

Abstract
Transcription factor c-Jun is proposed to control neuronal cell death and survival, but its activation by N-terminal phosphorylation and the underlying activity of the c-Jun N-terminal kinases (JNKs) remain to be elucidated in the adult mammalian brain. We generated a polyclonal antiserum that specifically recognizes c-Jun phosphorylated at its serine 73 (S73) residue after UV irradiation of 3T3 cells. Disruption of the c-jun locus in 3T3 cells abolished this reaction, and retransfection of the human c-jun at the c-jun−/− background restored it.
The phospho-c-Jun antiserum was used to visualize N-terminally phosphorylated c-Jun in the adult rat brain with cellular resolution. Prolonged c-Jun S73 phosphorylation was detected in affected neurons up to 5 d after transient occlusion of medial cerebral artery or up to 50 d after transection of central nerve fiber tracts. After cerebral ischemia–reperfusion, phosphorylation of c-Jun was linked with induced expression of Fas-ligand (APO-1, CD95-ligand), whose gene is a putative c-Jun/AP-1 target, and with terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) reactivity, a marker for apoptosis. After nerve fiber transection, however, lasting c-Jun phosphorylation occurred in axotomized neurons negative for Fas-ligand or TUNEL and regardless of degeneration or survival. In contrast to these lasting phosphorylation patterns, transient seizure activity by pentylenetetrazole provoked only a brief c-Jun phosphorylation and JNK activation.
In extracts from ischemic or axotomized brain compartments, c-Jun phosphorylation correlated with enhanced long-term JNK activity, and in-gel kinase assays visualized proteins with sizes corresponding to JNK isoforms as the only c-Jun N-terminally phosphorylating enzymes.
These results demonstrate that lasting c-Jun S73 phosphorylation and JNK activity are part of neuronal stress response after neurodegenerative disorders in the adult mammalian brain with Fas-ligand as a putative apoptotic effector.
c-Jun, a component of transcription factor AP-1, may serve a dual function in both cell death and protection–regeneration of neurons (Herdegen et al., 1997b). Suppression of c-Jun expression by antisense-oligonucleotides or functional blockade by microinjection of antibodies protects neonatal hippocampal and sympathetic neurons from neuronal cell death in culture (Schlingensiepen et al., 1993; Estus et al., 1994; Ham et al., 1995). Enhanced c-Jun expression occurs in degenerating and apoptotic neurons after ischemia, nerve fiber transection, and UV irradiation as well as in biopsies from patients suffering from multiple sclerosis, Alzheimer’s disease, and amyotrophic lateral sclerosis (Anderson et al., 1994; Ferrer et al., 1996a,b; Martin et al., 1996). c-Jun is also induced when damaged neurons are rescued by trophic supply and activation of survival programs, e.g., after conditioning ischemia (Sommer et al., 1995), or by regeneration of axotomized retinal ganglion neurons (Schaden et al., 1994) and axotomized rubrospinal neurons (Broude and Bregman, 1996; Giehl and Tetzlaff, 1996;Houlé et al., 1997). Recent findings suggest that the differential expression of AP-1 components, JNK activation (Karin et al., 1997), or modulatory transcription factors such as activating transcription factor 2 (ATF-2) (Herdegen et al., 1997a) could account for the actual outcome of c-Jun effects.
The ability of c-Jun to activate gene transcription is strongly potentiated by phosphorylation at serine (S) 73 and to a lesser extent at S63 (Pulverer et al., 1991; Smeal et al., 1991, 1994), executed by the c-Jun N-terminal kinases [JNKs; also known as stress-activated protein kinases (SAPKs)], which belong to the MAP kinase family (Hibi et al., 1993; Dérijard et al., 1994; Kallunki et al., 1994;Kyriakis et al., 1994). Moreover, JNKs are activated and participate in induction of c-jun transcription in cultured cells after stimulation by growth factors, proinflammatory cytokines, and environmental stressors including ultraviolet light or alkylating agents (Devary et al., 1992; Hibi et al., 1993; Dérijard et al., 1994; Kallunki et al., 1994; Kyriakis et al., 1994; Liu et al., 1996;Musti et al., 1997).
Recently, JNK activation was suggested to be important for apoptosis of neuronal-like PC12 cells after nerve growth factor (NGF) deprivation (Xia et al., 1995). Similarly, JNK activation was also observed during induction of apoptosis by NGF binding to the low-affinity p75 NGF-receptor (Casaccia-Bonnefil et al., 1996), stimulation by TNFα, Fas-ligand, lipid messengers, or hypoxia (Dérijard et al., 1994;Westwick et al., 1995; Chen et al., 1996; X. Yang et al., 1997). In the adult rat brain, disruption of the JNK-3 locus protected hippocampal neurons against excitotoxic neuronal cell death (D. Yang et al., 1997). JNK activation, however, does not inevitably lead to apoptosis, because JNKs are expressed in the untreated intact rat brain (Carletti et al., 1995) and activated after acquisition of novel information (Xu et al., 1997). In addition, apoptosis can occur in the absence of JNK activation (Liu et al., 1996; Goillot et al., 1997;Natoli et al., 1997).
Heretofore, it is not known to which extent the dichotomous role of c-Jun in survival or death depends on its N-terminal phosphorylation, which is exclusively catalyzed by the JNKs in non-neuronal cells (Smeal et al., 1991; Minden et al., 1994a,b). Therefore, we developed an antiserum that specifically recognizes c-Jun phosphorylated at S73 with cellular resolution. Moreover, kinase assays were performed to detect JNK activity in defined compartments after neurodegenerative stimuli such as ischemia, seizures, and axotomy. We also examined in which subpopulation of c-Jun-expressing neurons c-Jun becomes N-terminally phosphorylated by JNKs and whether its activation correlates with cell death and expression of the apoptotic mediator Fas-ligand (Nagata, 1997), a novel target gene of c-Jun/AP-1 (Kasibhatla et al., 1998). This study provides new insights into the involvement of the c-Jun/JNK-axis in the neuronal stress response of the adult mammalian brain.
MATERIALS AND METHODS
Phospho-c-Jun antibody
Generation of the [Cys66, P-Ser73]-h/c-Jun (67–79) peptide. A peptide corresponding to c-Jun aa 67–79 (GLLKLASPELERL) with a cysteine at its N terminus was synthesized manually using a solid phase-based Fmoc/Boc/t-Butyl approach (Otvos et al., 1989). The serine to be phosphorylated was incorporated with an unprotected hydroxyl group. Individual N 9-H-fluorenylmethoxycarbonyl (Fmoc)-protected amino acids were obtained from Bachem (Torrance, CA) with the exception of the unprotected Fmoc-serine, which was prepared in house. The cysteine was incorporated as the N-Boc, S-trityl derivative. Phosphorylation was accomplished postsynthetically by reacting the unprotected serine-OH of the resin-bound peptide with di-t-butyl N, N-diethylphosphoramidite followed by oxidation of the initially obtained P(III) species with t-butylhydroperoxide. After purification (Hoeger et al., 1987), all materials obtained gave satisfactory mass spectral results. The purity of the [Cys66, P-Ser73]-h/c-Jun (67–79) was >80% as assessed by reverse-phase HPLC and capillary zone electrophoresis.
Generation of the anti-phospho-c-Jun antiserum. The [Cys66, P-Ser73]-h/c-Jun (67–79) peptide was coupled to maleimide-activated keyhole limpet hemocyanin (Pierce, Rockford, IL). Two rabbits were immunized with phosphopeptide using complete Freund’s adjuvant (initial injection) or incomplete Freund’s adjuvant (booster injections). Bleeds from one rabbit that gave a much stronger ELISA signal with phosphorylated GST-c-Jun(1–79) than with unphosphorylated GST-c-Jun(1–79) were used in this study. Serum was diluted fourfold with PBS and passed through a first column of GST-c-Jun(1–223) bound to agarose followed by a column of nonphospho-c-Jun peptide (67–79) coupled to Sepharose 6B (Pharmacia, Piscataway, NJ). The flow-through was applied twice onto a column of phospho-c-Jun peptide coupled to Sepharose 6B. After three to four washes with PBS, bound IgG was eluted with 0.1 m glycine, pH 2.5, dialyzed immediately against PBS, and concentrated with Nanosep 10 (Pall Filtron). When necessary, the antiserum was further purified by preadsorption to a protein blot of nonstimulated cells or brain extract.
Surgical procedures
For transection of nerve fiber tracts, the medial forebrain bundle (MFB) and the mamillothalamic tract (MT) were together stereotaxically transected by a 1.5 mm razor blade at bregma −2.5 and 1 mm laterally from the midline in deeply anesthetized (pentobarbital, 60 mg/kg body weight, i.p.) male Sprague Dawley rats (250 gm). For immunocytochemistry, reanesthetized rats (pentobarbital, 100 mg/kg body weight, i.p.) were transcardially perfused with 4% paraformaldehyde after survival times of 12 hr, 24 hr, and 3, 10, 20, and 50 d (each n = 3). The brain was removed, fixed, and cryoprotected with 30% sucrose (Herdegen et al., 1993; Leah et al., 1993). For kinase assays from dorsal root ganglia homogenates, the sciatic nerve was exposed and ligated in anesthetized rats, and approximately 1 cm of the distal stump was removed close to the ligation site to prevent regeneration.
For cerebral ischemia–reperfusion, the left medial cerebral artery (MCA) was occluded for 90 min by siliconized nylon thread in deeply anesthetized (pentobarbital, 100 mg/kg body weight, i.p.) male Sprague Dawley rats. Thereafter, the thread was withdrawn for reperfusion as described elsewhere (Gavrieli et al., 1992). Rats were killed after 3, 12, and 24 hr, and 5 d by intracardial perfusion with 4% paraformaldehyde under deep anesthesia (see above).
After systemic application of the chemoconvulsant pentylenetetrazole (PTZ) (50 mg/kg, i.p.), rats were killed by transcardial perfusion with 4% paraformaldehyde after 15 min, 30 min, 2 hr, and 24 hr (eachn = 3).
Immunocytochemistry and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) reactivity
Cryostat sections (50 μm) of the rat brain were incubated with antisera against phospho-c-Jun (1:3000), c-Jun (1:20,000; a generous gift from Dr. R. Bravo, Bristol Myers Squibb) (Kovary and Bravo, 1991), or Fas-ligand [1:10,000 (Transduction Laboratories, Lexington, KY) and 1:500 (Alexis Corporation)] for 48 hr and visualized by the avidin–biotin complex system with diaminobenzidine as chromogen (Herdegen et al., 1991). For TUNEL staining, brain cryostat sections were incubated with 25 U of terminal transferase, 0.3 μl of Flu-dUTP (0.3 nmol), and 0.3 μl of dATP (3.0 nmol) as described previously (Kallunki et al., 1996).
Kinase assays
For kinase assay, crude tissue nuclear extracts (20 μg of protein) (Asanuma et al., 1995) in lysis buffer were precleared by protein-A Sepharose and immunoprecipitated with a monoclonal JNK-1 antiserum 333.8 (diluted 1:2000; PharMingen, San Diego, CA) and protein-A Sepharose beads. After 5 washes, kinase assays were performed (Hibi et al., 1993) with cold ATP (20 μm) and [γ-32P] ATP (5 μCi) for 20 min at 30°C and stopped by boiling in Laemmli buffer. Samples were separated electrophoretically, and the bands were visualized by autoradiography. For kinase assays from pooled axotomized compartments, rats underwent transection of sciatic nerve and MFB–MT with subsequent decapitation after 3 or 12 d (each n = 10); the respective compartments from untreated rats served as controls (n= 10).
For in-gel kinase assays, GST-c-Jun (1–79) was included in a polymerizing polyacrylamide gel. After electrophoretic separation of pooled nuclear extracts from six rats (20–30 μg per lane), proteins were denaturated in 6 m urea and gently renaturated, and in-gel kinase assay was performed (Hibi et al., 1993). Phosphorylation of the substrate was visualized by autoradiography.
RESULTS
Characterization of phospho-c-Jun antiserum
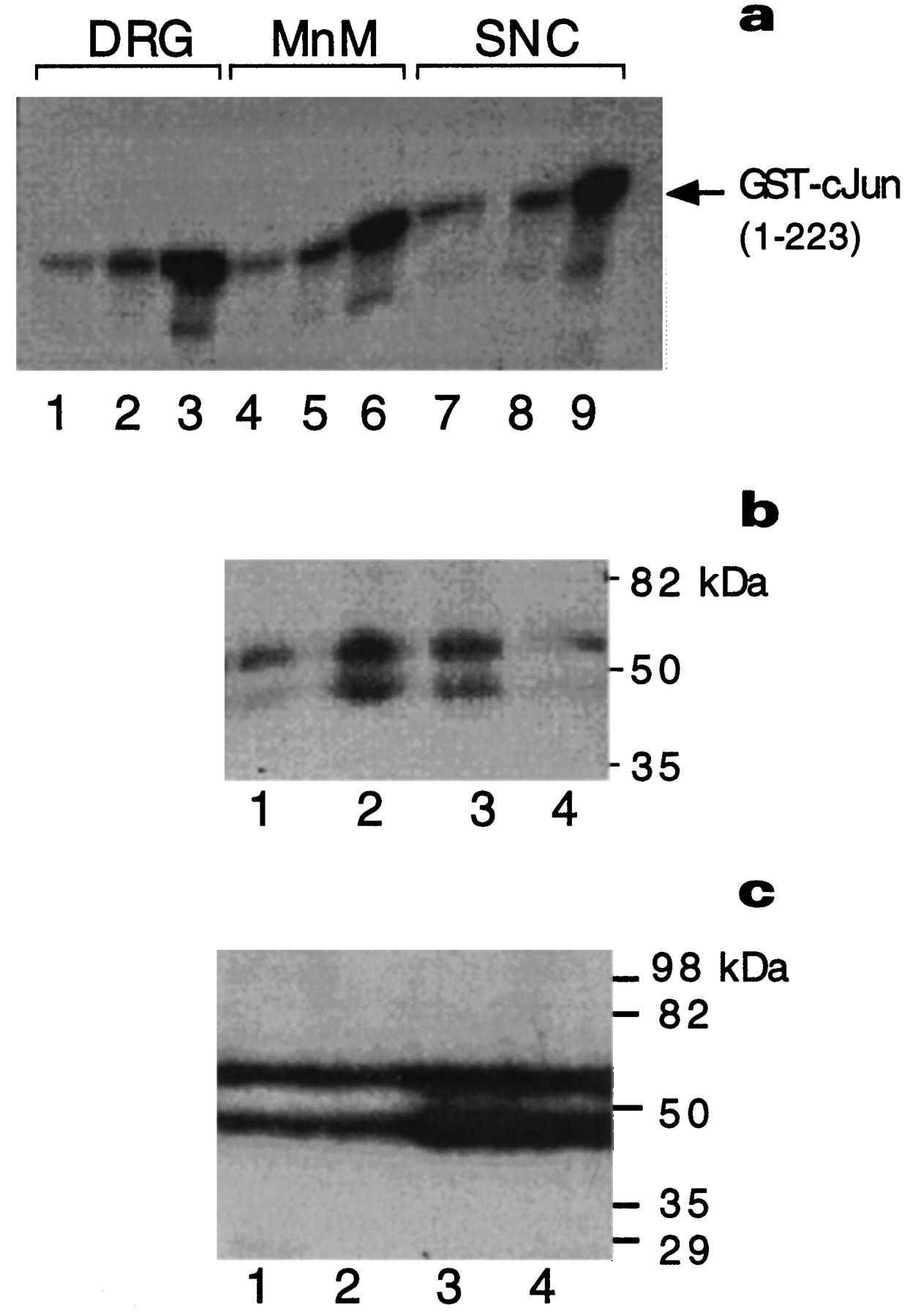
The anti-phospho-c-Jun antiserum specifically recognized the c-Jun protein phosphorylated at its serine 73 residue by activated JNK, whereas a variant containing alanine at position 73 [c-Jun(A73)] incubated with JNK (Fig.1a) or phosphorylated c-Jun treated with phosphatases (Fig. 1b) was not recognized.

Characterization of the anti-phospho-c-Jun antiserum. a, GST-c-Jun(1–223) (lanes 1, 2) or GST-c-Jun(1–223, A63/73) (lanes 3, 4) were (+) or were not (−) phosphorylated by recombinant JNK2 in the presence of [32P]ATP.(Top to bottom) first panel, Autoradiogram of32P-labeled proteins exposed either overnight (o/n) or (second panel) exposed only for 2 min. Third panel, The same blot was probed with affinity-purified phospho-c-Jun (α-P-cJun) antibody. Fourth panel, The blot was stripped and reprobed with the c-Jun antiserum (α-cJun).b, Samples (0.1 μg, lanes 2, 3; 1.0 μg, lanes 1, 4) of recombinant full-length c-Jun (Deng and Karin, 1992) were not (lanes 1, 2) or were phosphorylated (lanes 3, 4) with recombinant JNK2 in the presence of [32P]ATP (lanes 3, 4). (Top to bottom) first panel, Autoradiogram of the 32P-labeled proteins. Second panel, Immunoblotting with the phospho-c-Jun antiserum (α-P-cJun). Third panel, The blot was stripped and treated with buffer containing heat-inactivated calf intestinal phosphatase (
 ) or (fourth panel) native CIP (40 U/ml).Fifth panel, After final stripping, the blot was reprobed with the c-Jun antiserum (α-cJun).c, Immunodetection of phosphorylated c-Jun in immortalized 3T3 fibroblasts derived from wild-type (lanes 1, 2) or c-jun−/− mouse embryos (lanes 3, 4) (Hilberg et al., 1993) or c-jun−/−cells stably transfected with a human c-jun expression vector (lanes 5, 6), which were (+) or were not (−) UV-irradiated. The membrane was probed with the phospho-c-Jun antiserum (α-P-cJun) or c-Jun antibody (α-cJun). d, Detection of phosphorylated c-Jun in nuclear cortical extracts from untreated rats (lane 1) or after ischemia with 24 hr reperfusion (lane 2) by immunoblotting with the anti-phospho-c-Jun or anti-c-Jun antiserum.
) or (fourth panel) native CIP (40 U/ml).Fifth panel, After final stripping, the blot was reprobed with the c-Jun antiserum (α-cJun).c, Immunodetection of phosphorylated c-Jun in immortalized 3T3 fibroblasts derived from wild-type (lanes 1, 2) or c-jun−/− mouse embryos (lanes 3, 4) (Hilberg et al., 1993) or c-jun−/−cells stably transfected with a human c-jun expression vector (lanes 5, 6), which were (+) or were not (−) UV-irradiated. The membrane was probed with the phospho-c-Jun antiserum (α-P-cJun) or c-Jun antibody (α-cJun). d, Detection of phosphorylated c-Jun in nuclear cortical extracts from untreated rats (lane 1) or after ischemia with 24 hr reperfusion (lane 2) by immunoblotting with the anti-phospho-c-Jun or anti-c-Jun antiserum.
The phospho-c-Jun antiserum also specifically detected N-terminally phosphorylated c-Jun in whole-cell lysates from immortalized 3T3 fibroblasts after UV irradiation (Fig. 1c). Disruption of the c-jun locus abolished this reaction, and stable reexpression of human c-jun in c-jun−/− cells restored the appearance of phosphorylated c-Jun after UV irradiation (Fig. 1c). Phospho-c-Jun immunoreactivity was confined to a 40 kDa protein in those extracts that were UV-irradiated (Fig.1c).
The phospho-c-Jun antiserum also produced distinct immunoreactivity in extracts from rat brains subjected to cerebral ischemia. One day after reperfusion, the phospho-c-Jun antiserum detected a 40 kDa band in extracts of the piriform and entorhinal cortex ipsilateral to the site of ischemia, but not in extracts of the contralateral cortex or untreated brain (Fig. 1d) (for corresponding immunocytochemistry also see Fig. 5c). The presence of c-Jun in cell and tissue extracts was confirmed by immunoblotting with an antibody that recognizes c-Jun regardless on its phosphorylation state (Fig. 1c,d).
Specific immunoreactivity (IR) of c-Jun phosphorylation in the adult rat brain
The specificity of the immunoreactivity of the phospho-c-Jun antiserum was analyzed by immunocytochemistry in neurons of mamillary body (MnM) of adult rats subjected to nerve fiber lesions. In untreated rats, c-Jun-IR and phospho-c-Jun-IR were absent in the MnM (Fig.2a,e). After transection of the mamillothalamic tract, c-Jun and phospho-c-Jun reached maximal levels in the axotomized mamillary neurons after 5 d (Fig. 2b,f), and this signal was restricted to the nuclei of neurons. Preabsorption of phospho-c-Jun antiserum with the phosphorylated peptide or phosphatase treatment of the fixed sections abolished phospho-c-Jun-IR but did not affect c-Jun-IR (Fig.2c,d,g,h).

Immunodetection of c-Jun N-terminal phosphorylation in the adult rat brain. c-Jun-IR (a–d) and phospho-c-Jun-IR (e–h) in the mamillary nucleus (mm) are shown: a, e, untreated rats;b, f, 5 d after transection of the mamillothalamic tract; c, g, competition by preincubation of the antibodies with 100 pmol of the phosphorylated c-Jun peptide; d, h, preincubation of the section with 1.2 μU alkaline phosphatase before incubation with the antiserum; longitudinal (i) and (j) coronal aspect of the location site of the medial forebrain bundle (MFB) and mamillothalamic tract (MT) transection at bregma −2.3 and 1.5 mm lateral from midline. Scale bar, 200 μm.
c-Jun phosphorylation in untreated rats and after transection of central nerve fiber tracts
In untreated rats, a moderate phospho-c-Jun-IR was restricted to somatic and cranial motoneurons (data not shown) that express substantial amounts of c-Jun (Herdegen et al., 1991). In other areas with high c-Jun expression, such as dentate gyrus, phospho-c-Jun was virtually absent (also see Fig. 7c).
Transection of central nerve fiber tracts such as the MFB and MT axotomizes the neurons of the substantia nigra pars compacta (SNC) and MnM. This neuronal injury provokes an early and lasting expression of c-Jun that persisted in the MnM for up to 6 months (Herdegen et al., 1993; Leah et al., 1993). S73 phosphorylation of c-Jun appeared within 24 hr after axotomy in the SNC and MnM. Phospho-c-Jun-IR persisted for 5 d in the SNC and gradually disappeared during the next 2 weeks (Figs. 3,4). In the MnM, however, nuclear phospho-c-Jun-IR persisted for up to 50 d, the end of the observation period (Figs. 2f, 4). Phospho-c-Jun-IR was restricted to the nuclei of neurons in the SNC and MnM. Double-labeling with tyrosine hydroxylase, a marker enzyme of nigral dopaminergic neurons, and with NADPH-diaphorase, which marks axotomized MnM neurons (Herdegen et al., 1993), revealed that c-Jun was virtually N-terminally phosphorylated in axotomized neurons (data not shown). Onset, persistence, and distribution of phospho-c-Jun-IR were congruent with c-Jun-IR in the affected areas (Figs. 2b,f, 3). Staining of consecutive sections showed that phospho-c-Jun labeled approximately 75 and 30% of those neurons that were labeled by c-Jun-IR in the MnM and SNC, respectively (Figs.2b,f, 3). This indicates that the proportion of phosphorylated c-Jun is lower in degenerating neurons compared with surviving neurons.

Phospho-c-Jun-IR in the SNC. a–c, c-Jun-IR and (d–f) phospho-c-Jun-IR in the SNC of (a, d) untreated animals, (b, e) 5 d or (c, f) 20 d after transection of the medial forebrain bundle. The dotted line separates the pars compacta (p.c.) and the pars reticularis (p.r.). Arrows mark labeled nuclei. Scale bar, 100 μm.
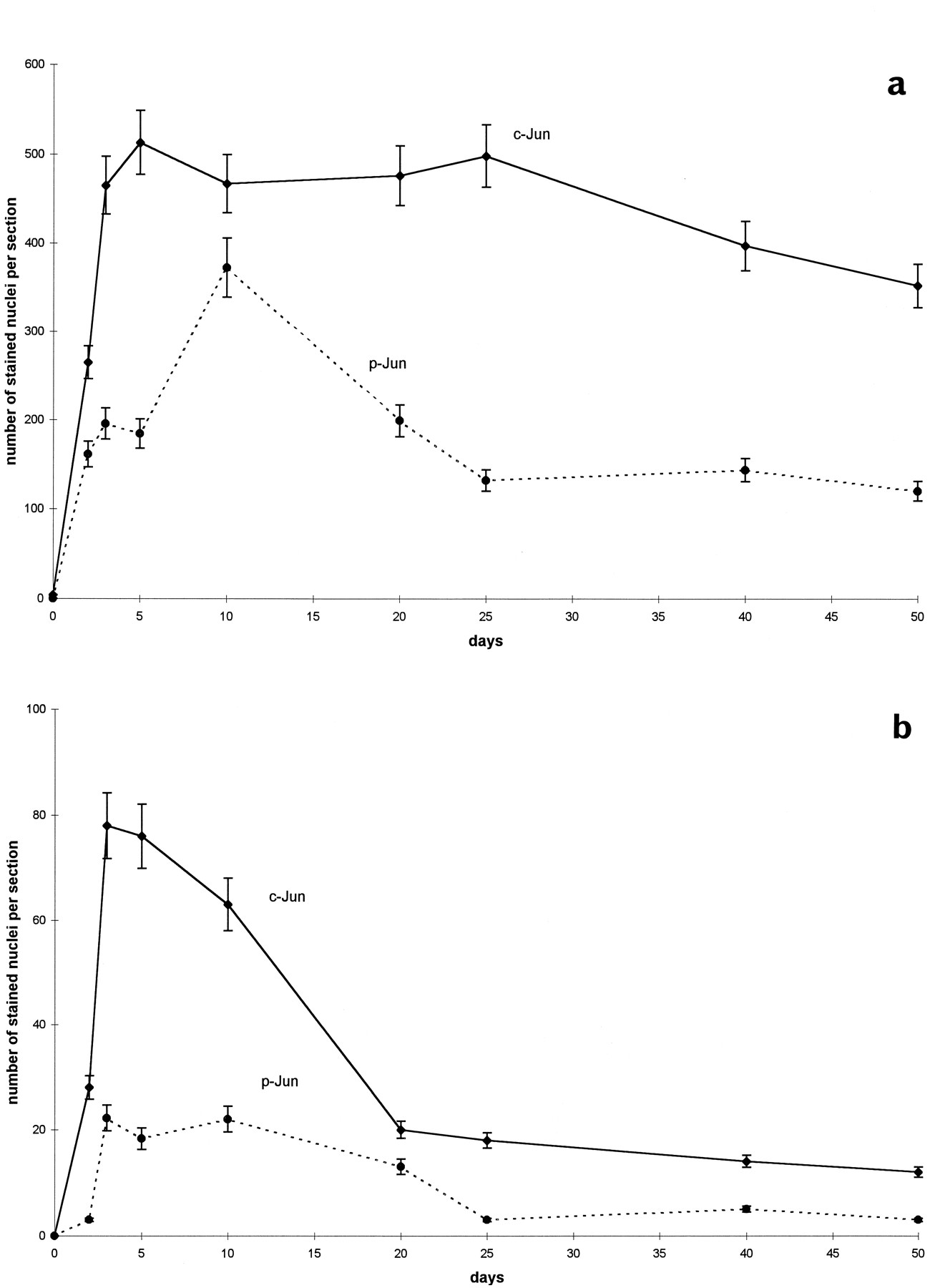
Time course of c-Jun and phosphorylated c-Jun in SNC and MnM after axotomy. Mean (±SD) of nuclei (per 50 μm section) labeled by (a) c-Jun and (b) phospho-c-Jun in the SNC (dotted line) and MnM (solid line) after transection of the medial forebrain bundle and mamillothalamic tract, respectively.
Lasting c-Jun phosphorylation after cerebral ischemia
MCA occlusion for 90 min provoked a strong expression of c-Jun around the necrotic infarcted area, e.g., the striatum or piriform and entorhinal cortex (Fig. 5a). High c-Jun expression, similar to that in the ipsilateral cortex, was also visible in the contralateral cortex (Fig. 5b), most likely because of impulse propagation via interhemispheric axon collaterals in the commissural tract. Numerous neurons displayed nuclear c-Jun phosphorylation in the ipsilateral cortex and striatum around the necrotic infarcted area that became detectable after 3 hr (Fig. 5c), reached its maximal level after 72 hr, and subsequently declined (Fig.6a,b,e). The presence of phospho-c-Jun in the ipsilateral cortex was also confirmed by immunoblotting (Fig. 1d). Importantly, phospho-c-Jun-IR remained absent throughout the observation period in the contralateral cortex (Fig. 5d).

Expression and phosphorylation of c-Jun, and TUNEL staining after MCA occlusion. Shown are (a, b) c-Jun-IR, (c, d) phospho-c-Jun-IR, and (e, f) TUNEL reaction in the ipsilateral (a, c, e) and contralateral (b, d, f) piriform cortex of consecutive sections after MCA occlusion with reperfusion for 3 d. Scale bar, 75 μm.

Co-labeling of c-Jun phosphorylation and TUNEL. Shown is double-immunofluorescence of (a, b) phospho-c-Jun and (c, d) TUNEL in the superficial layer of the ipsilateral piriform cortex 12 hr (a, c) and 3 d (b, d) after MCA occlusion.Arrows indicate some of the double-labeled nuclei.e, Numbers of neurons labeled by TUNEL (white bars) and phospho-c-Jun (black bars) in the piriform cortex ipsilateral to the site of ischemia (between bregma −1.30 and −2.30). The time course gives the reperfusion period after MCA occlusion, which lasted 90 min. The numbersrepresent mean (±SD) calculated from nine 35-μm-thick sections (three sections each of three rat brains per time point). Thegray bars give the proportion of TUNEL or phospho-c-Jun-labeled neurons that are co-labeled with phospho-c-Jun or TUNEL, respectively.
To study the relationship between c-Jun phosphorylation and neuronal cell death, sections were colabeled with phospho-c-Jun antiserum and TUNEL. The TUNEL reactivity was undetectable before 12 hr, reached a maximum between 24 and 72 hr after reperfusion (Figs. 5e,f,6c,d), and decreased after 5 d, the end of the observation period (Fig. 6e). After 12 hr, ∼81% of the phospho-c-Jun-positive neurons in the ipsilateral entorhinal cortex (EC) were TUNEL positive, and 23% of the TUNEL-positive neurons contained phospho-c-Jun. After 5 d, these values were still 42 and 34%, respectively (Fig. 6). Similar to phospho-c-Jun-IR, TUNEL reactivity was not detected in the contralateral EC (Fig.5f). Onset of neuronal c-Jun expression and N-terminal phosphorylation, which was detectable after 3 d, also preceded the appearance of TUNEL reactivity in the substantia nigra pars compacta (see Fig. 9a,c), which degenerates within 5 d because of lack of striatal neurotrophic supply.
Transient c-Jun phosphorylation after application of the chemoconvulsant PTZ
In contrast to the long-lasting c-Jun phosphorylation after axotomy or ischemia, injection of the chemoconvulsant PTZ resulted only in a transient appearance of phospho-c-Jun-IR in the dentate gyrus and superficial cortical layers (Fig. 7) that raised within 15 min, reached its maximal intensity after 30 min, when c-Jun expression was still at basal levels, and was no longer detectable after 2 hr. PTZ injections did not induce neuronal apoptosis, as indicated by the absence of TUNEL staining (data not shown).

c-Jun expression and phosphorylation after pentylenetetrazole-induced seizures. a, b, Expression of c-Jun and (c, d) phosphorylation of c-Jun in the dentate gyrus (dg) of (a, c) untreated rats and (b, d) 15 min after injection of PTZ. py, Pyramidal layer. Scale bar, 200 μm.
Partial correlation of c-Jun phosphorylation with expression of Fas-ligand
We examined whether induction of Fas-ligand, encoded by a putative c-Jun target gene (Kisabhatla et al., 1998), correlates with c-Jun N-terminal phosphorylation and neuronal apoptosis after focal ischemia and axotomy. With use of two different antibodies to rat Fas-ligand that yielded a similar immunoreactivity, no staining could be detected in untreated adult rat brain. However, after cerebral ischemia–reperfusion, Fas-ligand-IR appeared at the penumbra of the ipsilateral piriform cortex around the infarct site between 12 hr, but not 3 hr, and five d (Fig. 8) and in the SNC between 3 and 5 d (Fig.9b). The temporospatial pattern of neuronal Fas-ligand-IR paralleled that of TUNEL, whereas Fas-ligand-expressing neurons comprised only a subpopulation of c-Jun-expressing neurons. As shown in Figure 9, c-Jun was N-terminally phosphorylated in the SNC after ischemia and axotomy, but only ischemia induced Fas-ligand expression and apoptotic cell death as determined by TUNEL.

Expression of Fas-ligand in the penumbra after ischemia. Fas-ligand immunoreactivity in the piriform cortex (a) adjacent to the necrotic area that is marked by the dotted line and (b) in the contralateral intact cortex. Scale bar, 200 μm.

Phospho-c-Jun, Fas-ligand, and TUNEL in the SNC. Shown are (a, b) phospho-c-Jun immunoreactivity, (c, d) Fas-ligand immunoreactivity, and (e, f) TUNEL staining in the ipsilateral SNC 3 d after (a, c, e) MCA occlusion or (b, d, f) 10 d after transection of the MFB. Scale bar, 100 μm.
Activation of c-Jun N-terminal kinases
Finally we investigated whether the appearance of phospho-c-Jun-IR was paralleled by JNK activation by the use of a specific JNK-1 antiserum. Increased JNK-1 activity was observed in extracts of axotomized dorsal root ganglia (DRG), MnM, and SNC, 3 and 12 d after transection of sciatic nerve, MT, and MFB, respectively (Fig.10a). Compared with its activity in untreated tissues, JNK-2 activity was elevated 2.4- and 10.5-fold in the DRG, 1.5- and 5.2-fold in the MnM, and 1.3- and 6.8-fold in the SNC 3 and 12 d after axotomy.

Activation of JNK. a, JNK-1 assay with GST-c-Jun (1–223) as substrate from dorsal root ganglia (DRG) extracts after sciatic nerve cut (lanes 1–3), in MnM after MT transection (lanes 4–6), and in SNC after MFB transection (lanes 7–9). Tissues were isolated from untreated controls (lanes 1, 4, 7), 3 d (lanes 2, 5, 8), or 12 d (lanes 3, 6, 9) after axotomy.b, In-gel kinase assays using GST-c-Jun (1–79) as substrate were performed with nuclear extracts from hippocampus and cortex isolated from untreated rats (lane 1) or 5 min (lane 2), 10 min (lane 3), and 90 min (lane 4) after intraperitoneal injection of pentylenetetrazole. c, In-gel kinase assay using GST-c-Jun (1–79) as substrate from hippocampus or piriform cortex microdissected from untreated rats (lanes 1, 2) or 24 hr after ischemia–reperfusion (lanes 3, 4). The autoradiograms in b and c did not contain additional bands.
By in-gel kinase assay we determined the molecular masses of the kinases that phosphorylate c-Jun at S73. We detected only two bands of renaturable proteins with electrophoretic mobilities of JNK isoforms that range between 46 and 54 kDa (Fig. 10b,c) that can be produced by any of the JNK isoforms (Kyriakis et al., 1994; Gupta et al., 1996). Systemic application of the chemoconvulsant PTZ evoked only a transient increase in JNK activity with a maximum after 5–10 min and a return to basal levels after 90 min (Fig. 10b). By contrast, elevated JNK activity was still visible after 24 hr in cortical and hippocampal extracts after ischemia–reperfusion (Fig.10c). Both kinase assays (Fig. 10a) and in-gel kinase assays (Fig. 10b,c) revealed a minor but existent basal JNK activity in untreated rats.
DISCUSSION
Using an antiserum specific for c-Jun phosphorylated at S73 and biochemical measurements of JNK kinase activity, we demonstrated c-Jun N-terminal phosphorylation and JNK activation in specific brain areas of adult rats in response to neuronal injuries. Axotomy or ischemia with reperfusion led to lasting JNK activation and c-Jun N-terminal phosphorylation that persisted up to 50 d, i.e., much longer than observed in in vitro systems. Importantly, only a subset of c-Jun-expressing neurons revealed N-terminal c-Jun phosphorylation that appeared to correlate with the intensity and duration of the injury. Thus, nonphosphorylated c-Jun expressed in nonstressed neurons, e.g., in the dentate gyrus of untreated rats, might not be in its most active state, which requires N-terminal phosphorylation (Karin et al., 1995). Furthermore, we find that the appearance of N-terminally phosphorylated c-Jun and neuronal apoptosis in response to ischemia coincide with induced expression of Fas-ligand (also called APO-1, CD95-ligand), the activator of the cell surface receptor Fas, a potent mediator of apoptotic cell death (Mohit et al., 1995; Nagata and Goldstein, 1995;Nagata, 1997).
Specificity of the antiserum
A substantial part of our analysis relied on the use of an antiserum that is specific to the c-Jun protein phosphorylated at S73. Western blotting demonstrated that the antiserum reacts only with N-terminally phosphorylated c-Jun and that treatment with phosphatases abolished this reaction. After UV irradiation of immortalized 3T3 cells, recognition of the antigen strictly depended on the presence of c-jun, i.e., it was negative inc-jun−/− fibroblasts.
Distinct N-terminal phosphorylation of c-Jun in adult rat brain was restricted to areas that also expressed c-Jun. Preabsorption of the antiserum with the phospho-c-Jun peptide (67–79/S73) or treatment of brain sections with phosphatase abolished phospho-c-Jun-IR, whereas c-Jun-IR remained unchanged. In our hands, the nuclear signal produced by the phospho-c-Jun antiserum is rather reproducible and reliable. Because c-Jun is selectively phosphorylated at S73 (and to a minor extent at S63) by JNKs (for review, see Karin et al., 1997), the phospho-c-Jun antiserum also provides information regarding thein vivo activation of JNKs. It is mandatory to achieve cellular resolution, because expression and phosphorylation of c-Jun are independently regulated (Karin, 1995).
Phosphorylation of c-Jun
Elevated c-Jun expression and JNK activation have been shown to be tightly associated with induction of apoptosis in cultured neonatal neurons or neuronal cell lines after trophic factor deprivation (Estus et al., 1994; Ham et al., 1995; Xia et al., 1995; Ferrer et al., 1996a,b; D. Yang et al., 1997; Eilers et al., 1998; Watson et al., 1998). After ischemia–reperfusion, c-Jun expression is bilaterally induced in the cortical hemispheres, but N-terminal phosphorylation was only detectable in neurons of the infarcted areas that were also positive for TUNEL, an indicator of apoptosis (Gavrieli et al., 1992). This finding strongly suggests that N-terminal phosphorylation of c-Jun is involved in programmed cell death in the adult brain. Recent experiments demonstrated that apoptosis of hippocampal neurons after kainate excitotoxicity is closely linked to c-Jun phosphorylation, and knock-out of the JNK-3 locus with inhibition of c-Jun phosphorylation prevents this neuronal death (D. Yang et al., 1997). Finally, the apoptotic action of c-Jun requires an intact N terminus suggesting a role for c-Jun phosphorylation in certain forms of cell death (Bossy-Wetzel et al., 1997; Watson et al., 1998).
The findings on the apoptotic role of c-Jun in cultured neonatal neurons might not always be representative for the adult brain. This is particularly true for sympathetic neurons (Estus et al., 1994; Ham et al., 1995; Eilers et al., 1998) that downregulate c-Jun expression during survival of axotomy-induced degeneration (Blottner and Herdegen, 1997) whereas other neuronal populations of the CNS increase c-Jun expression after trophic support and regeneration (Schaden et al., 1994; Herdegen et al., 1997b; Houlé et al., 1997).
Additionally, phosphorylation of c-Jun is not strictly linked to the onset of apoptosis. c-Jun was phosphorylated for up to 50 d in nondegenerating MnM neurons after axotomy that show an ongoing coexpression of trophic factors such as galanin (Brecht et al., 1997) and protective enzymes such as nitric oxide synthase (Herdegen et al., 1993). Because c-Jun expression after nerve fiber transection is restricted almost exclusively to axotomized neurons (for review, seeHerdegen et al., 1997b), phosphorylation of c-Jun has to occur in the same population as also shown after sciatic nerve cut (Kenney and Kocsis, 1998). The persistent N-terminal phosphorylation of c-Jun and lack of TUNEL reactivity [which might be a general feature of axotomy-triggered cell death (Hughes et al., 1997)] strongly argues against the assumption that phosphorylation of c-Jun inevitably leads to neuronal cell death. c-Jun is also phosphorylated in the dentate gyrus after kainate excitotoxicity without subsequent cell death (D.Yang et al., 1997). Finally, the activation of c-Jun participates in cell cycle control without apoptosis (Bossy-Wetzel et al., 1997). Phosphorylation at S73 may protect c-Jun against ubiquitin-dependent degradation (Musti et al., 1997) and is the major mechanism for the positive autoregulation of c-jun transcription by c-Jun (Karin, 1995; Eilers et al., 1998).
The expression of c-Jun and its N-terminal phosphorylation can be regulated independently as shown in the present study and inJNK-3−/− mice after kainate application that preserves c-Jun expression without N-terminal phosphorylation (D. Yang et al., 1997). In cerebellar granule cells, c-Jun is expressed and phosphorylated after survival signal withdrawal, whereas the high prewithdrawal levels of JNK activity do not change (Eilers et al., 1998). These findings also argue against a major role of JNKs in the induction of c-jun expression in the adult brain.
Taken together, the protective or apoptotic function of c-Jun does not depend merely on its phosphorylation state but may be determined by cofactor proteins such as Jun activation domain binding protein (Claret et al., 1996) or CREB binding protein (Arias et al., 1994; Kamei et al., 1996) (for review, see Karin et al., 1997) or by the dimerization partners, such as the neuroprotective ATF-2 transcription factor (Reimold et al., 1996; Herdegen et al., 1997a).
Activation of JNK
Inhibition of JNK activation protects post-mitotic PC12 and sympathetic neurons from apoptosis (Xia et al., 1995; Eilers et al., 1998; H. Le-Niculescu, Y. Kasuya, F.-X. Claret, and M. Karin, unpublished results). Similar to c-Jun phosphorylation, however, the linkage between JNK activation and neuronal apoptosis is not simple. Both nerve fiber transection and cerebral ischemia–reperfusion led to lasting c-Jun phosphorylation and JNK activation in the SNC, but only ischemia–reperfusion resulted in apoptotic cell death as detected by TUNEL staining. On the basis of this and other studies (Le-Niculescu, Kasuya, Claret, and Karin, unpublished results), we find a more critical correlation between apoptosis and induction of Fas-ligand expression. Recently, transcription of the gene that codes for Fas-ligand was suggested to be controlled by c-Jun (Kasibhatla et al., 1998).
Apart from apoptosis, JNKs exert a role in neuronal plasticity as suggested by their expression and activity in the brain of untreated rats (Carletti et al., 1995), during neuronal differentiation of PC12 cells (Eilers et al., 1998) and after axotomy in affected neurons and the transected nerve stump (Kenney and Kocsis, 1998). It remains to be clarified to which extent the various JNK isoforms exert different functions. Thus, knockout of the JNK-3 locus, but not theJNK-1 or JNK-2 loci, prevents excitotoxic cell death of hippocampal neurons (D. Yang et al., 1997), but it is not known whether loss of JNK-1 and JNK-2 also results in reduction of c-Jun phosphorylation in the adult mammalian brain.
Our in-gel kinase assays indicate that proteins in the range of electromobility of the JNK isoforms are the only mediators of c-Jun phosphorylation in the adult nervous system. These isoforms could be produced by any of the JNK genes; the activity of these products is regulated very similarly (Kyriakis et al., 1994; Gupta et al., 1996).
The final action of JNK as mediator of apoptosis or plasticity might depend on the selective activation of its nuclear substrates c-Jun, ATF-2, or Elk-1. For example, expression of ATF-2 is downregulated after various neurodegenerative stimuli (Herdegen et al., 1997a), and consequently c-Jun does not have to compete with ATF-2 for JNK binding (Kallunki et al., 1996), which may result in an increased number of phosphorylated c-Jun molecules. Such an out-competing of JNK targeting has been observed for p53 and c-Jun (S. Fuchs, V. Adler, and Z. Ronai, unpublished observations).
Fas-ligand
Ischemic injury is a strong inducer of the apoptotic cytokine Fas-ligand in neurons around the ischemic core and in delayed dying neurons of the substantia nigra compacta as shown by immunocytochemistry. Importantly, neurons upregulate the respective Fas receptor after ischemia (Matsuyama et al., 1995) or other neurological disorders, such as Alzheimer’s disease (de la Monte et al., 1997). In the neuronal-like PC12 cell line, JNK activation and c-Jun N-terminal phosphorylation were supposed to be crucial components in the pathway leading to apoptosis after either expression of MEKK1, a potent upstream activator of the JNK cascade (Minden et al., 1994a,b; Eilers et al., 1998), or deprivation of NGF from differentiated PC12 neurons (Xia et al., 1995; Le-Niculescu, Kasuya, Claret, and Karin, unpublished results). It appears that one likely function of N-terminally phosphorylated c-Jun is to induce Fas-ligand expression via several AP-1 sites in the fas-ligand promoter (Kasibhatla et al., 1998). However, we found certain situations, such as after axotomy, in which c-Jun N-terminal phosphorylation does not result infas-ligand induction. These findings allow the hypothesis that phosphorylation of c-Jun is a necessary but not sufficient prerequisite for fas-ligand induction.
In summary, our data demonstrate that the expression patterns and function of JNK and c-Jun in the adult brain are not simply related to neuronal cell death. Here, we have identified a coincident signaling response that involves prolonged JNK activation, selective c-Jun N-terminal phosphorylation, and Fas-L induction that is triggered by ischemia–reperfusion with subsequent apoptosis. After axotomy, c-Jun phosphorylation and JNK activity also occur as part of the neuronal stress response, but no apoptosis and no Fas-ligand expression ensue. It remains to be elucidated which modulators of c-Jun transactivation contribute to the propagation of apoptosis and induction of Fas-ligand.
Footnotes
This work was supported by grants from Deutsche Forschungsgemeinschaft (Zi 110/22, He 1561), University of Heidelberg (72/96), and National Institutes of Health (HL 35018, ES 06376, and CA 54418). T.He. was a visiting scientist at the University of California San Diego, Department of Pharmacology, supported by a Heisenberg-Fellowship of the Deutsche Forschungsgemeinschaft. F.X.C. and T.K. were supported by postdoctoral fellowships from the French National League against Cancer and the American Heart Association California affiliate, respectively. T.Hu. is an American Cancer Society Research Professor. We thank R. Bravo for providing the c-Jun antibody, M. Ellisman for discussion and use of his equipment, and H. Brendel and J. M. Tian for the purified recombinant JNK2 and recombinant c-Jun, respectively. We also thank C. Hoeger for synthesizing the purified c-Jun phosphopeptide, J. Vaughan for immunizing the rabbits, E. Wagner for WTc-jun+/+ and c-jun−/− mouse fibroblast cell lines, F. Piu for c-jun−/− cells stably expressing a human c-Jun, and D. Green and H. Le-Niculescu for sharing unpublished data.
Correspondence should be addressed to Dr. Herdegen at his present address: University of Kiel, Institute of Pharmacology, Hospitalstrasse 3, 24105 Kiel, Germany.
Dr. Claret’s present address: M. D. Anderson Cancer Center, University of Texas, Department of Molecular Oncology, 1515 Holcombe Boulevard, Houston, TX 77030-4095.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}