

Abstract
Potassium (K+) deprivation-induced apoptosis of cerebellar granule neurons requires new mRNA and protein synthesis. Using a fluorogenic substrate for interleukin-1β converting enzyme (ICE), we show that K+ deprivation of cerebellar granule neurons induces cycloheximide-sensitive ICE-like protease activity. A peptide inhibitor of ICE-like protease activity, Ac-YVAD-chloromethylketone (Ac-YVAD-CMK), prevents K+ deprivation-induced apoptosis. Further, reactive oxygen species (ROS) are essential mediators of K+ deprivation-induced apoptosis of cerebellar granule neurons because neuronal death is also blocked by superoxide dismutase, N-acetyl-l-cysteine, and free radical spin traps. Using fluorescent assays, we show that ROS production after K+ deprivation is blocked by actinomycin D, cycloheximide, and Ac-YVAD-CMK, suggesting that ROS act downstream of gene transcription, mRNA translation, and ICE activation. Taken together, we show that new mRNA and protein synthesis, activation of ICE-like proteases, and ROS production are sequential events in K+ deprivation-induced apoptosis of cerebellar granule neurons.
- apoptosis
- reactive oxygen species
- interleukin-1β converting enzyme
- ICE inhibition
- cerebellar granule neurons
- spin traps
- superoxide dismutase
Programmed cell death refers to a spatially and temporally reproducible loss of cells that occurs during the normal development of the CNS. Neuronal programmed cell death is thought to serve the removal of neuronal precursors that fail to establish appropriate synaptic connections (Oppenheim, 1991; Johnson and Deckwerth, 1993). Morphologically, apoptosis underlies some, but not all, forms of developmental programmed cell death in the mammalian brain (Wood et al., 1993). Inappropriate apoptosis has been suggested to be involved in neuronal loss in various human neurodegenerative diseases, such as Alzheimer’s disease (Loo et al., 1993), Huntington’s disease (Portera-Cailliau et al., 1995), amyotrophic lateral sclerosis (Rabizadeh et al., 1995), and spinal muscular atrophy (Roy et al., 1995).
In vitro, mature cerebellar granule neurons deprived of depolarizing levels (25 mm) of extracellular potassium (K+) undergo apoptosis characterized by chromatin condensation, pyknosis, and nucleosomal size DNA fragmentation (D’Mello et al., 1993; Yan et al., 1994; Galli et al., 1995). Apoptosis of cerebellar granule neurons after K+ deprivation is blocked by inhibitors of macromolecular synthesis, forskolin, and insulin-like growth factor.
The intracellular events that result in apoptosis are often genetically controlled by proapoptotic and antiapoptotic genes. The CED3 protein, which is required for cell death during the development ofCaenorhabditis elegans, shows structural (Walker et al., 1994; Wilson et al., 1994) and functional (Miura et al., 1993; Yuan et al., 1993) homology to mammalian interleukin-1β converting enzyme (ICE). ICE is the first member of a family of cysteine proteases with the distinguishing feature of a near-absolute specificity for aspartate in the S1 subsite (Thornberry et al., 1992). Subsequently, further proteases of the ICE familiy were identified as ICE-CED-3 homolog 1 (ICH-1/NEDD2), ICH-2, or CPP32/YAMA (Miura et al., 1993; Kumar et al., 1994; Wang et al., 1994; Nicholson et al., 1995;Tewari et al., 1995).
The mammalian homolog of the C. elegans ced-9 gene, which is a potent suppressor of cell death, is the bcl-2 gene. It has been suggested that bcl-2 regulates an antioxidant pathway to prevent apoptosis in lymphocytes (Hockenbery et al., 1993; Kane et al., 1993). Events that lead to oxidative stress, including exposure to β-amyloid (Loo et al., 1993; Behl et al., 1994) and transient ischemia (Héron et al., 1993; Rosenbaum et al., 1994; Li et al., 1995), may trigger neuronal apoptosis.
Reactive oxygen species (ROS) have been implicated as mediators of excitotoxic (Coyle and Puttfarcken, 1993; Schulz et al., 1995a,b) and apoptotic (Hockenbery et al., 1993; Kane et al., 1993; Greenlund et al., 1995; Rabizadeh et al., 1995) neuronal death. Overexpression of glutathione peroxidase inhibits apoptosis (Hockenbery et al., 1993), and the overexpression of copper–zinc superoxide dismutase (SOD) has been shown to inhibit apoptosis both in neural cell lines (Rabizadeh et al., 1995) and in primary neurons in culture (Greenlund et al., 1995). Free radicals may serve either as effectors of cell death, resulting in oxidative damage of DNA, lipids, and proteins, or as signaling molecules via redox-sensitive cellular factors such as c-junor NFκB (Bredesen, 1995)
Here we report that new mRNA and protein synthesis, induction of ICE-like activity, and formation of ROS are sequential steps in K+ deprivation-induced apoptosis of cerebellar granule neurons.
MATERIALS AND METHODS
Materials. Actinomycin D, cycloheximide,N-tert-butyl-α-phenylnitrone (PBN), SOD,N-acetyl-l-cysteine, vitamin E, glutathione, catalase, cytosine-β-d-arabinofuranoside (Ara-C), phenylmethylsulfonyl fluoride (PMSF),N-tosyl-l-lysyl chloromethylketone (TLCK), N-tosyl-l-phenylalanyl chloromethylketone (TPCK), E-64, aprotinin, pepstatin, poly-(l-lysine), and fluorescein diacetate were obtained from Sigma (St. Louis, MO). Biotinylated anti-rabbit IgG, control rabbit IgG, peroxidase, and alkaline phosphatase-conjugated anti-rabbit IgG were from Dakopatts (Glostrup, Denmark). RNase, avidin-alkaline phosphatase, fluorescein isothiocyanate-streptavidin, nitroblue tetrazolium chloride, and 5-bromo-4-chloro-3-indolyl phosphate were purchased from Boehringer Mannheim (Mannheim, Germany). Ac-YVAD-chloromethylketone (Ac-YVAD-CMK) and DABCYL-YVADAPV-EDANS were purchased from Bachem (Heidelberg, Germany). 2′,7′-Dichlorodihydrofluorescein diacetate (DCF-H2) and dihydrorhodamine 123 were obtained from Molecular Probes (Groningen, Netherlands). ICE p10 (M20) rabbit polyclonal antibody raised against the p10 subunit of murine ICE was purchased from Santa Cruz Biotechnology (Heidelberg, Germany). p53-antibody (AB-1) was obtained from Oncogene Science (Cam- bridge, MA) .
Neuronal cultures. Cerebellar granule neurons were prepared from 8-d-old Sprague–Dawley rat pups (Interfauna, Tuttlingen, Germany) as described previously (Novelli et al., 1988; Marini and Paul, 1992;Weller et al., 1992, 1994c). Cells were dissociated from freshly dissected cerebella by mechanical disruption in the presence of trypsin and DNase and then plated in poly-l-lysine-precoated 35 or 100 mm culture plates (Nunc, Wiesbaden, Germany) or 24-well plates. Cells were seeded at a density of 2.1 × 105cells/cm2 in basal modified Eagle’s (BME) medium supplemented with 10% fetal calf serum, 2 mmglutamine, and 20 μg/ml gentamycin. Cells maintained in depolarizing conditions were supplemented with 20 mm KCl to achieve a final concentration of 25 mmK+. Ara-C (10 μm) was added to the culture medium after 24 hr to arrest the growth of non-neuronal cells. Cultures were fed 5 mmd-glucose on day in vitro (DIV) 7. Cultures generated by this method have been characterized and shown to contain >95% granule neurons (Nicoletti et al., 1986; M. Weller, unpublished data).
Treatment of cultures. All experiments were performed using cerebellar granule neurons at DIV 8. Neuronal viability was determined 24 hr later. Culture medium was replaced with serum-free BME medium containing 5 mm KCl and supplemented with glutamine and gentamycin as indicated above. Control cells were treated identically but maintained in serum-free BME medium supplemented with 25 mm KCl (final concentration). All drug solutions were sterilized by filtration and added in a volume of 2–20 μl.
Morphometric analysis of cell viability. Neurons plated in 35 mm dishes were used for assessment of viability. Neuronal viability was assessed by the capability of cells to diesterify and retain fluorescein diacetate in their cytoplasm (Marini and Paul, 1992; Weller et al., 1992). Cells were washed with Locke’s buffer (in mm): 154 NaCl, 5.6 KCl, 2.3 CaCl2, 1 MgCl2, 3.6 NaHCO3, 5 HEPES, and 20 glucose, incubated for 3 min at 37°C with 5 μg/ml fluorescein diacetate dissolved in Locke’s buffer, washed with Locke’s buffer again, and then examined under ultraviolet light microscopy. Three randomly chosen fields from each dish were digitized by a blinded observer using a CCD camera connected to an image processor (MCID-IV, Imaging Research, St. Catharine’s, Ontario, Canada). Images were averaged, filtered, and the total number of cells was estimated automatically by MCID-IV computer software. Parameters for the detection of granule neurons were the shape of cells (round) and the average diameter of the granule neurons (7–12 μm). We confirmed the accuracy of computerized granule-neuron counting after switch to either high K+ or low K+ serum-free medium by comparing the counts of a blinded observer with the counts of the computer in several experiments. In high K+ controls, 1500–1800 cerebellar neurons were counted per field. Viability is expressed as percentage of cerebellar granule neurons retaining fluorescein compared with high K+ control cultures treated with the same drug.
Analysis of production of ROS. Cerebellar granule neurons were seeded in 24-well plates at a density of 2.1 × 105 cells/cm2 as described above. After the switch to medium containing low concentrations of KCl (5 mm) at DIV 8, the cultures were incubated for 30 min with 2 μg/ml dihydrorhodamine 123 or 1 μg/ml DCF-H2 at the time points indicated. Cells were washed with Locke’s buffer and read on a CytoFluor 2350 plate reader (Millipore, Bedford, MA) at 485 nm excitation and 530 nm emission. Neurons that were not switched did not show any fluorescence and were used for background readings.
Fluorometric quantification of DNA fragmentation. For quantitative DNA fluorometry (Weller et al., 1994a,b), detached cerebellar granule neurons were harvested and pooled with the attached cells. All cells were lysed in 10 mm Tris-HCl, pH 7.5, 10 mm EDTA, and 0.2% Triton X-100 for 10 min on ice. Fragmented DNA was separated from nucleus-attached DNA by high-speed centrifugation. After disruption of the pellets by brief sonication and RNase A digestion, fragmented and pelleted DNA was measured by ethidium bromide (0.5 μg/ml) fluorometry using 530 nm excitation and 620 nm emission wavelengths (Cyto- Fluor 2350). The linear range was between 0.05 and 3 μg/ml DNA. Percent fragmentation was calculated by dividing fragmented DNA by the total sum of fragmented and pelleted DNA.
Detection of ICE and p53 expression. Expression of ICE was detected by Western blot (Weller et al., 1994b, 1995). Cerebellar granule neurons were seeded on 100 mm dishes. Soluble protein was harvested from cells lysed for 15 min on ice in 50 mm Tris-HCl, pH 8, containing 120 mm NaCl, 0.5% NP-40, 2 μg/ml aprotinin, 100 μg/ml PMSF, 10 μg/ml leupeptin, 50 mm sodium fluoride, and 200 μm sodium vanadate followed by high-speed centrifugation at 4°C. Twenty micrograms protein per lane were separated by 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electroblotted to nitrocellulose. Immunodetection involved blocking for 1 hr in 10 mm Tris-HCl, pH 7.5, containing 150 mm NaCl, 0.1% Tween 20, 5% skim milk, and 2% BSA; incubation with antibodies to ICE p10 (2 μg/ml) or p53 (2 μg/ml) overnight at 4°C; incubation with biotinylated anti-rabbit IgG (1:20,000 in PBS/0.1% Tween 20), streptavidin-alkaline phosphatase (1:1,000), and nitroblue tetrazolium chloride (0.41 mm), and 5-bromo-4-chloro-3-indolyl phosphate (0.38 mm) in 200 mm Tris-HCl, pH 9.5, containing 10 mm MgCl2 as substrate.
ICE activity. Cerebellar granule neurons were seeded in 24-well plates at a density of 2.1 × 105cells/cm2 as described above. After the switch to medium containing low (5 mm) or high (25 mm) concentrations of KCl at DIV 8, cerebellar granule neurons were washed with Locke’s buffer and made permeable by 0.03% digitonin for analysis of ICE-like protease activity at time points indicated. After 10 min, the fluorogenic ICE substrate DABCYL-YVADAPV-EDANS (20 μm) was added. Fluorescence was determined in 10 min intervals for 1 hr using 360 nm excitation and 480 nm emission wavelengths (CytoFluor 2350) in the 24-well plates. The fluorometric intensity peaked at 20 min. This time point was used for statistical analysis.
Statistics. Data are expressed as mean ± SEM. Statistical significance was assessed by two-tailed Student’st test (comparison of 2 groups) or one-way ANOVA followed by Scheffe’s post hoc test (comparison of more than 2 groups). All experiments reported here represent at least three independent replications performed in triplicate.
RESULTS
K+ deprivation-induced apoptosis of cerebellar granule neurons
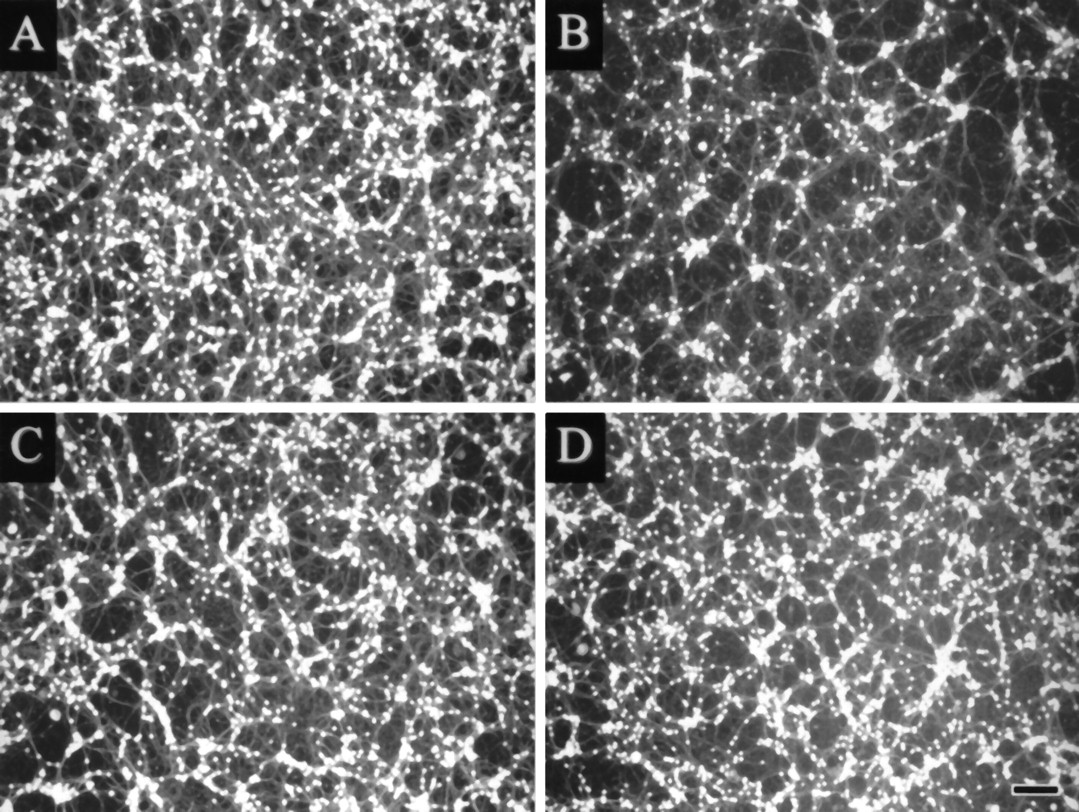
Apoptosis of differentiated cerebellar granule neurons can be induced by lowering the extracellular K+concentration from 25 to 5 mm DIV 8 (D’Mello et al., 1993; Yan et al., 1994; Galli et al., 1995). Switch to low K+ concentrations decreases viability of cerebellar granule neurons by >50% when measured after 24 hr. As previously reported (Yan et al., 1994), exposure to the transcriptional inhibitor actinomycin D and the translational inhibitor cycloheximide at the time of K+ deprivation protected against low K+-induced neuronal death as assessed by staining with fluorescein diacetate at 24 hr (Fig. 1, Table 1). Quantitative DNA fluorometry showed that at this time point 28 ± 3% of the total DNA was fragmented in neurons maintained in low K+ versus 10 ± 2% maintained in high K+ medium. Treatment with actinomycin D and cycloheximide prevented DNA fragmentation induced by K+ withdrawal (Table 1). Thus, new mRNA and protein synthesis appear to be required for K+deprivation-induced apoptosis of cerebellar granule neurons.

Cycloheximide and PBN prevent low K+ deprivation-induced apoptosis. Neurons at DIV 8 were switched from high K+ (25 mm) to low K+ (5 mm) serum-free culture medium and stained for viable cells with fluorescein diacetate 24 hr later. Stained cells were visualized under ultraviolet illumination and digitized using a CCD camera. Culture A was maintained in 25 mm, cultures B–D were kept at 5 mm K+. Low K+ neurons were either untreated (B) or treated with 10 μg/ml cycloheximide (C) or 100 μm PBN (D). Scale bar, 50 μm.
Effects of actinomycin D and cycloheximide on K+ deprivation-induced apoptosis of cerebellar granule neurons
p53 and ICE protein expression in low K+-induced apoptosis of cerebellar granule neurons
We studied the role of two putative mediators of apoptotic cell death by Western blot. The tumor suppressor gene product p53 mediates apoptosis after DNA damage in certain cell types (Clarke et al., 1993; Lowe et al., 1993a,b). Although DNA breaks and DNA fragmentation are characteristic features of K+deprivation-induced apoptosis of cerebellar granule neurons, we did not find any expression of p53 protein by Western blot up to 24 hr after K+ deprivation (Fig.2A). T98G human malignant glioma cells, which express abundant mutant p53, were used as a positive control (Ullrich et al., 1992).
Effects of protease inhibitors on K+deprivation-induced apoptosis of cerebellar granule neurons

p53 and ICE protein expression of cerebellar granule neurons during K+ deprivation-induced apoptosis. Protein expression was determined at the time points indicated after switch to serum-free medium with high (25 mm) or low (5 mm) concentrations of K+. A, p53 protein expression was not detected in cerebellar granule neurons after switch to low or high K+ medium. T98G human malignant glioma cells served as a positive control. B, The 45 kDa ICE precursor protein, but not the cleaved 10 kDa protein, was detected in cerebellar granule neurons.
The cysteine protease ICE is the mammalian homolog of CED-3, a C. elegans gene product required for cell death (Yuan et al., 1993). Although we detected the 45 kDa ICE precursor (proICE) protein by immunoblot analysis with an antibody directed against the p10 subunit of murine ICE, we did not observe a band corresponding to the cleaved and active 10 kDa subunit or a decrease in expression of the 45 kDa proICE-protein, arguing against an activation of ICE (Fig.2B).
Inhibition of ICE-like proteases protects against apoptosis of cerebellar granule neurons
To screen for other ICE-like proteases that might be involved in low K+-induced apoptosis of cerebellar granule neurons, we used Ac-YVAD-CMK, an irreversible and specific peptide inhibitor of ICE and ICE-like proteases (Thornberry et al., 1992;Lazebnik et al., 1994; Enari et al., 1995). Figure 3shows that Ac-YVAD-CMK attenuated low K+-induced apoptosis of cerebellar granule neurons in a concentration-dependent manner when added at the time of K+ withdrawal. Additional preincubation with Ac-YVAD-CMK for 1 hr resulted in almost complete survival of cerebellar granule neurons as assessed 24 hr after K+ deprivation (Fig. 3A). The peptidic nature of the ICE inhibitor limits cell penetration, which may explain the requirement of micromolar concentrations and preincubation with this agent for complete inhibition of apoptosis. Treatment with Ac-YVAD-CMK had no effect on granule neurons maintained at high K+ concentrations, indicating that the ICE inhibitor is not toxic at concentrations tested. The cysteine protease inhibitors E-64 and leupeptin, the aspartic protease inhibitor pepstatin, the serine protease inhibitor PMSF, the chymotrypsin inhibitor TPCK, and the trypsin inhibitor TLCK had no effect on K+ deprivation-induced apoptosis at concentrations tolerated by the granule neurons (Table 2).
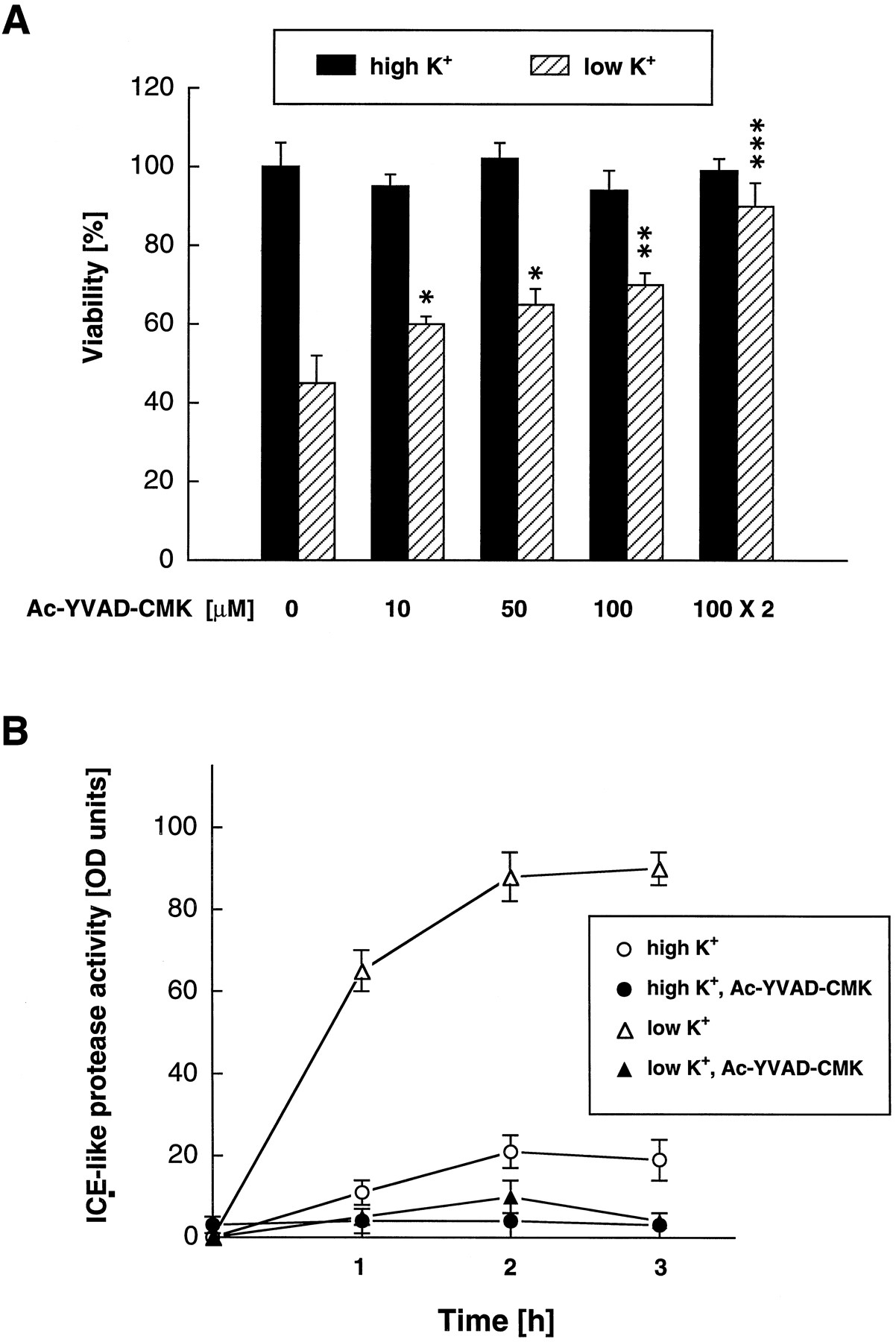
The role of ICE-like proteases in K+ deprivation-induced apoptosis. A, Inhibition of ICE-like protease activity abrogates K+ deprivation-induced apoptosis of cerebellar granule neurons. Apoptosis was induced by K+deprivation. Neurons maintained in high K+ medium were used to control for possible toxic effects of the ICE inhibitor. Neuronal viability was assessed by fluorescein diacetate staining and interactive counting by an image analysis system. The ICE inhibitor tetrapeptide Ac-YVAD-CMK was added at the time point of medium switch. Additional preincubation with Ac-YVAD-CMK for 1 hr before switch showed the best protection (100 μm × 2). ***p < 0.001, **p < 0.01, *p < 0.05 compared with untreated granule neurons maintained at low K+. B, ICE-like protease activity is induced after K+deprivation. ICE-like protease activity was measured with the fluorogenic ICE substrate DABCYL-YVADAPV-EDANS in the absence or presence of Ac-YVAD-CMK in low or high K+ medium at defined time points after medium switch.
To confirm that K+ deprivation induces ICE-like proteases, ICE-like proteolytic activity was measured using the fluorogenic ICE substrate 4-(4-dimethylaminophenylazo)benzoyl-YVADAPVU-5-[(2-aminoethyl)amino]-naphthalene-1-sulfonic acid (DABCYL-YVADAPV-EDANS), which contains the cleavage site of the ICE-like enzymes (Pennington and Thornberry, 1994). Measurements at 1, 2, and 3 hr after switch to low K+ confirmed an increase in ICE-like protease activity that was blocked by Ac-YVAD-CMK (Fig. 3B).
Antioxidants and free radical spin traps block K+deprivation-induced apoptosis of cerebellar granule neurons
ROS have been implicated as important effector molecules in apoptotic cell death (Hockenbery et al., 1993; Kane et al., 1993;Zamzami et al., 1995). Therefore, we studied the effects of the antioxidants SOD, N-acetyl-l-cysteine, catalase, glutathione, vitamin E, and PBN on apoptosis of cerebellar granule neurons induced by K+ deprivation at DIV 8. PBN is a free radical spin trap that reacts with free radicals to form stable adducts and can therefore serve as a free radical scavenger (Knecht and Mason, 1993; Schulz et al., 1995a). Exposure to PBN, SOD, and N-acetyl-l-cysteine concentration dependently inhibited cerebellar granule neuron death at 24 hr (Fig.4). Treatment with glutathione (100 μm: 88 ± 5% vs 51 ± 4% viability normalized to 25 mm K+;p < 0.01), vitamin E (1 mg/ml: 78 ± 6% vs 51 ± 4%; p < 0.05), and catalase (1000 U/ml: 72 ± 5% vs 51 ± 4%; p < 0.05) was also effective.

K+ deprivation-induced apoptosis requires ROS formation. Effects of treatment withN-tert-butyl-α-phenylnitrone (PBN) (A), superoxide dismutase (SOD) (B), and N-acetyl-l-cysteine (C). Neuronal viability was assessed by fluorescein diacetate staining and interactive counting by an image analysis system at 24 hr after switching to low K+. ***p < 0.001, **p < 0.01, *p < 0.05 compared with untreated cells switched to low K+.D shows the time course of ROS production as detected by dihydrorhodamine 123 fluorescence. Because the switch to serum-free high K+ medium led to a minor increase in the fluorescence of both dyes compared with nonswitched controls, the difference in fluorescence between high K+- and low K+-treated cultures at every time point is given.
To confirm the generation of ROS directly and to follow the time course of ROS production, we used the oxidation-sensitive indicators dihydrorhodamine 123 and DCF-H2. These lipophilic nonfluorescent indicators are easily deacetylated to their active forms after penetrating cells. They are oxidized by ROS to the fluorescent dyes rhodamine 123 and DCF, respectively. Because the fluorescent derivative rhodamine 123 is positively charged, it moves to the inside-negative mitochondrial environment (Johnson et al., 1980). Staining with dihydrorhodamine 123 showed a gradual increase in fluorescence over time with a maximum at 4 hr after K+ deprivation and a slow decline thereafter (Fig. 4D). A similar time course of ROS production with a maximum at 4 hr was obtained by staining with DCF-H2 with the exception of an additional minor peak of DCF fluorescence at 1 hr.
Four hours after switch to low K+ concentrations, we used confocal laser microscopy to confirm that ROS were generated intracellularly. Culture dishes were loaded with 4 μg/ml dihydrorhodamine 123 or 2 μg/ml DCF-H2 for 30 min. After this incubation period, cellular fluorescence was imaged using a laser scanning confocal microscope at 485 nm excitation and 530 nm emission. For both dyes, fluorescence was strictly localized within cerebellar granule neurons (data not shown).
DNA fragmentation and survival
Fragmentation of nuclear DNA is a typical feature of apoptosis. A 17% increase of DNA fragmentation was observed at 24 hr after switch to low K+ medium compared with switch to high K+ medium (Fig. 5A). This increase was blocked by treatment with actinomycin D and cycloheximide, but not with PBN or Ac-YVAD-CMK. Yet, these drugs greatly enhanced neuronal viability as assessed by staining with fluorescein diacetate at the same time point. To confirm that PBN- and Ac-YVAD-CMK-mediated rescue of staining with fluorescein diacetate at 24 hr after K+ deprivation truly reflected survival compared with untreated neurons, and that DNA fragmentation at 24 hr did not inevitably predict neuronal death, cerebellar granule neurons were switched back to their initial conditioned medium 24 hr after K+ deprivation and cultured for another 48 hr. According to staining with fluorescein diacetate, reexposure to the original medium arrested the process of neuronal apoptosis induced by K+ deprivation in that neuronal counts exceeded 35% in cultures switched back to high K+ medium, whereas <10% of the neurons survived 72 hr culture in low K+ medium (Fig. 5B). Further, staining with fluorescein diacetate at 72 hr confirmed near-complete rescue from K+ deprivation-induced apoptosis in neurons exposed to either cycloheximide, ICE-inhibitor, or PBN during K+ deprivation from 0 to 24 hr. At 72 hr, DNA fragmentation in neurons treated with PBN and Ac-YVAD-CMK was reduced compared with measurements at the end of K+deprivation (24 hr). In fact, DNA fragmentation of PBN- or Ac-YVAD-CMK-treated neurons at 72 hr was not different from that of cycloheximide-treated neurons (Fig. 5A), suggesting that DNA repair takes place during high K+-mediated rescue of the neurons between 24 and 72 hr. Because there was no decrease in total DNA harvests between 24 and 72 hr, a selective loss of fragmented DNA into the culture medium is unlikely to account for the decrease of DNA fragmentation.

DNA fragmentation and survival after K+ deprivation. Cell viability and specific DNA fragmentation were measured 24 (A) and 72 (B) hr after switch to low K+ medium. Specific DNA fragmentation was calculated by subtracting baseline DNA fragmentation before calculating ratios of fragmented versus pelleted DNA in experimental samples. In these experiments, baseline DNA fragmentation never exceeded 9% in cells switched to high K+concentrations. A, Treatment with cycloheximide (CHX), Ac-YVAD-CMK, and PBNgreatly enhanced neuronal viability at 24 hr after K+ withdrawal as assessed by staining with fluorescein diacetate. However, only treatment with cycloheximide prevented DNA fragmentation (***p < 0.001).B, When cerebellar granule neurons were switched back to their initial conditioned medium 24 hr after K+deprivation and cultured for another 48 hr, significantly more neurons remained viable compared with neurons continuously maintained in low K+ medium for 72 hr (p < 0.01). Furthermore, treatment with cycloheximide, ICE inhibitor, and PBN was protective. After switch back to preconditioned high K+ medium, DNA fragmentation was markedly reduced in treated and untreated cells compared with neurons at the end of K+ deprivation (24 hr) and with neurons maintained in low K+ for 72 hr (**p < 0.01).
Sequential mechanisms of apoptosis in cerebellar granule neurons
To characterize the sequential mechanisms during K+ deprivation-induced apoptosis of cerebellar granule neurons, we determined the point of commitment to death as the latest time at which neurons can be rescued after switch to low K+ (5 mm) by addition of a given survival agent (Fig. 6). Neurons were shifted to low K+ medium and, at different time points, actinomycin D, cycloheximide, or PBN was added to the culture medium. At 24 hr, neuronal survival was evaluated by fluorescein diacetate staining. Neurons were rescued by addition of PBN up to 3 hr after the initial switch to low K+. The therapeutic window for the transcriptional and translational inhibitors actinomycin D and cycloheximide, respectively, was shorter. To obtain a significant protection, actinomycin D had to be added within 40 min and cycloheximide within 60 min after K+ deprivation. These results suggest that, within 1 hr, most granule neurons have transcribed and translated the message for one or more putative “suicide proteins” and that ROS are produced downstream of this protein synthesis. Because the peptide ICE inhibitor required prolonged exposure for cell protection, no such experiment for the kinetics of ICE inhibitors could be performed.

Effects of delayed treatment to prevent apoptosis triggered by K+ deprivation of cerebellar granule neurons. Rescue treatments included actinomycin D (1 μg/ml), cycloheximide (10 μg/ml), and PBN (100 μm) at the time points indicated. Neuronal viability was evaluated by staining with fluorescein diacetate and automated counting by image analyzing software. A significant protection (p < 0.01, ANOVA) was provided by actinomycin D up to 40 min, cycloheximide up to 1 hr, and PBN up to 3 hr.
To further elucidate the sequential mechanisms of apoptosis in cerebellar granule neurons, we studied the effects of cycloheximide, Ac-YVAD-CMK, and antioxidants on ICE-like protease activity as measured with the fluorogenic ICE substrate DABCYL-YVADAPV-EDANS. As expected, Ac-YVAD-CMK completely blocked ICE-like activity (Fig.7A). In addition, neurons treated with cycloheximide at the time of switching did not exhibit ICE-like activity, suggesting that the putative killer proteins are an upstream positive regulator of ICE-like protease activity. In contrast, PBN, SOD, and N-acetyl-l-cysteine did not affect ICE-like protease activity. In another experiment, we studied the effects of cycloheximide, Ac-YVAD-CMK, and PBN on ROS production with the oxidation-sensitive indicators DCF-H2 and dihydrorhodamine 123 at 4 hr after K+ deprivation (Fig. 7B,C). As expected, PBN prevented the increase of DCF and rhodamine 123 fluorescence. However, cycloheximide and Ac-YVAD-CMK had a similar effect, suggesting that both agents interfere with the apoptotic cascade upstream of ROS formation.

Interaction of RNA translation, induction of ICE-like protease activity, and formation of ROS. A, Cycloheximide (CHX) and Ac-YVAD-CMK, but not PBN, SOD, orN-acetyl-l-cysteine (N-AC) prevent the induction of ICE-like activity as measured with the ICE-substrate DABCYL-YVADAPV-EDANS at 3 hr after K+ deprivation. Cycloheximide (CHX), Ac-YVAD-CMK, and PBNattenuate the production of ROS as detected by 2′,7′-DCF (B) and rhodamine 123 (C) fluorescence at 4 hr after switch to low K+ medium. **p < 0.01, ***p < 0.001 compared with untreated granule neurons switched to low K+ medium.
DISCUSSION
The regulation of programmed cell death in the developing nervous system involves target-derived survival factors, afferent synaptic activity, and hormone- and cytokine-dependent signaling. Cerebellar granule neurons undergo extensive cell death characterized by nuclear DNA fragmentation between postnatal days 5 and 9 (P5 and P9) in vivo (Wood et al., 1993). Mossy fiber input to the granule cell layer begins at P5, and synapse formation occurs by P12 (Burgoyne and Cambray-Deakin, 1988). Similarly, formation of synaptic contacts between parallel fibers and Purkinje cell dendrites is prominent between the second and third postnatal weeks, corresponding to the maximal rate of synaptogenesis (Burgoyne and Cambray-Deakin, 1988). Therefore, cell death before synaptogenesis may help to regulate granule neuron number.
In the absence of exogenously added growth or survival factors other than those present in fetal calf serum, cerebellar granule neurons can be differentiated and maintained in vitro for weeks in the presence of high concentrations of K+. These neurons undergo highly synchronous apoptosis when deprived of depolarizing concentrations of K+ (D’Mello et al., 1993; Yan et al., 1994; Galli et al., 1995). We have previously suggested that the functional innervation of postmigratory granule neurons during cerebellar development by mossy fibers and climbing fibers may prevent further elimination of these neurons by blocking their programmed death (Yan et al., 1994). Therefore, K+ deprivation-induced apoptosis of differentiated cerebellar granule neurons may be a model of neuronal death after deafferentation. On the other hand, studies that examined the postnatal histogenesis of the cerebellum in normal mice, neurologically mutant mice, and chimeras between normal and mutant mice show that there is a numerical matching of granule neurons with Purkinje cells. They indicate that a target-related cell death of granule cells occurs during development of the cerebellum. The survival-promoting effects of K+ in cortical neurons are mediated by a specific neurotrophin, brain-derived neurotrophic factor (Ghosh et al., 1994), suggesting that K+ deprivation-induced apoptosis of cerebellar granule neurons may also involve specific unidentified growth factors.
A requirement for new mRNA and protein synthesis is a typical feature of neuronal apoptosis (Johnson and Deckwerth, 1993). Apoptosis of several non-neuronal cells, including lymphoid cells and neoplastic cells, in contrast, is induced or augmented by inhibitors of mRNA and protein synthesis (Weller et al., 1994a,b). Here we report that new mRNA and protein synthesis, ICE-like proteases, and production of ROS are necessary and sequential events of apoptosis in this paradigm of neuronal apoptosis. Time-kinetic studies revealed that gene activation required for apoptosis is an early event, because exposure to actinomycin D and cycloheximide >60 min after K+withdrawal failed to enhance neuronal survival when assessed at 24 hr. This is in contrast to nerve growth factor (NGF) deprivation-induced apoptosis of sympathetic neurons, 50% of which are rescued when protein synthesis was blocked within 22 hr of NGF deprivation (Martin et al., 1992) .
ICE-like proteases have recently been identified as important mediators of apoptotic cell death, e.g., in Fas/APO-1-mediated apoptosis (Enari et al., 1995; Kuida et al., 1995; Los et al., 1995) and programmed cell death of motor neurons (Milligan et al., 1995). The detection of ICE-like activity by a substrate peptide and the protective effects of an inhibitor of ICE-like proteases strongly suggest that a member of the ICE family of proteases is also involved in K+ deprivation-induced apoptosis of cerebellar granule neurons. Treatment with other chloromethylketone inhibitors of proteases (TPCK and TLCK) that lack aspartate in the P1 position and with other protease inhibitors did not result in enhanced neuronal survival. We believe that ICE-like protease activity operates downstream of the putative killer genes because the induction of enzymatic activity was blocked by cycloheximide.
The fluorogenic ICE substrate DABCYL-YVADAPV/EDANS and the peptide ICE inhibitor Ac-YVAD-CMK do not differentiate between members of the ICE family (Lazebnik et al., 1994; Nicholson et al., 1995; Tewari et al., 1995). To examine whether ICE or other ICE-like proteases mediate K+ deprivation-induced apoptosis, we studied the role of ICE by Western blotting. Mammalian ICE is synthesized as an inactive 45 kDa precursor processed proteolytically to generate active enzyme that comprises polypeptides of 20 kDa (p20) and 10 kDa (p10) (Thornberry et al., 1992). We detected the expression of the 45 kDa precursor protein in cerebellar granule neurons but did not observe a 10 kDa protein or a decrease of the precursor protein up to 8 hr after K+ deprivation, indicating that the precursor protein is not cleaved to the active ICE subunits. The p10 antibody we used was directed against the mouse p10 subunit. The mouse and rat p10 subunits show 95% homology (Keane et al., 1995). The detection of the 45 kDa precursor protein appears to prove that the antibody detects rat ICE. In contrast, the p10 subunits of other members of the ICE-like protease family show less homology (Kumar, 1995). Our results suggest that not ICE itself, but one or more other members of the ICE family are important regulators of apoptosis in cerebellar granule neurons.
ROS have been implicated as mediators of excitotoxic (Coyle and Puttfarcken, 1993; Schulz et al., 1995a,b) and apoptotic (Hockenbery et al., 1993; Kane et al., 1993; Greenlund et al., 1995; Rabizadeh et al., 1995) neuronal cell death. One major aim of this study was to examine the role of ROS in K+ deprivation-induced apoptosis of cerebellar granule neurons. Using redox-sensitive dyes, we found that ROS are generated during apoptosis and that inhibitors of ROS formation prevented neuronal death. Scavengers of superoxide (SOD, PBN) were equally effective as scavengers of peroxides (glutathione, catalase), antioxidants (vitamin E), andN-acetyl-l-cysteine, a thiol antioxidant and glutathione precursor. These pharmacological data indicate that superoxide, hydrogen peroxide, and hydroxyl radicals are produced in K+-deprived cerebellar granule neurons. Measurements with the redox-sensitive dyes DCF-H2and dihydrorhodamine 123 indicate that there is an increase in the formation of ROS that peaks at 4 hr and slowly declines thereafter. The time course of the production of free radicals correlates well with the ability of PBN to protect against apoptosis. ROS formation is a downstream event in granule neuron apoptosis because both cycloheximide and the ICE inhibitor prevented ROS formation.
The antiapoptotic properties of the bcl-2 oncogene product have been attributed to the detoxification of ROS (Hockenbery et al., 1993). Although cerebellar granule neurons maintained in high K+ medium express bcl-2 mRNA (Montpied et al., 1993), we have not been able to detect the BCL-2 protein in cerebellar granule neurons maintained in either high K+ or low K+ medium (de Luca et al., 1996). Thus, cerebellar granule neurons may be specifically vulnerable to ROS-induced toxicity.
ROS formation is a downstream event in the intracellular cascade leading to apoptosis of cerebellar granule neurons. We show that gene induction and activation of ICE-like proteases are necessary for the generation of ROS. The mechanism of ROS-induced cytotoxicity is unclear. Targets of cell injury by ROS are cellular macromolecules including DNA. ROS-mediated DNA damage is unlikely to be responsible for cell death because p53 activity was not induced and because antioxidants and inhibitors of ICE-like proteases inhibited cell death but not DNA fragmentation. Apoptosis of neuronal precursors in the cerebellum of transgenic mice lacking functional p53 is similar to that in wild-type mice, arguing for a p53-independent apoptotic pathway of physiological cerebellar granule cell loss during development (Wood and Youle, 1995). In contrast, p53 expression is required for gamma irradiation-induced apoptosis of cerebellar granule neuron precursorsin vivo.
In contrast to their recognized role as damaging molecules, ROS have recently been implicated as important signal transduction molecules. Sympathetic neurons show an increase in ROS that peaks at 3 hr after deprivation of NGF. If NGF was added back to the culture medium after the period of peak ROS generation, apoptosis was completely prevented, suggesting that ROS production serves as an early signal, rather than a toxic agent, to mediate apoptosis (Greenlund et al., 1995). Commitment to cell death is defined as the time after which readdition of trophic support or other treatment will no longer prevent cell death. Fifty percent of sympathetic neurons can be rescued when protein synthesis was blocked 22 hr after NGF deprivation (Martin et al., 1992). Therefore, it was argued that ROS may signal by modulating transcription either directly or through known redox-sensitive proteins or transcription factors. This cascade is different in K+ deprivation-induced apoptosis of cerebellar granule neurons, because (1) neurons can only be rescued after K+ deprivation when inhibitors of transcription and translation are added during the first hour (whereas free radical scavengers protect when added up to 3 hr after switch to low K+) and (2) generation of ROS is a downstream event of protein synthesis.
Apoptosis of cerebellar granule neurons induced by K+ deprivation is associated with DNA fragmentation that is prevented by inhibition of protein synthesis. However, treatment with the peptide inhibitor of ICE-like proteases, Ac-YVAD-CMK, and a free radical scavenger did not block DNA fragmentation at 24 hr after K+ deprivation. Although 90% of cells were viable at that time point with either treatment, almost 20% of the total DNA was fragmented. When switched back to conditioned medium, this fragmentation did not lead to further cell death, but DNA appeared to be partially repaired. These results argue against the hypothesis that the degree of DNA fragmentation observed in cerebellar granule neurons will necessarily lead to apoptotic death. The finding that apoptosis can be induced in enucleated cells (cytoblasts) provided evidence that events occurring independently or upstream of nuclear alterations have a major impact in the regulation and execution of apoptosis (Jacobson et al., 1994;Schulze-Osthoff et al., 1994). Our data indicate that DNA fragmentation is an epiphenomenal event in K+deprivation-induced apoptosis of cerebellar granule neurons.
Conclusions
Previous studies have shown that K+deprivation-induced apoptosis of cerebellar granule neurons requires new mRNA and protein synthesis. Here we present data that provide a framework for the understanding of the apoptotic cascade in these neurons. Our results suggest that an ICE-like protease is a critical mediator of this apoptotic cell death. Although we cannot identify the specific protease of the ICE family involved in apoptosis of cerebellar granule neurons, it is unlikely to be ICE itself. Further, ROS are essential mediators of apoptotic cell death. Because inhibition of transcription, translation, and of ICE-like activity prevents production of ROS, ROS formation is a downstream event in the intracellular cascade leading to apoptotic cell death. The mechanisms by which ROS induce cell death remain to be elucidated.
Footnotes
We thank L. Dumitrescu and I. Müller for excellent technical assistance and Dr. P.-A. Löschmann for valuable discussions.
Correspondence should be addressed to Dr. Jörg B. Schulz, Department of Neurology, University of Tübingen, Hoppe-Seyler-Strasse 3, D-72076 Tübingen, Germany.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}