but clearly has a different explanation from that seen at the human β2-adrenoceptor (cf. Table 3 and Baker et al., 2003a). However, it appears that all of these agonist responses are occurring primarily via the catecholamine site rather than a separate third conformation of the human β1-adrenoceptor. One explanation could be that some agonists (e.g., cimaterol) purely activate the catecholamine site and others purely activate the secondary site (e.g., CGP 12177), whereas the others activate both sites to differing degrees. Clearly, more investigation into the nature of the two sites of the β1-adrenoceptor is needed.
Footnotes
-
J.G.B. is a Wellcome Trust Clinician Scientist Fellow.
-
doi:10.1124/jpet.104.082875.
-
ABBREVIATIONS: CGP 12177, (–)-4-(3-tert-butylamino-2-hydroxypropoxy)-benzimidazol-2-one; LY 362884, 6-{4-[2-({2-hydroxy-3-[(2-oxo-2,3-dihydro-1H-benzimidazol-4-yl)oxy]propyl}amino)-2-methylpropyl]phenoxy}}nicotinamide; ICI 118551, (–)-1-(2,3-[dihydro-7-methyl-1H-inden-4-yl]oxy)-3-([1-methylethyl]-amino)-2-butanol; CHO, Chinese hamster ovary; CRE, cyclic AMP response element; SPAP, secreted placental alkaline phosphate; DMEM/F-12, Dulbecco's modified Eagle's medium/nutrient mix F-12; CGP 20712A, 2-hydroxy-5-(2-[{hydroxy-3-(4-[1-methyl-4-trifluoromethyl-2-imidazolyl]phenoxy)propyl}amino]ethoxy)benzamide; SR 59230A, 1-(2-ethylphenoxy)-3-[[(1S)-1,2,3,4-tetrahydro-1-naphthalenyl]amino]-(2S)-2-propanol hydrochloride; BRL 37344, (R*,R*)-(±)-4-[2-[(2-(3-chlorophenyl)-2-hydroxyethyl)amino]propyl]phenoxyacetic acid; ZD 7114, (S)-4-[2-hydroxy-3-phenoxypropylaminoethoxy]-N-(2-methoxyethyl)phenoxyacetamide.
- Received December 30, 2004.
- Accepted February 14, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}