

Abstract
We report the molecular cloning and characterization of 4.1N, a novel neuronal homolog of the erythrocyte membrane cytoskeletal protein 4.1 (4.1R). The 879 amino acid protein shares 70, 36, and 46% identity with 4.1R in the defined membrane-binding, spectrin-actin–binding, and C-terminal domains, respectively. 4.1N is expressed in almost all central and peripheral neurons of the body and is detected in embryonic neurons at the earliest stage of postmitotic differentiation. Like 4.1R, 4.1N has multiple splice forms as evidenced by PCR and Western analysis. Whereas the predominant 4.1N isoform identified in brain is ∼135 kDa, a smaller 100 kDa isoform is enriched in peripheral tissues. Immunohistochemical studies using a polyclonal 4.1N antibody revealed several patterns of neuronal staining, with localizations in the neuronal cell body, dendrites, and axons. In certain neuronal locations, including the granule cell layers of the cerebellum and dentate gyrus, a distinct punctate-staining pattern was observed consistent with a synaptic localization. In primary hippocampal cultures, mouse 4.1N is enriched at the discrete sites of synaptic contact, colocalizing with the postsynaptic density protein of 95 kDa (a postsynaptic marker) and glutamate receptor type 1 (an excitatory postsynaptic marker). By analogy with the roles of 4.1R in red blood cells, 4.1N may function to confer stability and plasticity to the neuronal membrane via interactions with multiple binding partners, including the spectrin-actin–based cytoskeleton, integral membrane channels and receptors, and membrane-associated guanylate kinases.
The red blood cell (RBC) protein 4.1 (4.1R) is a critical structural component of the erythrocyte membrane cytoskeleton. The spectrin-actin–binding domain (SABD) of 4.1R potentiates the interactions of spectrin tetramers with F-actin (Correas et al., 1986), and the membrane-binding domain (MBD) links this cytoskeletal scaffold to the plasma membrane via interactions with band 3 (Pasternack et al., 1985) and glycophorin C (Reid et al., 1990). The importance of 4.1R to the structural integrity of the membrane cytoskeleton is underscored by the aberrant RBC phenotype seen in patients with 4.1R mutations. In the disease hereditary elliptocytosis, which is caused by 4.1R deficiency, RBCs are elliptical rather than biconcave in shape, have increased osmotically fragility, and are prone to fragmentation, leading to various degrees of hemolytic anemia (Palek and Lux, 1983; Conboy et al., 1986b; Shi et al., 1999). Thus, the protein–protein interactions of 4.1R in the RBC membrane cytoskeleton are integral to cell morphology, membrane stability, and RBC survival.
Although the function of 4.1R has been predominantly characterized in RBCs, immunological methods have identified protein 4.1 with molecular weights of 30–210 kDa in numerous nonerythroid tissues (Goodman et al., 1984; Spiegel et al., 1984; Constantinescu et al., 1986; Anderson et al., 1988; De Cesaris et al., 1989; Sihag et al., 1994). The molecular weight diversity of 4.1 proteins among tissues and within individual cells has been attributed to complex alternative splicing of the 4.1R gene (Tang et al., 1990). We have recently obtained evidence that the diversity of 4.1 isoforms also derives from the expression of distinct 4.1 genes. In situ hybridization studies with 4.1R-specific probes revealed that 4.1R is not ubiquitous and is predominantly expressed in hematopoietic tissues and specific neuronal populations (Walensky et al., 1998b). We have recently cloned a novel 4.1 homolog that is generally expressed throughout the body and designated 4.1G (Parra et al., 1998a; Walensky et al., 1998a). Whereas 4.1R is localized to human chromosome 1, 4.1G is found on chromosome 6 (Parra et al., 1998a). The cytoskeletal proteins ankyrin and β-spectrin also have erythroid and generally expressed homologs encoded by distinct genes (Lux et al., 1990; Winkelmann et al., 1990;Hu et al., 1992; Kordeli et al., 1995). In addition, neuron-specific homologs/isoforms of these cytoskeletal proteins occur in the brain (Riederer et al., 1986; Tse et al., 1991). In the present study, we have cloned and characterized a third homolog of 4.1 that is specifically enriched in neurons and thus designated 4.1N.
MATERIALS AND METHODS
Molecular cloning of mouse 4.1N. We screened the dBest database [National Center for Biotechnology Information (NCBI), Bethesda, MD] for novel clones with homology to 4.1R and 4.1G, and a human clone was identified (accession number R67624) that shared 72% identity with the C-terminal 36 amino acids of mouse 4.1R (m4.1R) and 62% identity with the C-terminal 39 amino acids of m4.1G. A single mouse clone (accession number W41934) shared 70% nucleic acid identify with sequence in the 3′-untranslated region of the human clone. To obtain the mouse coding sequence, we used a nondegenerate 3′ primer to a portion of the 3′-untranslated region (GTGACAGTACGTCGCAGC) and a degenerate 5′ primer to the conserved CVEHHTF motif of 4.1 proteins (TGTGT[A/G]GA[A/G]CATCA-CACGTT) in PCR experiments with mouse brain cDNA as template. A 1674 bp product was obtained, and the coding sequence shared ∼40% identity with 4.1R and 4.1G, with conservation in the defined membrane-binding, spectrin-actin–binding and C-terminal domains. Two distinct probes (MVKDF to QHPDM, 387 bp; SRSLDG to LKEPNS, 252 bp) were generated from the PCR product and used to screen double lifts of a mouse brain cDNA library (Stratagene, La Jolla, CA). The DNA probes were radiolabeled by nick translation (Boehringer Mannheim, Indianapolis, IN), and the library screening was performed according to the manufacturer’s protocol. The PCR product and full-length clones were double strand sequenced using the fluorescent terminator method of cycle sequencing on an Applied Biosystems (Foster City, CA) 373a automated DNA sequencer at the DNA Analysis Facility of the Johns Hopkins University (Smith et al., 1986; McCombie et al., 1992). Oligonucleotides used for sequencing were synthesized on an ABI 394 synthesizer following ABI protocols. DNA sequence data were analyzed using Sequencher software from Gene Codes (Ann Arbor, MI). The 4.1 sequence alignment was generated using the MacVector program (Oxford Molecular Group, Oxford, UK).
In situ hybridization. In situ hybridization experiments were conducted using digoxigenin-labeled probes corresponding to amino acids 428–523 of m4.1N (285 bp) and amino acids 481–842 of m4.1R (1083 bp) (Huang et al., 1993). Fresh-frozen 20 μm cryostat sections of whole-mount embryos (embryonic days 11.5, 13.5, 15.5, 17, and 18.5), whole-mount postnatal mice (postnatal days 4, 7, and 21), and adult mouse tissues were cut onto Superfrost Plus slides (Fisher Scientific, Pittsburgh, PA), allowed to air dry 1–3 hr, and post-fixed for 5 min in 4% paraformaldehyde in PBS. The slides were washed three times for 3 min each in Tris-buffered saline (TBS), treated 10 min in 0.25% acetic anhydride and 0.1m triethanolamine, pH 8.0, washed three times for 3 min each in TBS, and prehybridized for 2 hr at room temperature in hybridization buffer (50% formamide, 5× SSC, 5× Denhardt’s solution, 500 mg/ml sonicated herring sperm DNA, and 250 mg/ml yeast tRNA). The tissue was incubated with 0.1 ml of buffer containing 40 ng of cRNA probe under a siliconized coverslip at 65°C overnight. After removal of the coverslips in 5× SSC at 65°C, sections were washed as follows: twice for 1 hr each in 0.2× SSC, once for 5 min in 0.2× SSC, and once for 5 min in TBS. After a 1 hr block at room temperature in 4% normal goat serum (NGS) in TBS, the slides were incubated overnight at 4°C in a 1:5000 dilution of anti-digoxigenin Fab fragment (Boehringer Mannheim) in 4% NGS in TBS. Sections were then washed three times for 5 min each in TBS and once for 5 min in AP buffer (0.1 m Tris-Cl, pH 9.5, 0.1 m NaCl, and 50 mm MgCl2). The color signal was developed in AP buffer containing 3.375 mg/ml nitro blue tetrazolium (Boehringer Mannheim), 3.5 mg/ml 5-bromo-4-chloro-3-indolyl phosphate (Boehringer Mannheim), and 0.24 mg/ml levamisole. The color reaction was performed in the dark for 24–72 hr at room temperature and then terminated with a TE wash. The developed slides were coverslipped with Aquapolymount (Polysciences, Warrington, PA). Control sections were hybridized with identical quantities of sense cRNA, and no signal was observed. The in situ hybridization protocol has been used to distinguish the localizations of transcripts sharing as much as 85–90% nucleic acid identity (Vassar et al., 1993;Blackshaw and Snyder, 1997).
Northern analysis. A 200 bp probe derived from the human 4.1N (h4.1N) 3′-untranslated DNA sequence was used to probe a multiple tissue Northern blot (Clontech, Palo Alto, CA) according to the manufacturer’s protocol. The primers used to generate the 200 bp probe were (5′) CCTCTGTGAAGAGATCCTGG and (3′) AAGGATGCGGTGTTTCCTGT. The blot was probed with ubiquitin as a control for RNA integrity.
4.1N antibody production. Nucleotide sequence corresponding to amino acids (aa) 5–100 of the m4.1N N terminal was subcloned into the pet22b expression vector (Novagen, Madison, WI). The plasmid was transformed into Escherichia coli BL21(DE3) bacteria (Novagen), and the fusion protein was expressed and then purified over nickel columns (Novagen) according to the manufacturer’s protocol. New Zealand white rabbits were immunized with the fusion protein according to established protocols (Covance, Denver, PA). Production bleeds were affinity purified by passing the serum [diluted 1:1 in 20 mm Trizma (Sigma, St. Louis, MO), pH 7.5] over affigel-10 (Bio-Rad, Hercules, CA) columns containing the m4.1N fusion protein. After extensive washing with 10 mm Trizma, pH 7.5, and 10 mm Trizma, pH 7.5, plus 500 mm NaCl, the antibodies were eluted with 100 mm glycine, pH 2.5, and 100 mm triethylamine, pH 11.5, and dialyzed against PBS and then PBS plus 40% glycerol for storage.
COS-7 cell transfection. COS-7 cells were transfected by the calcium phosphate precipitation method (Sambrook et al., 1989) with 10 μg of a cytomegalovirus promoter-driven mammalian expression vector containing the full-length m4.1N-coding sequence; a mock transfection was run in parallel to serve as a control for Western analysis. After 24 hr, the cells were washed with 3 ml of PBS and treated with 1 ml of lysis buffer (50 mm Trizma, pH 7.4, 40 mm NaCl, 0.5% Triton X-100, 1% SDS, 4 μg/ml leupeptin, 2 μg/ml antipain, 2 μg/ml chymostatin, 2 μg/ml pepstatin, and 1 mm PMSF) on ice for 10 min. Supernatants were isolated after centrifugation at 14,000 × g for 10 min at 4°C, and protein was assayed using DC reagents (Bio-Rad, Hercules, CA). Twenty micrograms of each sample were diluted in 5× protein load buffer and electrophoresed on 4–12% gradient Tris–glycine minigels (Novex, San Diego, CA) for Western analysis (see below).
Western analysis. Mouse tissues were homogenized in 2% SDS, 1% Triton X-100, 50 mm Trizma, pH 7.4, 100 mmNaCl, 1 mm EGTA, and protease inhibitors (4 μg/ml leupeptin, 2 μg/ml antipain, 2 μg/ml chymostatin, 2 μg/ml pepstatin, and 1 mm PMSF) and incubated on ice for 20 min, with intermittent vortexing. After table-top centrifugation at maximum speed, the supernatants were collected, and protein was assayed using DC reagents (Bio-Rad). Twenty micrograms of each sample were diluted in 5× protein load buffer and subjected to electrophoresis using 4–12% gradient Tris-glycine minigels (Novex). Proteins were wet transferred to polyvinylidene fluoride membrane (Millipore, Bedford, MA), blocked for 1 hr in 5% nonfat dry milk, and incubated with a 1:2000 dilution of the rabbit m4.1N antibody or a 1:2000 dilution of the antibody preadsorbed with the m4.1N fusion protein in 3% BSA plus PBS overnight at 4°C. Blots were then washed in 5% milk once for 15 min and then twice for 5 min each, followed by a 1 hr incubation at room temperature with a 1:5000 dilution of anti-rabbit secondary antibody (Amersham, Arlington Heights, IL). After washing once in 5% milk for 15 min and then twice in 0.1% Tween in PBS for 10 min each, the blots were developed by chemiluminescence using the Renaissance kit (NEN Dupont, Boston, MA) according to the manufacturer’s protocol.
Immunohistochemistry. Adult mice were perfused with PBS followed by 4% paraformaldehyde and PBS. Tissues were removed, post-fixed in 4% paraformaldehyde in PBS for 2 hr, and then cryoprotected overnight in 15% sucrose plus PBS. Brains were frozen on dry ice, and 40 μm sections were cut on a sliding microtome. The floating sections were collected in TBS (1.5% NaCl and 50 mm Tris-HCl, pH 7.4), washed four times in TBS, and then blocked and permeabilized for 45 min in room temperature TBS containing 4% NGS, 0.2% Triton X-100, and 0.02% sodium azide. The sections were then incubated overnight at 4°C with the m4.1N antibody diluted 1:500 in TBS containing 2% NGS, 0.1% Triton X-100, and 0.02% sodium azide. Control sections were incubated with primary antibody preabsorbed with the m4.1N fusion protein. The sections were subsequently washed three times in TBS containing 1% NGS and then incubated with a biotinylated anti-rabbit antibody (Vector Laboratories, Burlingame, CA) diluted 1:200 in TBS containing 1.5% NGS for 1 hr at room temperature. After two 10 min washes in TBS plus 1% NGS and two additional 10 min washes in TBS, the sections were incubated in a 1:50 dilution of the streptavidin-peroxidase reagent (Vectastain elite kit; Vector Laboratories) in TBS for 45 min at room temperature. The sections were then subjected to three 10 min washes in TBS and developed in 0.5 mg/ml diaminobenzidine (Life Technologies, Gaithersburg, MD) in TBS for 1–4 min at room temperature. The reaction was stopped in 0.05% sodium azide, and then the sections were mounted onto Superfrost Plus slides (Fisher Scientific), air dried, washed under a stream of distilled water, air dried again, dehydrated in graded alcohols, cleared in xylenes, and then coverslipped in Permount (Fisher Scientific). All other tissues were frozen in TissueTek (Sakura Finetek, Torrance, CA), and 20 μm sections were cut onto Superfrost Plus slides using a cryostat. The slides were processed as follows: three 5 min washes in PBS, 15 min permeabilization in 0.1% Triton X-100, three 5 min washes in PBS, 15 min quenching of endogenous peroxidase activity in 0.05% hydrogen peroxide, three 5 min washes in PBS, 1 hr block with 2% NGS plus 1% BSA in PBS, and overnight incubation with a 1:500 dilution of m4.1N antibody or antibody preadsorbed with the m4.1N fusion protein in 0.5% BSA and PBS in a moist chamber at 4°C. Slides were developed using the avidin–biotin–peroxidase system (Vectastain kit; Vector Laboratories) with diaminobenzidine as the chromogen, according to the manufacturer’s protocol.
Indirect immunofluorescence. m4.1N indirect immunofluorescence was performed on paraformaldehyde-fixed adult rat brain sections and primary hippocampal cultures. Adult brain sections were processed as described above for immunohistochemistry, except that a streptavidin-FITC antibody (Jackson Immunoresearch, West Grove, PA) was used in place of the streptavidin-peroxidase antibody to detect m4.1N labeling and, after washing, the sections were mounted on glass slides and coverslipped with vectashield containing 4,6-diamidino-2-phenylindole (DAPI; Vector Laboratories). The brain sections were examined by immunofluorescence and confocal microscopy. Hippocampal cultures were prepared as described previously (Allison et al., 1998) and grown on coverslips. The samples were fixed with 4% paraformaldehyde in PBS for 20 min at 4°C and permeabilized in 0.2% Triton X-100 for 10 min at 4°C. The coverslips were treated with primary antibodies [1:200 dilution of m4.1N antibody with either a 1:200 dilution of the postsynaptic density protein of 95 kDa (PSD95) antibody or a 1:50 dilution of glutamate receptor type 1 (GluR1) antibody (Craig et al., 1993)] for 1 hr at room temperature and then secondary antibodies [1:100 dilution of FITC anti-rabbit and Texas Red anti-mouse (Jackson Immunoresearch)] for 1 hr at room temperature. The coverslips were mounted on glass slides using vectashield (Vector Laboratories) and examined by immunofluorescence microscopy.
RESULTS
Identification and molecular cloning of 4.1N
A search of the dBest database identified a human clone (accession number R67624) that shared 72% identity with the C-terminal 36 amino acids of m4.1R and 62% identity with the C-terminal 39 amino acids of m4.1G. A single mouse clone (accession number W41934) shared 70% nucleic acid identity with the sequence in the 3′-untranslated region of the human clone. To obtain the mouse coding sequence, we used a nondegenerate 3′ primer to a portion of the 3′-untranslated region and a degenerate 5′ primer to the conserved CVEHHTF motif of 4.1 proteins (Huang et al., 1993; Walensky et al., 1998a) in PCR experiments with mouse brain cDNA as template. A 1674 bp product was obtained, and the coding sequence shared ∼40% identity with the corresponding region of 4.1R and 4.1G, with strong conservation in the defined MBD and C-terminal domain (CTD). The novel sequence was used to generate a digoxigenin-labeled probe for in situ hybridization experiments in 4-d-old mouse whole mounts. These studies revealed a striking enrichment of mRNA in the brain compared with the rest of the body (Fig. 1A). Furthermore, high levels of mRNA were present in a distinctly neuronal distribution throughout the adult mouse brain (Fig.1B). Sections treated with the sense probe were completely negative (Fig. 1C) (data not shown). The almost universal expression of the novel protein 4.1 homolog in neurons of the brain contrasts with the discrete localization of m4.1R mRNAs to granule cells of the cerebellum and dentate gyrus (Walensky et al., 1998b). Thus, the new protein 4.1 homolog was designated 4.1N, because of its striking neuronal distribution.

Neuronal distribution of the novel 4.1 homolog.In situ hybridization was performed on a 4-d-old whole-mount mouse and on adult mouse brain using a digoxigenin-labeled antisense probe corresponding to 285 bp of the novel 4.1 clone (aa 428–523 of m4.1N). A, There is marked enrichment of the novel 4.1 clone in the brain compared with elsewhere in the body.B, The mRNAs are found in almost all neurons of adult mouse brain. C, No signal was evident when sections were incubated with the corresponding sense cRNAs. Scale bars:A, 2.7 mm; B, C, 1.25 mm.
To identify a full-length m4.1N clone, two probes were generated from the PCR product and used to screen a mouse brain cDNA library. We obtained a clone that encoded an 879 amino acid protein with 70, 36, and 46% identity to the MBD, SABD, and CTD of m4.1R and with 74, 37, and 49% identity to the corresponding domains of 4.1G (Fig.2). The starting methionine was assigned on the following basis: (1) it is located in the context of a primary Kozak translation initiation sequence (−6 to +1 = GTCACC ATGA), (2) it encodes the MTTE motif that initiates 4.1G and the 135 kDa isoform of 4.1R (Huang et al., 1993; Parra et al., 1998a; Walensky et al., 1998a), and (3) a human cDNA later identified in the NCBI database (accession number 2224617) shares 87% nucleic acid identity with m4.1N in the coding region but diverges sharply just upstream of the Kozak sequence.

Amino acid sequence of m4.1N. m4.1N is an 879 amino acid protein with sequence homology to 4.1R and 4.1G in the defined membrane-binding, spectrin-actin–binding, and C-terminal domains (domain boundaries are indicated by arrows). The amino acid alignment highlights identical sequence shared by the three proteins (boxed and shaded) and residues with conservative amino acid changes (shaded only). Sequence identities shared by 4.1R and 4.1G are boxedand shaded, whereas sequence identities specific to 4.1G and 4.1N are boxed only. Whereas there is additional m4.1R sequence between the MBD and SABD, the region between the SABD and CTD is expanded in m4.1N. The recently identified Ca2+-dependent calmodulin (CD-CaM)– and Ca2+-independent calmodulin (CI-CaM)–binding sites of 4.1R are located within the MBD. The original m4.1N PCR product obtained from mouse brain cDNA (start site marked by an arrowhead) did not contain two stretches of sequence present in the full-length clone (marked by + symbols and asterisks). m4.1N and h4.1N (accession number 2224617) share 95% amino acid identity. m4.1N sequence data are available from GenBank (accession number AF061283).
An alignment of m4.1N, m4.1R, and m4.1G demonstrates that the strongest homology occurs in the MBD, which is highly conserved among members of the 4.1 superfamily, including the ezrin, radixin, and moesin family of proteins. Within this domain, 4.1N contains two conserved peptide motifs found in 4.1R and 4.1G that have recently been shown to function as distinct Ca2+-independent and Ca2+-dependent calmodulin-binding sites (Nunomura et al., 1997a). Compared with m4.1R and m4.1G, m4.1N has significantly less homology in the SABD, suggesting that m4.1N may interact with distinct neuronal homologs of spectrin and actin or with entirely different proteins in the neuronal cytoskeleton. m4.1N shares 73% identity with m4.1R and 4.1G in the second one-half of the CTD, which corresponds to exons 20–21 of m4.1R. Within this region, m4.1N contains the histidyl-proline moiety that has been shown to account for the interaction of m4.1G with the 13 kDa FK506-binding protein FKBP13 (Walensky et al., 1998a). There is little homology in the N-terminal region and the first one-half of the CTD. The length of unique regions between the conserved MBD, SABD, and CTD is variable among members of the 4.1 family. Whereas there is additional m4.1R sequence between the MBD and SABD, the region between the SABD and CTD is expanded in m4.1N. A comparison of the original PCR product from mouse brain (starting at aa 481) with the corresponding region in the full-length clone revealed that aa 484–495 (Fig. 2, marked with +) and aa 557–692 (marked with *) are absent in the PCR product, implying that, like m4.1R and m4.1G, m4.1N has splice forms.
The determination that 4.1N represents a distinct gene has also been confirmed by chromosomal localization studies. Whereas 4.1R and 4.1G are found on human chromosomes 1 and 6 and mouse chromosomes 4 and 10, respectively (Conboy et al., 1986b; Parra et al., 1998a), 4.1N has been mapped to human chromosome 20 (Kim et al., 1998) and mouse chromosome 2 (Peters et al., 1998).
Tissue distribution of 4.1N mRNAs
Multiple tissue Northern analysis was performed using a probe derived from the 3′-untranslated DNA of h4.1N (Fig.3). The most prominent hyridization occurred in the brain, where two major species of 7.5 and 4.1 kb were detected; a minor 8.8 kb product was also seen. Substantial levels of h4.1N mRNA were also detected in the heart, kidney, and pancreas, with lesser levels in the placenta, lung, and skeletal muscle; no hybridization was evident in the liver. Interestingly, the 7.5 kb band was most enriched in the brain, whereas the 4.1 kb species predominated in peripheral tissues. The several h4.1N species detected by Northern analysis likely represent alternative splice forms; extensive alternative splicing of 4.1R, for example, has been well documented (Conboy et al., 1988, 1991). A ubiquitin probe was used as a control for mRNA integrity (Fig. 3).

Northern analysis of h4.1N. A, A multiple tissue Northern blot was incubated with a probe derived from the 3′-untranslated DNA of h4.1N. The most intense band was a 7.5 kb species identified in brain, which also contained 8.8 and 4.1 kb bands. h4.1N mRNAs were also detected in the heart, kidney, and pancreas, with lesser levels in the placenta, lung, and skeletal muscle; no hybridization was evident in the liver. Whereas the 7.5 kb band is enriched in brain, the 4.1 kb species predominates in peripheral tissues. B, A ubiquitin probe was used as a control for mRNA integrity.
High-power views of m4.1N in situ hybridization in adult mouse brain confirmed the association of m4.1N mRNA with neuronal cell bodies in a variety of brain regions including the cerebellum, cortex, hippocampus, and striatum (Fig.4A–D). Although in most brain regions m4.1N mRNAs occurred in all neurons, notable exceptions included the Purkinje cells of the cerebellum (located at the boundary between the granule cell layer and the molecular layer in Fig. 4A) and the majority of thalamic nuclei (Fig. 4E). High levels of m4.1N mRNAs also occurred in sensory tissues, including the olfactory epithelium (Fig. 4F), the retina (data not shown), and sensory ganglia (data not shown). In the gastrointestinal tract, m4.1N mRNAs were exclusively localized to the enteric neurons, which lie between the smooth muscle layers (Fig. 4G). Substantial levels of m4.1N mRNAs were found in the kidney, associated with convoluted and collecting tubule epithelia (Fig. 4H,white, black arrowheads, respectively). Focal expression was also seen in the lung, with m4.1N mRNAs detected only in select bronchioles (data not shown). In the adrenal gland, the medullary chromaffin cells, which are derived from neural crest cells, were also robustly positive for m4.1N mRNAs (data not shown).

Localization of m4.1N mRNAs in the brain. A–D, High-power views of m4.1N in situ hybridization performed on adult mouse brain reveal robust neuronal labeling in the granule cell layer (GCL) of the cerebellum [note that the molecular layer (ML) is negative] and in deep cerebellar nuclei (DCN) (A), in all layers of the cerebral cortex (B), in the dentate gyrus (DG) and CA1–CA3 regions of the hippocampus (C), and in the striatum [note that the white matter tracts (indicated by black arrowheads) are negative] (D). E, Interestingly, no m4.1N mRNAs are detected in the thalamus except for neurons in the reticular thalamic nucleus (RTN).F, There is intense labeling of olfactory neurons in the olfactory epithelium (black arrow). G, m4.1N mRNAs are detected in enteric neurons (black arrowhead) throughout the gastrointestinal tract.H, The major non-neuronal localization of m4.1N mRNAs is in the kidney, where transcripts are detected in the convoluted and collecting tubule epithelia (white, black arrowheads, respectively). Scale bars:A–F, 200 μm; G, 50 μm; H, 100 μm.
Developmental in situ hybridization studies identified m4.1N mRNAs at the earliest stage of neuronal differentiation, occurring first in the postmitotic neurons of the spinal cord (data not shown) and then in the postmitotic neurons of the subcortical plate (Fig.5). At embryonic days 11.5 and 13.5, m4.1N mRNAs localized to the thin layer of postmitotic neurons that occupy the subcortical plate; proliferating neuronal progenitor cells in the ventricular zone were negative (Fig.5A,B). By embryonic days 17 and 18.5, successive layers of neurons migrate beyond the subcortical plate to form the cortical plate; whereas m4.1N mRNAs were abundant in the postmitotic neurons of the cortical plate, the migrating cells of the intermediate zone were negative (Fig. 5C,D).In situ hybridization studies in 4- and 21-d-old mice demonstrated the association of m4.1N mRNAs with neurons in all six layers of the cerebral cortex; the white matter of the corpus callosum was completely negative (Fig.5E,F).

Distribution of m4.1N RNAs in the cerebral cortex during development. A, By embryonic day 11.5 (E11.5), m4.1N mRNAs are detected in the first group of postmitotic neurons that reach the subcortical plate (SP); proliferating progenitor cells in the ventricular zone (VZ) are negative. B, As more postmitotic neurons reach the subcortical plate byE13.5, the thickness of the cell layer increases, and there is likewise more m4.1N labeling. C,D, By E17 (C) andE18.5 (D), successive layers of neurons migrate beyond the subcortical plate to form the cortical plate (CP); whereas m4.1N mRNAs are abundant in the differentiating neurons of the cortical plate, the migrating cells of the intermediate zone (IZ) are negative.E, F, Sections of cortex from 4-d-old (P4; E) and 21-d-old (P21;F) mice reveal m4.1N mRNAs in neurons of all six cortical layers; the white matter of the corpus callosum (CC) is completely negative.
Localization of 4.1N protein
A fusion protein containing the unique N terminal of m4.1N was used to generate an m4.1N-specific polyclonal antibody. Western blot analysis of COS-7 cells transfected with the full-length 879 aa m4.1N clone revealed a doublet, with the predominant band migrating at 135 kDa (Fig. 6, left); the less prominent higher molecular weight band of the doublet may represent an alternative post-translational modification of m4.1N (e.g., phosphorylation and/or glycosylation) that may occur in the transfected cells. The m4.1N antibody did not recognize any proteins in control lysates from mock-transfected cells. The observed molecular weight of m4.1N is greater than the predicted value, which has likewise been documented for 4.1R (Conboy et al., 1991) and 4.1G (Parra et al., 1998a). The major band identified in brain comigrates with the 135 kDa–expressed protein, confirming that the m4.1N clone encodes a protein of similar molecular weight to that specifically detected by the m4.1N antibody in brain. A very faint band of 165 kDa was also detected and may represent a larger splice form of 4.1N that is of low abundance. A prominent 100 kDa band was detected in the kidney and large intestine; longer exposures revealed bands of 100 kDa in the heart, lung, testis, spleen, and brain. The 100 kDa form likely corresponds to an alternatively spliced species that excludes intervening sequence between the SABD and CTD, such as that identified by PCR in mouse brain (Fig. 2). In a control experiment, preabsorption of the antibody with the m4.1N fusion protein completely abolished the m4.1N immunoreactivity (Fig. 6, right).
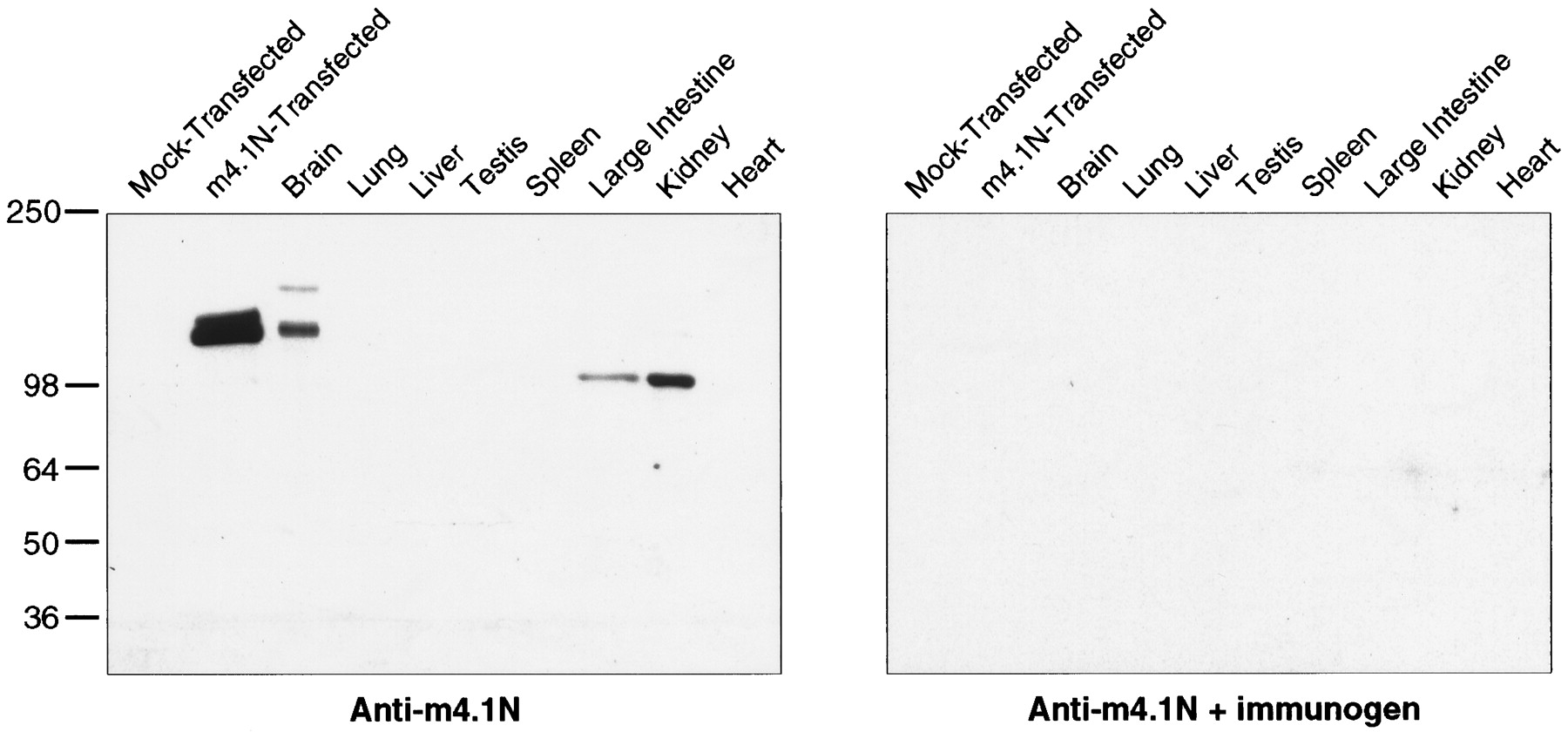
m4.1N Western analysis. A polyclonal antibody was generated against a fusion protein containing the unique N terminal of m4.1N. Left, Western blot analysis of COS-7 cells transfected with the full-length m4.1N clone revealed a major band of ∼135 kDa. No proteins were recognized in control lysates from mock-transfected cells. In the brain, there is a faint 165 kDa band and a major 135 kDa band that comigrates with the expressed protein. Bands of 100 kDa are detected in the kidney and large intestine.Right, When a parallel blot is incubated with antibody preabsorbed with the m4.1N fusion protein, no bands are evident.
Immunohistochemistry using the m4.1N antibody revealed several patterns of neuronal staining. In certain neuronal populations, including the mitral cells of the olfactory bulb (Fig.7A–C), the bipolar cells of the retina (Fig. 7J), deep cerebellar nuclei (data not shown), and motor neurons of the spinal cord (data not shown), there was intense staining of cell bodies and dendrites. Indirect immunofluorescence confocal microscopy of mitral cell dendritic cross sections demonstrated a ring-shaped pattern consistent with a juxta-membrane localization (Fig. 7C,inset). m4.1N was also present in axons; because granule cells were the only cerebellar cells containing m4.1N mRNAs (Fig.4A), the robust labeling of the cerebellar molecular layer must derive from m4.1N in granule cell axons. In other neuronal locations, including the granule cell layers of the cerebellum (Fig.7D–F) and dentate gyrus (Fig.7G–I), the cerebral cortex (data not shown), and the enteric neurons of the intestine (Fig.7K), a distinct punctate-staining pattern was observed. This speckled immunoreactivity was further demonstrated by indirect immunofluorescence confocal microscopy of the granule cell layers of the cerebellum (Fig. 7F) and dentate gyrus (Fig. 7I). In the cerebellum, the discrete immunoreactive spots (Fig. 7F, white arrow) were associated with the synaptic glomeruli of the granule cells; the nonreactive areas corresponded to the location of cell bodies, as determined by DAPI staining of the granule cell nuclei (data not shown). In the dentate, there was punctate labeling that outlined granule cell bodies (Fig. 7I, white arrow), whereas in the molecular layer, which contains the granule cell dendrites, there was a densely speckled pattern (white arrowheads). The predominant non-neuronal localization of m4.1N was in the kidney, where there was robust staining of select epithelial cells of the convoluted and collecting tubules (Fig.7L). In all cases, preincubation of the antibody with the m4.1N fusion protein eliminated the immunoreactivity (Fig.7B,E,H) (data not shown).

Immunolocalization of m4.1N protein. Immunoperoxidase and indirect immunofluorescence staining was performed on floating 40 μm sections of adult rat brain using the m4.1N antibody; immunohistochemistry was also conducted on select peripheral tissues. A, Mitral cells of the olfactory bulb exhibit intense staining of the cell bodies and dendritic processes (black arrowhead); the nuclei are distinctly negative. Among the m4.1N-positive dendritic processes, discrete varicosities exhibit robust staining. B, The olfactory bulb is devoid of staining when incubated with antibody preabsorbed with the m4.1N fusion protein. C, An indirect immunofluorescence confocal image of the same region of the olfactory bulb demonstrates the dendritic labeling of mitral cells with areas of speckling.Inset, Profiles of dendrites in cross section indicate that m4.1N is found at the outer rim of the processes, consistent with a juxta-membrane localization. D, The granule cell layer (GCL) of the cerebellum exhibits a punctate pattern of immunostaining; there is also robust labeling of the molecular layer (ML). E, No signal is detected in the cerebellum when the preabsorbed antibody is used. F, An indirect immunofluorescence confocal image of the cerebellar granule cell layer demonstrates discrete spots of immunofluorescence in the synaptic glomeruli; a white arrow points to one of the punctate structures. G, In the hippocampus, there is a prominent reticular pattern seen in the granule cell layer of the dentate gyrus (DG) and among the pyramidal cells of CA1–CA3; there is also homogeneous speckling of the molecular layer of the dentate and CA3.H, No immunostaining is detected in the hippocampus with the preabsorbed antibody. I, An indirect immunofluorescence confocal image of the granule cell layer of the dentate gyrus reveals discrete spots of immunofluorescence outlining the cell bodies; a white arrow points to one of the punctate structures. The molecular layer (white arrowheads) exhibits a densely speckled pattern of immunofluorescence. J, Within the retina, there is prominent labeling of the apical dendrite and the cell body of bipolar cells. K, Between the smooth muscle layers of the gastrointestinal tract, there is specific immunostaining of the enteric neurons (black arrowheads). Inset, The punctate pattern of labeling is evident at high magnification. L, In the kidney, there is intense immunostaining of the convoluted tubule epithelia; the collecting tubule epithelia stain in a “checkerboard” pattern, such that alternating cells are positive. The cells of the glomeruli are completely negative (asterisks). Inset, The robust immunostaining of a tubular epithelial cell is shown at higher magnification. Scale bars, A, B,K, 40 μm; C, F, 10 μm;D, E, G, H, 160 μm; I, 12.5 μm; J, 16 μm;L, 80 μm.
The punctate pattern of immunostaining in regions of axosomatic and axodendritic contact, coupled with areas of intense dendritic labeling, suggested that one of the roles of m4.1N may be to serve as a component of the synaptic architecture. Double-labeling indirect immunofluorescence was performed on primary hippocampal cultures using the m4.1N antibody with PSD95 (postsynaptic marker) and GluR1 (excitatory postsynaptic marker) antibodies (Fig.8). Indirect immunofluorescence microscopy demonstrated enrichment of m4.1N at discrete foci along the neuronal dendrites (Fig. 8B,E). The colocalization with GluR1 and PSD95 at these foci indicated that m4.1N was indeed present at the discrete sites of synaptic contact (Fig.8C,F, respectively).

Enrichment of m4.1N at sites of synaptic contact in primary hippocampal cultures. Double-labeling indirect immunofluorescence of primary hippocampal cultures was performed using the m4.1N antibody and PSD95 (postsynaptic marker) and GluR1 (excitatory postsynaptic marker) antibodies. B,E, m4.1N is enriched at discrete foci along the neuronal dendrites. A–F, An overlay of the FITC-m4.1N immunolabeling (B, E) with the Texas Red GluR1 (A) and PSD95 (D) labeling reveals colocalization of m4.1N with the synaptic proteins at these sites (yellow foci in C,F). Scale bar, 5.3 μm.
DISCUSSION
In the present study we have identified a neuronal homolog of protein 4.1, which is expressed in almost all central and peripheral neurons of the body. The association of 4.1N with neurons is underscored by the presence of m4.1N mRNAs in embryonic neurons at the earliest stage of postmitotic differentiation. 4.1N is the third member of a gene family of membrane cytoskeletal proteins that share homology with the prototypic RBC protein 4.1 (4.1R) in the defined membrane-binding, spectrin-actin–binding, and C-terminal domains. The RBC protein (4.1R), the generally expressed homolog (4.1G), and the neuronal homolog (4.1N) each occur on distinct human chromosomes, namely, chromosomes 1 (Conboy et al., 1986a), 6 (Parra et al., 1998a), and 20 (Kim et al., 1998), respectively. Although the presence of protein 4.1 in the brain has been well documented (Goodman et al., 1984; Bennett et al., 1986), our results indicate that the novel 4.1N homolog, rather than the prototypic 4.1R protein, is the major isoform expressed in neurons.
Like 4.1R and 4.1G, 4.1N exists in multiple splice forms as evidenced by PCR and Western analysis. The 135 kDa m4.1N is the predominant isoform identified in the brain, whereas a smaller 100 kDa isoform is enriched in peripheral tissues, such as the enteric neurons of the intestinal tract and the tubular epithelium of the kidney. The kidney is the major non-neuronal site of m4.1N expression, presumably reflecting the existence of a common cytoskeletal structure found in neurons and in kidney epithelium. An interesting connection between neurons and kidney cells is illustrated by the finding that mice with targeted deletion of glial-derived neurotrophic factor have specific neuronal deficits in addition to completely lacking the enteric nervous system, ureters, and kidneys (Moore et al., 1996).
Neuron-specific and generally expressed homologs/isoforms have been identified for several other erythrocyte cytoskeletal proteins, including ankyrin (Otto et al., 1991; Kordeli et al., 1995) and β-spectrin (Riederer et al., 1986; Hu et al., 1992). For a given protein family, the distinct expression patterns of its homologs throughout the body presumably reflect cell-specific functions. Whereas 4.1R, 4.1G, and 4.N are all expressed in the brain, 4.1R is found in only select neuronal populations (Walensky et al., 1998b), 4.1G is predominantly associated with glial elements (Walensky et al., 1998a), and 4.1N occurs in almost all neurons. This complementary pattern of 4.1 expression is further exemplified by our recent identification and localization of another homolog of 4.1 in the brain, named 4.1B (Parra et al., 1998b; Peters et al., 1998). Whereas m4.1N is found in all neurons of the brain except Purkinje cells and most thalamic neurons, high levels of m4.1B mRNAs are discretely localized to those neurons that lack m4.1N (Parra et al., 1998b).
Immunohistochemical studies reveal that different homologs of a given cytoskeletal protein occurring in the same cell type often have distinct subcellular localizations. For example, the generally expressed β-spectrin occurs in axons and presynaptic terminals of neurons, whereas the erythroid β-spectrin is present in neuronal cell bodies and dendrites (Riederer et al., 1986; Siman et al., 1987). In agreement with a report localizing 4.1R to the nucleus of nonerythroid cells in culture (Krauss et al., 1997), we have recently identified m4.1R in the nuclei of mitral cells of the olfactory bulb (L.D. Walensky, S. Blackshaw, and S.H. Snyder, unpublished observations); m4.1N is also found in mitral cells but localizes instead to the cell body and dendrites. Conceivably, combinations of specific homologs of membrane cytoskeletal proteins, such as 4.1, spectrin, and ankyrin, assemble to form tissue-specific and intracellular compartment–specific scaffolds.
Among the multiple intracellular localizations of 4.1N in neurons (e.g., cell body, dendrites, and axons), a synaptic localization was suggested by the punctate pattern detected in neuropil by immunohistochemistry and indirect immunofluorescence confocal microscopy. 4.1N was enriched at discrete foci along the dendrites of hippocampal neurons in culture. The association of m4.1N with synaptic sites was further supported by the colocalization of m4.1N with PSD95 and GluR1 in cultured hippocampal neurons. The colocalization of m4.1N with GluR1, the major postsynaptic excitatory neurotransmitter receptor, suggests that one of the roles of m4.1N may be to function as a component of the cytoskeletal architecture of excitatory synapses. The architecture of the synapse is based on an F-actin cytoskeletal network and contains numerous actin-binding proteins such as spectrin, myosin, adducin, ankyrin, and neurabin (Harris and Kater, 1994; Garner and Kindler, 1996; Matsuoka et al., 1997; Nakanishi et al., 1997). Analogous to the stability and flexibility afforded to circulating RBCs by their actin-based cytoskeleton, the corresponding neuronal proteins are believed to be involved in the morphological development, stability, and dynamic plasticity of the synapse (Morales and Fifkova, 1991; Matsuoka et al., 1997; Nakanishi et al., 1997). In addition, structural proteins of the synapse are implicated in the selective targeting of channels and receptors to synaptic membranes (Burns and Augustine, 1995; Allison et al., 1998) [e.g., interactions of NMDA receptors with the PDZ domain–containing protein PSD95 (Kornau et al., 1995)].
Insight into the potential functions of 4.1N as a component of the neuronal membrane cytoskeleton may be derived from our understanding of the multiple protein–protein interactions of 4.1R in RBCs. The SABD of 4.1R potentiates the interaction of spectrin with F-actin, thereby stabilizing the RBC cytoskeleton (Correas et al., 1986). 4.1R participates in tethering the cytoskeleton to the plasma membrane via interactions between its MBD and the integral membrane protein glycophorin C (Reid et al., 1989). The MBD of 4.1R also interacts with p55, a member of the membrane-associated guanylate kinase (MAGUK) family of proteins that contain PDZ domains, an SH3 motif, and a guanylate kinase motif (Alloisio et al., 1993); the PDZ domain of p55 interacts with the C terminal of glycophorin C, and thus 4.1R, glycophorin C, and p55 exist as a ternary complex (Marfatia et al., 1997). The binding of 4.1R to the anion transporter band 3 (Pasternack et al., 1985) decreases the association of ankyrin with band 3, resulting in the modulation of RBC mechanical properties. These 4.1R-binding interactions are not static. 4.1R is phosphorylatedin vitro and in vivo by protein kinase A, protein kinase C, and casein kinase II (Cohen and Gascard, 1992); in each case, 4.1R phosphorylation leads to a marked decrease in its ability to bind spectrin and promote the spectrin–actin interaction. In addition, protein kinase C phosphorylation of 4.1R inhibits its binding to membranes by blocking the interaction with band 3 (Danilov et al., 1990). Recent work has shown that the binding of Ca2+-calmodulin to specific conserved sites within the MBD also reduces the interaction of 4.1R with integral membrane proteins (Nunomura et al., 1997a,b). Like 4.1R in RBCs, 4.1N may have multiple binding partners in neurons, including components of the neuronal membrane cytoskeleton, integral membrane channels and receptors, and MAGUKs. Furthermore, the regulation of these 4.1N interactions by phosphorylation and Ca2+-calmodulin may play important roles in membrane stability and plasticity.
The critical role of 4.1R in conferring stability and flexibility to the membrane cytoskeleton of circulating RBCs exemplifies the relevance of cell structure to cell function. Recently, targeted deletion of 4.1R in mice confirmed that 4.1R-deficient RBCs are dysmorphic and mechanically unstable (Shi et al., 1999). In addition, 4.1R knock-out mice have movement, balance, coordination, and spatial-learning defects that fit with the discrete localization of 4.1R in the brain (Walensky et al., 1998b). The potential importance of 4.1N in neuronal physiology is underscored by its almost universal expression in central and peripheral neurons and by analogy to the known impact of 4.1R deficiency in knock-out mice and in patients with hereditary elliptocytosis.
Footnotes
This work was supported by National Institute of Mental Health Grant GM-07309 to L.D.W. and S.H.S., United States Public Health Service Grant MH-18501 and Research Scientist Award DA-00074 to S.H.S, and National Institutes of Health Grant DK-32094 to N.M. We thank Roxann Ashworth for DNA sequencing and analysis, Clark Riley for sequence alignments, Michael Dellannoy for expert technical guidance with confocal microscopy, Edward Benz, Jr., and Shu Huang for providing the mouse 4.1R cDNA, and Patrick Burnett, Lit-fui Lao, and Colin Collins for technical assistance.
Correspondence should be addressed to Dr. Solomon H. Snyder, Department of Neuroscience, The Johns Hopkins University School of Medicine, 813 Wood Basic Science Building, 725 North Wolfe Street, Baltimore, MD 21205.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}