

Abstract
Several scorpion toxins have been shown to exert their neurotoxic effects by a direct interaction with voltage-dependent sodium channels. Both classical scorpion α-toxins such as Lqh II from Leiurus quiquestratus hebraeus and α-like toxins as toxin III from the same scorpion (Lqh III) competitively interact for binding on receptor site 3 of insect sodium channels. Conversely, Lqh III, which is highly toxic in mammalian brain, reveals no specific binding to sodium channels of rat brain synaptosomes and displaces the binding of Lqh II only at high concentration. The contrast between the low-affinity interaction and the high toxicity of Lqh III indicates that Lqh III binding sites distinct from those present in synaptosomes must exist in the brain. In agreement, electrophysiological experiments performed on acute rat hippocampal slices revealed that Lqh III strongly affects the inactivation of voltage-gated sodium channels recorded either in current or voltage clamp, whereas Lqh II had weak, or no, effects. In contrast, Lqh III had no effect on cultured embryonic chick central neurons and on sodium channels from rat brain IIA and β1 subunits reconstituted in Xenopus oocytes, whereas sea anemone toxin ATXII and Lqh II were very active. These data indicate that the α-like toxin Lqh III displays a surprising subtype specificity, reveals the presence of a new, distinct sodium channel insensitive to Lqh II, and highlights the differences in distribution of channel expression in the CNS. This toxin may constitute a valuable tool for the investigation of mammalian brain function.
- scorpion α-toxin
- scorpion α-like toxin
- sodium channel subtypes
- receptor site 3
- hippocampus slices
- expression in oocytes
- chick central neurons
- insect sodium channels
- rat brain synaptosomes
Intoxication by scorpion venom is mainly caused by the presence of a homologous family of polypeptides 60–70 amino acids long cross-linked by four disulfide bridges that specifically affect voltage-gated sodium channels in excitable tissues (for review, see Martin-Eauclaire and Couraud, 1995). Contrary to β-toxins, scorpion α-toxins (ScαTxs) induce a prolongation of action potentials caused by selective inhibition of sodium current inactivation. According to their binding properties and their preferential toxicity to mammals or insects, the α-toxin class has been further divided into three major groups (for review, see Gordon et al., 1998). Toxins highly toxic to mammals, such as α-toxins fromAndroctonus australis hector (Aah II) and Leiurus quinquestratus hebraeus (Lqh II) encompass the classical α-toxin group, whereas α-toxins highly toxic to insects (such as LqhαIT) comprise a second group. The recently discovered toxin III from the venom of Leiurus quinquestratus hebraeus (Lqh III) has been shown to belong to a third group, the so-called scorpion α-like toxins (Gordon et al., 1996, 1998), based on its high toxicity to both mammals and insects (Table 1) and its low potency in competition for Aah II binding in rat brain synaptosomes (Sautière et al., 1998; Krimm et al., 1999).
Activity of some scorpion α and α-like toxins on mammals and insect
Neurotoxins that target sodium channels bind to at least seven topologically distinct receptor sites on the α-subunit (for review, see Gordon, 1997). Receptor site 3, where ScαTxs bind, includes the short external loop between transmembrane segments S3 and S4 on domain IV of rat brain sodium channel II and the large external loops between transmembrane segments S5 and S6 in domains I and IV (Thomsen and Catterall, 1989; Rogers et al., 1996). Receptor site 3 has been suggested to be homologous (but not identical) in insect and rat brain sodium channels. This is supported by the fact that the sea anemone toxin ATX II binds to an overlapping site with ScαTxs on both channels (Gordon and Zlotkin, 1993; Gordon et al., 1996; Rogers et al., 1996). Furthermore, all toxins competing for binding to receptor site 3 induce similar inhibition of inactivation of the sodium current in different neuronal preparations from insect and mammals (Catterall, 1992; Martin-Eauclaire and Couraud, 1995; Gordon et al., 1996;Cestèle et al., 1999).
To shed light on the peculiar behavior of the α-like toxin Lqh III, which is highly toxic to mice but competes very weakly with α-toxin binding in rat brain synaptosomes, we have analyzed the binding and effects of Lqh III in several CNS sodium channel preparations. Our results suggest that in contrast to insect sodium channels, in rat brain Lqh III does not bind to the same sodium channel subtypes as the classical ScαTx and especially not to the RIIA, the main α-subunit expressed in rat brain (Gordon et al., 1987;Auld et al., 1988; Mandel, 1992). As a consequence, Lqh III appears able to discriminate between mammalian CNS sodium channel subtypes expressed in neural somata versus nerve terminals.
MATERIALS AND METHODS
Toxins. Lqh II (LTX001), Lqh III (LTX002), and LqhαIT from Leiurus quinquestriatus hebraeus scorpion were from Latoxan (Rosans, France; A.P. 1724, 05150) and, in part, were a generous gift from Dr. Pierre Sautière, Institut Pasteur, Lille, France. The toxins from Buthus occitanus mardochei(Bom III and Bom IV) were purified as described in Vargas et al. (1987)and were a kind gift of Dr. Martin-Eauclaire, Marie-France, Faculty of Medecine Nord, Biochimie, Marseille, France. ATX II, the isoleucine isotoxin from Anemonia sulcata, was purchased from Calbiochem (Novabiochem International, San Diego, CA). The reverse-phase C18 (250 × 4.6 mm; 30 nm, 5 μm particle size) HPLC column was from Vydac (Mojave, CA). Iodogen was from Pierce (Rockford, IL). Carrier-free Na125I was from Amersham (Buckinghamshire, UK). All other chemicals were of analytical grade. Filters for binding assays were glass fiber GF/C (Whatman, Maidstone, UK) preincubated in 3% polyethylenimine (Sigma, Steinhem, Germany).
Neuronal membrane preparations. Rat brain synaptosomes were prepared from adult albino Sprague Dawley rats (∼300 gm, laboratory bred), according to the method described by Kanner (1978). Mice brains were homogenized in ice-cold 0.3 m mannitol buffer containing 10 mm EDTA and 10 mm HEPES–Tris, pH 7.4. After centrifugation at 1000 × g for 10 min, the supernatant was recentrifuged at 27,000 × g for 30 min (P2 fraction). All buffers contained a cocktail of proteinase inhibitors composed of: phenylmethylsulphonyl fluoride (50 μg/ml), pepstatin A (1 μm), iodoacetamide (1 mm), and 1 mm of 1,10-phenanthroline. Insect synaptosomes were prepared from whole heads of adult cockroaches, Periplaneta americana, according to the method previously described (Krimm et al., 1999). The membranes were kept at −80°C until used. No loss of binding activity was observed for at least 6 months. Membrane protein concentration was determined using a Bio-Rad (Hercules, CA) protein assay, with BSA as standard.
Radioiodination. Lqh III and Lqh II were radioiodinated by iodogen (Pierce) using 5 μg toxin and 0.5 mCi carrier-free Na125I, as previously described for LqhαIT (Gordon et al., 1996). The monoiodotoxins were purified using a Vydac RP C18 column and a gradient of acetonitrile from 5 to 90% B (A = aqueous 0.1% trifluoroacetic acid (TFA); B = 0.085% TFA, 50% acetonitrile; 0.2% B per min) at a flow rate of 1 ml/min. The peak of the monoiodotoxin came out just after the peak of nonmodified toxin at ∼27% of acetonitrile. The concentration of the radiolabeled toxin was determined according to the specific activity of the 125I corresponding to 2500–3000 dpm/fmol of monoiodotoxin, depending to the age of the radiotoxin and by estimation of its biological activity (usually 50–80%).
Binding assays. Equilibrium competition and saturation assays were performed using increasing concentrations of the unlabeled toxin in the presence of a constant low concentration of the radioactive toxin. To obtain saturation curves (“cold” saturation), the specific radioactivity and the amount of bound toxin were calculated and determined for each toxin concentration. Equilibrium saturation experiments were analyzed by the iterative computer program Ligand (Elsevier Biosoft, Cambridge, UK) using cold saturation analysis. Competition binding experiments were analyzed by the computer program KaleidaGraph (Synergy Software, Reading, PA) using a nonlinear Hill equation (for IC50 determination), and theKi values were calculated by the Cheng and Prusoff (1973) equation (Ki = IC50/1 + [L*/Kd] whereL* is the concentration of the hot ligand andKd is its dissociation constant). The kinetic data for ligand association and dissociation rates were subjected to the analysis of Weiland and Molinoff (1981).
Standard binding medium composition was (in mm): choline Cl 130, CaCl2 1.8, KCl 5.5, MgSO4 0.8, HEPES 50, glucose 10, and BSA 2 mg/ml. Wash buffer composition was (in mm): choline Cl 140, CaCl2 1.8, KCl 5.4, MgSO40.8, HEPES 50, pH 7.4, and BSA 5 mg/ml.
Rat brain synaptosomes (0.1–1.0 mg of protein/ml) or cockroach synaptosomes (3–8 μg/ml) were suspended in 0.2 ml binding buffer, containing 125I-Lqh III. After incubation for the designated time periods, the reaction mixture was diluted with 2 ml ice-cold wash buffer and filtered through GF/C filters under vacuum. Filters were rapidly washed with an additional 2 × 2 ml buffer. Nonspecific toxin binding was determined in the presence of 1 or 5 μm Lqh III, for binding to insect or rat brain synaptosomes, respectively, and consisted typically of 15–20% of total binding for 125I-Lqh III using cockroach membranes or 50–70% using rat brain synaptosomes, and ∼10–20% using 125I-Lqh II and rat brain membranes.
Electrophysiology on chick neurons. The culture medium for chick spinal and cortical neurons was L15 medium, supplemented with sodium bicarbonate (0.19% w/v), glucose (20 mm), insulin (5 μg/ml), sodium selenite (30 nm), conalbumin (0.1 mg/ml), progesterone (20 nm), putrescine (0.1 mm), penicillin (100 IU/ml), and chick serum (5% v/v).
Chick spinal neurons were isolated essentially as described byHenderson et al. (1995). Briefly, spinal cords from 4- to 6-d-old embryos (E4–E6) were dissected in Ca2+- and Mg2+-free PBS. They were cut into small pieces and incubated first in 0.05% trypsin in PBS for 15 min at 37°C, then in culture medium supplemented with 4 mg/ml DNase I for 2 min. After mechanical dissociation, the cell suspension was layered onto a BSA cushion (4% w/v in L15 medium) and centrifuged at 300 × g for 10 min. The cells from the pellet were then resuspended in culture medium. In some cases, the cells from the whole spinal cord were submitted to a motoneuron enrichment procedure: the cell suspension was gently layered onto a metrizamide cushion (6.8% w/v in L15) and centrifuged at 500 × g for 15 min with the brake off. “Large cells” (essentially motoneurons) concentrated in a sharp band on top of the metrizamide cushion were collected and resuspended in culture medium. The cell suspension was layered again onto a BSA cushion, centrifuged at 300 ×g for 10 min, and the cells from the pellet were resuspended in culture medium.
Chick cortical neurons were isolated similarly to the neurons of the whole spinal cord except that they were obtained from E16–E18 cortical pieces incubated in 0.25% trypsin, instead of 0.05%.
Spinal or cortical cells in culture medium were allowed to settle in 35 mm Petri dishes previously coated with 5 μg/ml poly-d,l-ornithine and maintained at 37°C in a humidified 5% CO2 atmosphere. Cells were cultured for 1–14 d. Culture medium was thoroughly replaced immediately before experiments by a salt solution containing (in mm): 120 NaCl, 5 KCl, 2 MgCl2, 2 CaCl2, 25 glucose, and 10 HEPES, pH adjusted to 7.4 with NaOH. Whole-cell recordings were performed at room temperature (20–22°C) on clearly identified neurons. Patch pipettes were pulled from borosilicate glass capillaries and had resistances ranging from 2 to 6 MΩ when filled with the “CsCl” internal solution of the following composition (in mm): 120 CsF, 10 CsCl, 5 NaCl, 2 MgCl2, 0.1 CaCl2, 10 BAPTA, 43 CsOH, and 10 HEPES, pH 7.3. Currents were amplified using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA), low-pass filtered at 1 kHz, digitized at 5 kHz, and stored on a personal computer equipped with an analog-to-digital converter (ATMO-16D; National Instruments, Austin, TX) and the DATAC package (Bertrand and Bader, 1986). Electrode and whole-cell capacitance were compensated as much as possible, and series resistance (4–11 MΩ) was electronically compensated at 80%. Cell membrane potential was maintained at −100 mV throughout the experiments; voltage steps were triggered from this value and spaced by at least 1 sec. Cells were continuously superfused at a rate of ∼1 ml/min by means of a custom-made multibarrel with a 200–300 μm tip opening placed ∼400 μm away from the recorded cell.
Electrophysiology on rat hippocampal slices. Rat hippocampal slices were prepared from 2- to 3-week-old Sprague Dawley rats. After stunning and decapitation, the brain was dissected, and coronal slices (300- to 400-μm-thick) were cut on a vibrating microtome (Campden Instruments, Loughborough, UK). Two to three slices of the hippocampus were kept in an artificial cerebrospinal solution containing (in mm): 135 NaCl, 5 KCl, 15 NaHCO3, 1 MgCl2, 2 CaCl2, and 10 glucose, saturated with 95% O2 and 5% CO2, pH 7.3–7.4, and allowed to recover for at least 1 hr at 37°C. Neurons were visualized using a Zeiss axioscop, equipped with a 40 × 0.75 NA water immersion objective and DIC optics, and an infrared (IR)-sensitive video camera (range, 400–800 nm; type C25400–07; Hamamatsu, Tokyo, Japan). Visible (<700 nm) and IR (>1000 nm) wavelength were filtered (RG9; Schott). Whole-cell recordings were performed on pyramidal cells of the CA1 region. Patch pipettes had resistances ranging from 2 to 6 MΩ when filled with either the same CsCl internal solution as the one used for experiments with chick neurons (for voltage clamp) or of the following composition (for current-clamp and TTX experiments; “KGlu”; in mm): 140 K-gluconate, 10 KCl, 4 MgCl2, 2 Na2ATP, 0.4 Na2GTP, 0.1 BAPTA, and 10 HEPES, pH adjusted to 7.2–7.3 with NaOH. Current and voltage signals were amplified using an Axopatch 200A amplifier, low-pass filtered at 1 kHz, digitized at 2–10 kHz, and stored on a personal computer equipped with an analog-to-digital converter (Digidata 1200; Axon Instruments) and the pClamp6 package. Electrode and whole-cell capacitance were compensated as much as possible, and series resistance (10–20 MΩ) was electronically compensated up to 60–80%. Cell membrane potential was maintained at −80 mV throughout the experiments, and voltage steps were triggered from this value and spaced by at least 2 sec. The slices were continuously superfused at a rate of ∼1–4 ml/min and kept at 37°C.
Lqh II and Lqh III were dissolved in the extracellular solution used at the desired concentration from stock solution (1 μm for Lqh II; 10 or 20 μm for Lqh III) kept at −20°C. To prevent adsorption on plastics, stock and test solutions were supplemented with 1 and 0.05 mg/ml BSA, respectively. BSA by itself caused no detectable effects on the cell properties.
cRNA injection into Xenopus oocytes and electrophysiology. Rat brain IIA sodium channel α-subunit (RIIA) cRNA was generated from pVA2580 construct, linearized by ClaI and transcribed in vitro with T7 RNA-polymerase as described in Gershon et al. (1992). Oocytes were coinjected with 0.85 ng of RIIA cRNA and with 0.4 ng of β1-subunit cRNA (Wallner et al., 1993). Injected oocytes were incubated at 22°C for 3–4 d in ND96 solution (in mm: 96 NaCl, 2 KCl, 1 MgCl2, and 5 HEPES, pH 7.5) supplemented with 1.8 mm CaCl2, 2.5 mm sodium pyruvate, and 100 μg/ml gentamicin (NDE solution), as described (Levin et al., 1996).
Sodium currents were recorded using a Dagan 8500 two-electrode voltage-clamp amplifier with a series resistance compensation circuit and low-resistance (0.2–0.5 MΩ) electrodes (Levin et al., 1996). Data acquisition and analysis were performed out with pClamp software (Axon Instruments). Net current was estimated by subtraction of scaled leak current. Experiments were made in ND96 solution supplemented with 1 mm CaCl2 at pH 6.5 or at pH 7.67, at 20–22°C. Sodium currents were measured before and after application of the relevant toxin.
RESULTS
α-Like toxin 125I-Lqh III binds to insect sodium channel receptor site 3
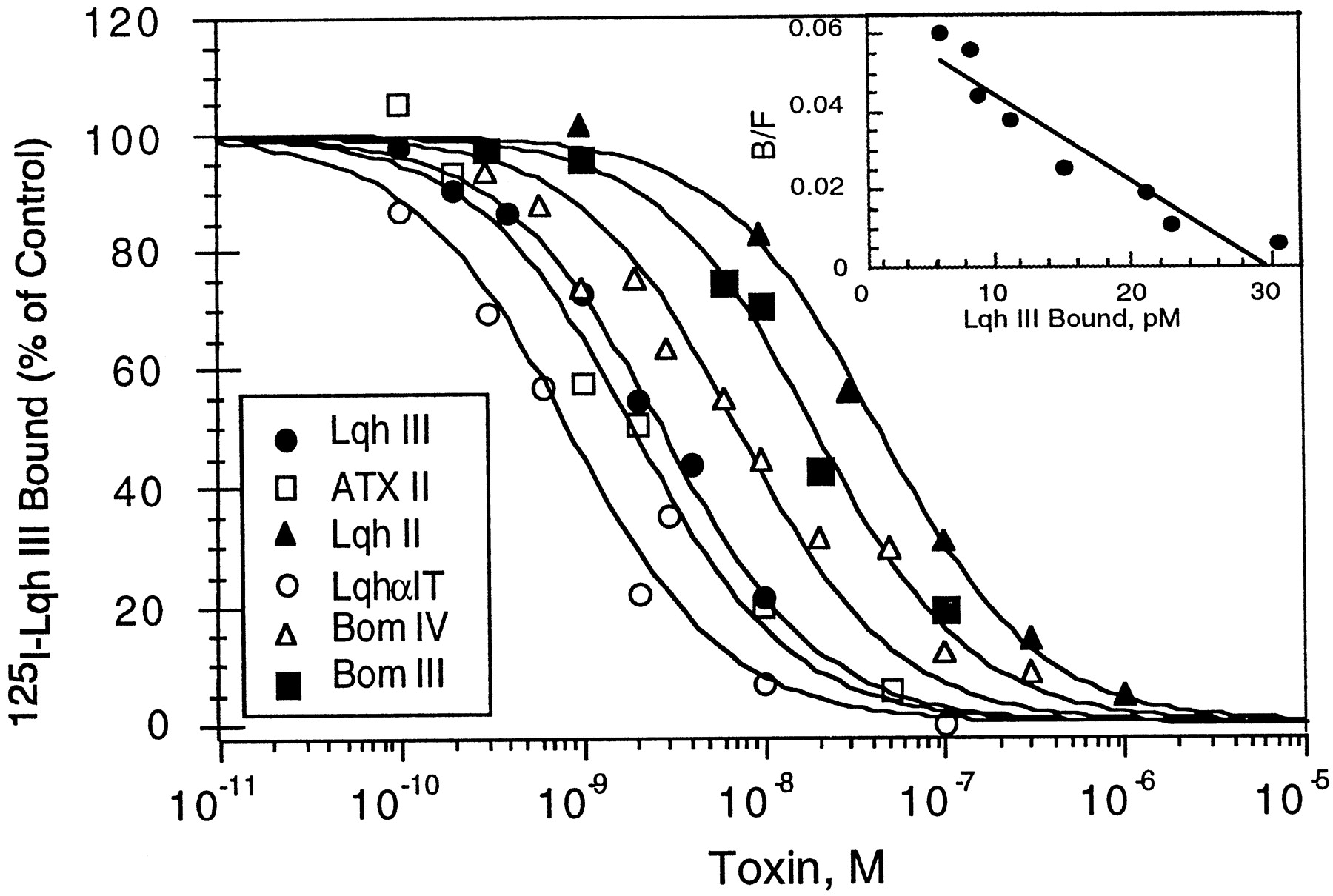
The scorpion α-like toxin Lqh III has been shown to bind to cockroach neuronal membranes (Krimm et al., 1999). To examine the receptor site of Lqh III on the insect sodium channels, competition binding studies with α- and α-like toxins were performed. Figure1 indicates that125I-Lqh III binds with high affinity (Kd = 1.43 ± 0.37 nm;n = 5) to cockroach sodium channels (Fig. 1, inset). Kinetic analysis of the binding interaction confirms the results obtained from the equilibrium studies (data not shown). The dissociation constant calculated from the kinetic rate constants (Kd =koff/kon) is 1.48 nm (association rate constant,kon = 3.37 × 106 ± 0.85 × 106M−1/sec−1; dissociation rate constant, koff = 4.72 × 10−3 ± 0.8 × 10−3 sec−1;n = 3).

Binding interaction of 125I-Lqh III in cockroach neuronal membranes. A, Competition curves for125I-Lqh III binding inhibition by various neurotoxins in cockroach synaptosomes. Cockroach neuronal membranes (5 μg/ml) were incubated for 60 min at 22°C with 120 pm125I-Lqh III and increasing concentrations of the indicated toxins. Nonspecific binding, determined in the presence of 1 μm Lqh III, was subtracted. The amount of125I-Lqh III bound is expressed as the percentage of the maximal specific binding without additional toxin. The competition curves are fitted by the nonlinear Hill equation (with a Hill coefficient of 1) to determine IC50 values (see Materials and Methods). The Ki values are (in nm): Lqh III, 1.93 ± 0.90; Bom III, 12.3 ± 4.0; Bom IV, 5.3 ± 1.0; LqhαIT, 0.7 ± 0.5; Lqh II, 44.5 ± 9.0; and ATX II, 1.4 ± 0.5. The values represent mean ± SE (n = 3). Inset, Scatchard transformation of competition binding curves of 125I-Lqh III by increasing concentrations of Lqh III (cold saturation). Membranes are incubated with 160 pm125I-Lqh III under conditions as in the main panel. The equilibrium binding parameters are calculated by the program Ligand (see Materials and Methods) and are (mean ± SE; n = number of experiments): Kd = 1.43 ± 0.37 nm; Bmax = 1.9 ± 0.5 pmol/mg protein (n = 5).
Toxins shown to interact with receptor site 3 on sodium channels, such as Lqh II, a classical α-toxin highly active on mammals (Little et al., 1998; Sautière et al., 1998), LqhαIT, a typical α-toxin most efficient on insects and the sea anemone toxin ATX II (Catterall and Beress, 1978; Gordon and Zlotkin, 1993; Gordon et al., 1996;Sautière et al., 1998) as well as the α-like toxins Bom III and Bom IV (Gordon et al., 1996; Cestèle et al., 1999) inhibit the specific binding of Lqh III at low concentrations (Fig. 1, Table 2). The similarity between theKi values of Lqh II and ATX II, the known ligands of receptor site 3 on mammalian and insect sodium channels (Catterall and Beress, 1978; Gordon and Zlotkin, 1993; Gordon et al., 1996; Rogers et al., 1996) and the other α- and α-like toxins for both 125I-Lqh III and125I-LqhαIT binding (Table 2) indicate that all these toxins bind to receptor site 3 area.
Inhibitory dissociation constants (Ki) of several toxins competing with125I-Lqh III and 125I-LqhαIT binding on cockroach sodium channels
Binding interactions of 125I-Lqh III with rat brain sodium channels
Since the α-like toxins are highly toxic by direct injection into mice brain (Table 1), we examined the interaction of125I-Lqh III with rat brain synaptosomes. Surprisingly, no specific binding was detected to either rat or mouse brain synaptosomes under conditions previously used for α-toxin binding, such as Aah II and Lqh II (Cestèle et al., 1995, 1999;Little et al., 1998). In insect synaptosomes, lowering the pH from 7.5 to 6.5 decreased the Kd of Lqh III by almost fivefold (N. Gilles and D. Gordon, unpublished observation). Accordingly, significant binding was detected in rat brain synaptosomes when the pH of the medium is lowered to 6.5 at 4°C. These conditions were thus used to examine the binding of 125I-Lqh III to rat brain synaptosomes.
The binding of 125I-Lqh III was inhibited by increasing concentrations of Lqh III, with aKi of ∼500 nm(Fig. 2A). Transformation of such competition curves yields a single class of low-affinity and high-capacity binding sites with aKd and Bmax of 479 ± 24 nm and 28 ± 11 pmol/mg of protein, respectively (n = 3; Fig.2A, inset). The receptor site capacity is at least 10-fold higher than expected from sodium channels in rat brain synaptosomes (Ray et al., 1978; Jover et al., 1980), suggesting that the low-affinity—high-capacity binding of Lqh III represents interactions with receptors other than sodium channels. Dissociation kinetics revealed a very fast drop of ∼60–70% of the displaceable binding immediately after addition of unlabeled Lqh III (data not shown). These results argue in favor of a displaceable binding of nonspecific nature for 125I-Lqh III.

Binding interaction of Lqh III with rat brain synaptosomes. A, Competition for 125I-Lqh III binding by increasing concentration of native Lqh III. Rat brain synaptosomes (0.9 mg protein/ml) were incubated with 1.2 nm125I-Lqh III in binding medium at pH 6.5 at 4°C for 60 min with increasing concentrations of Lqh III. Nonspecific binding was determined in the presence of 5 μm Lqh III and subtracted from the data. Inhibition of 125I-Lqh III binding was assessed relative to the maximal specific binding without native toxin.Inset, Scatchard transformation of a competition curve. Rat brain synaptosomes (1 mg/ml) were incubated with 1 nm125I-Lqh III at pH 6.5 at 4°C for 60 min. Nonspecific binding, determined with 5 μm Lqh III, was subtracted. The data were analyzed by Ligand (see Materials and Methods) to give the equilibrium dissociation constantKd = 499 nm and receptor site capacity Bmax = 39 pmol/mg. B,Competition of Lqh III for the binding of the α-toxin125I-Lqh II in rat brain synaptosomes. Rat brain synaptosomes (17.5 μg/ml) were incubated with 84 pm125I-Lqh II for 30 min at 20°C in the presence of increasing concentrations of Lqh II or Lqh III. Nonspecific binding determined in the presence of 300 nm Lqh II was subtracted. The inhibition of specific 125I-Lqh II binding is presented as percent of the control with no native toxins. The data points were fit with the nonlinear Hill equation (Hill number for Lqh II and Lqh III curves are 1.27 ± 0.023 and 0.87 ± 0.06, respectively). The calculated Ki values are: Lqh II, 0.18 ± 0.06 nm; Lqh III, 2.0 ± 0.5 μm.
Because the apparent affinity of Lqh III to rat brain synaptosomes may be too low to be detected by direct binding studies, we examined its ability to compete with125I-Lqh II, the classical α-toxin that binds to receptor site 3 of rat brain sodium channels. Figure2B demonstrates that125I-Lqh II binds with high affinity to rat brain synaptosomes (Kd = 0.18 ± 0.06 nm). Lqh III is able to inhibit all the high-affinity binding sites of 125I-Lqh II at high concentration (Ki = 2.0 ± 0.5 μm), consistent with its identification as an α-like toxin (Gordon et al., 1996; Sautière et al., 1998) and suggesting a very low-affinity interaction with receptor site 3 in synaptosomes. Such low-affinity binding cannot be detected by direct binding studies of 125I-Lqh III to rat brain synaptosomes.
Effect of Lqh III in rat brain slices
As indicated in Table 1, Lqh III is only 25 times less toxic to mice than Lqh II, the α-toxin highly active on mammals. However, Lqh III binds to rat brain synaptosomes with nearly four orders of magnitude less affinity than Lqh II (Fig. 2). We therefore attempted to clarify this apparent discrepancy by examining electrophysiologically the effects of both toxins on CA1 pyramidal neurons in acute hippocampal slices of 2- to 3-week-old rats.
Three CA1 pyramidal cells were studied in current-clamp conditions. Current injection in these cells triggered slowly accommodating trains of independent action potentials. In control conditions, only the first spike of a train was followed by a prolonged afterdepolarizing potential (ADP) that lead to a second and sometimes a third spike with very close intervals. Perfusion with 10 nm Lqh III gradually and reversibly provoked the appearance of a marked ADP after each spike of a train and led to the triggering of a reactivating spike (Fig. 3A). Because ADP depends on the ratio between a calcium-activated potassium current and a noninactivating sodium current, the Lqh III enhancement of ADP may be caused by additional noninactivation of sodium channels.
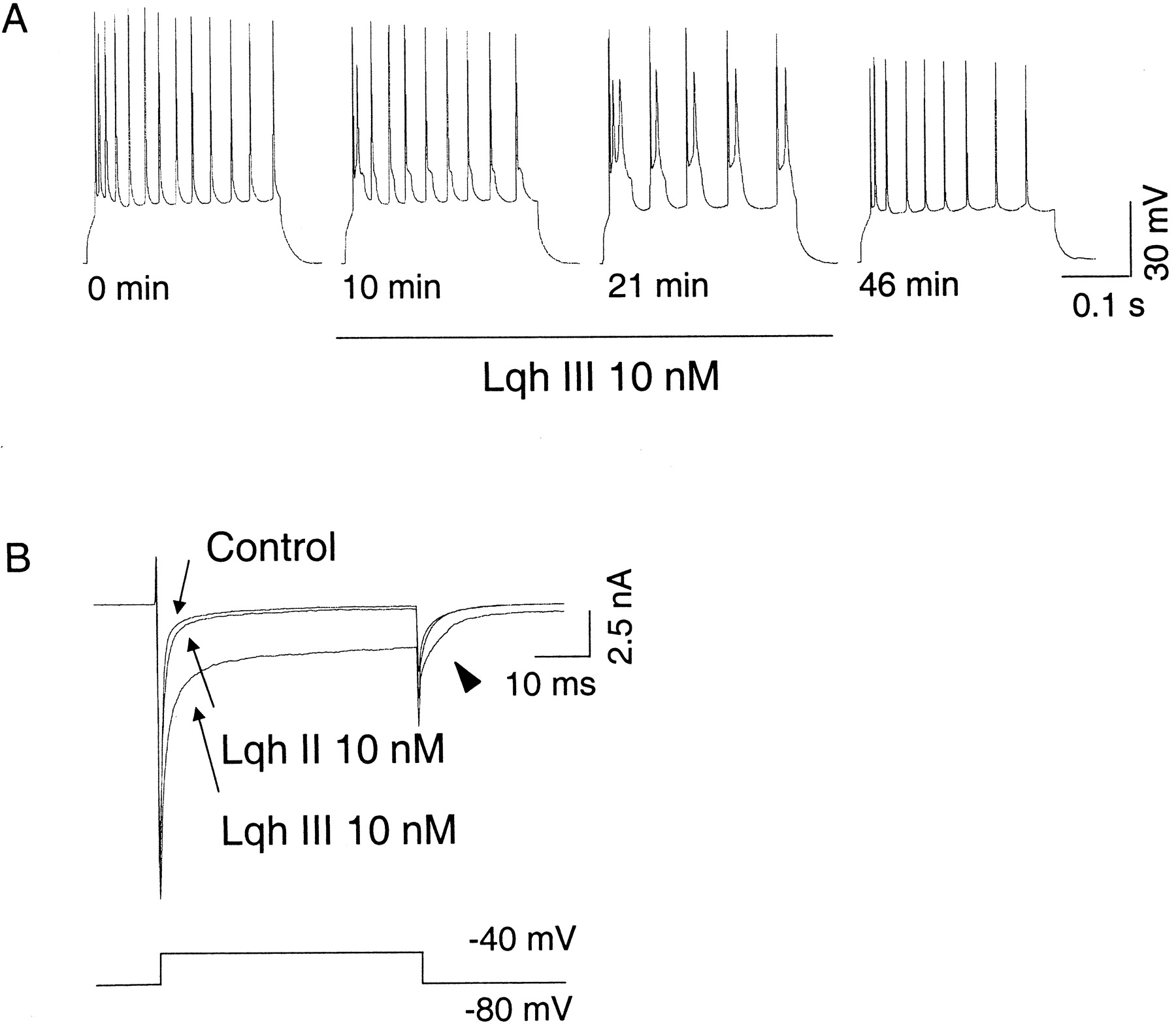
Rat CA1 pyramidal cells are very sensitive to Lqh III. A, A 10 nm concentration of Lqh III reversibly affects excitability of a CA1 pyramidal cell recorded in an acute rat hippocampal slice. Trains of action potentials were evoked every minute by 100 pA current injection for 300 msec before, during 21 min perfusion of 10 nm Lqh III (from 1 to 22 min), and after washout. Lqh III gradually enhances APD, leading to a dramatic alteration of the cell firing. B, Lqh III, but not Lqh II, strongly inhibits sodium current inactivation in voltage-clamp conditions. Traces of currents recorded during voltage steps from −80 to −40 mV before, after 10 min of 10 nm Lqh II perfusion, and after subsequent 10 min of 10 nm Lqh III perfusion are superimposed. Lqh II inhibits sodium current inactivation very weakly compared to Lqh III. Full recovery of Lqh III effect is not shown for clarity. In addition to sodium current inactivation inhibition, Lqh III is responsible for the appearance of a marked tail current (arrowhead) that, in this case, is followed by a sustained inward residual current.
Lqh III was also tested in voltage-clamp conditions on 11 other CA1 pyramidal cells dialyzed with CsCl internal solution to block most potassium channels. As indicated in Table3 and Figure 3B, Lqh III inhibited voltage-dependent Na+ current inactivation. Furthermore, in addition to inhibiting Na+ current inactivation, 10 nm Lqh III provoked the appearance of a marked tail current sometimes followed by a residual sustained current (Fig.3B, arrowhead).
In contrast, exposure to 10 nm Lqh II caused only partial effects in some but not all the cells tested (n = 7; Table 3). Variability of the effects of this α-toxin suggests that CA1 pyramidal cells may be divided into subpopulations depending on their susceptibility to Lqh II. However, one must keep in mind that this observed variability may depend on the localization of Lqh II-sensitive Na+ channels and their electrophysiological access either because of the space clamp or to the loss of processes consecutive to the slice cut.
As illustrated in Figure 3B, the effects of 10 nm of both toxins have been compared on four hippocampal neurons. Lqh II, which was always tested first, had a very weak effect on three and a more pronounced effect on the fourth neuron, whereas Lqh III strongly inhibited sodium current inactivation of all cells (Table 3). Although additivity of both toxins could not be tested in the last cell, the absence or very weak effect of Lqh II on the three first cells strongly suggests that both toxins target distinct sodium channel subtypes. It should be noted, however, that because of the limited number of cells tested, the absence or very weak effect of Lqh II presented in Figure 3B might not be representative of the α-toxin sensitivity of most CA1 pyramidal cells.
Finally, we verified that Lqh III modifies the voltage-gated sodium channels exclusively and does not affect other voltage-dependent channels in the brain. To test this possibility, two pyramidal neurons, dialyzed with the KGlu internal solution to preserve K+ channels activity, were challenged with 100 nm Lqh III after the complete inhibition of voltage-dependent Na+ channels with 1 μm TTX. Under these conditions, Lqh III had no apparent effect on the residual voltage-dependent currents (data not shown).
Effect of Lqh III on chick central neurons in culture
To further compare the effects of Lqh II and Lqh III on neurons from the CNS, we examined their effect on voltage-dependent Na+ currents of chick spinal neurons maintained in culture (Fig. 4). Surprisingly, efficiencies of both toxins were opposite to those found on rat hippocampal neurons (Table 3). Lqh III, at 50 nm or more, displayed almost no effect on chick spinal neurons (n = 14). In contrast, 10 nm Lqh II strongly inhibited the sodium current inactivation of the seven neurons tested, five of which were previously found insensitive to Lqh III. Figure 4 illustrates the effects of both toxins on one of these neurons and suggests that Lqh III does not compete with Lqh II because Lqh II effects were not altered by the coapplication of Lqh III (100 nm).

Cultured chick central neurons are sensitive to Lqh II but not to Lqh III. A, Typical effects of Lqh III and Lqh II on voltage-gated sodium current recorded in a voltage-clamped chick spinal neuron. Currents recorded during voltage steps from −100 to −10 mV before, during 100 nm Lqh III perfusion, and during 10 nm Lqh II perfusion are superimposed. Lqh III has very weak, if any, effect on sodium current inactivation, whereas Lqh II strongly inhibits it. Thearrow indicates the time at which the current amplitude reported in B was measured (10 msec after the onset of the voltage step). B, Effects of Lqh III and Lqh II on sodium current inactivation, measured as indicated in A,as a function of time. Bars indicate the time and duration of application of 10 nm Lqh II and 100 nm Lqh III. Asterisks are placed above the measurement taken from records shown in A.
To assess the specificity of both toxins on other chick CNS neurons, effects of Lqh II and Lqh III were also examined on cortical neurons maintained in culture. As shown for the spinal neurons, Lqh III at 50 (n = 3) or 100 nm(n = 2) displayed no effect, whereas the three cortical neurons that could be challenged subsequently with 10 nm Lqh II were all strongly affected (Table 3). Interestingly, for one of these neurons, we could observe that 100 nm Lqh III did not affect the onset, the steady state, or the offset of Lqh II inhibition of sodium current inactivation (data not shown). This supports the hypothesis that Lqh III does not target Lqh II binding site on embryonic chick central neurons.
Effects of Lqh II (10 nm) and Lqh III (50 or 100 nm) on current–voltage relationships of the sodium current were studied on eight (five spinal, three cortical) or 11 (seven spinal, four cortical) neurons, respectively. None of the toxins significantly affected half activation and inactivation potentials, but Lqh II clearly increased peak current amplitude at all potentials tested (data not shown).
Lqh III has no effect on rat IIA sodium channel subtype expressed in Xenopus oocytes
Although the activity of Lqh III in rat brain slices corresponds well with its high toxicity in mice brain, the enigma of the low-affinity interaction with rat brain synaptosomes still remains to be solved.
To examine whether Lqh III affects the major sodium channel α-subunit II/IIA expressed in adult rat brain (Gordon et al., 1987; Auld et al., 1988; Beckh et al., 1989; Sarao et al., 1991; Mandel, 1992), the effect of Lqh III was examined on rat IIA sodium channels reconstituted inXenopus oocytes. Coinjection of cRNA encoding for RIIA together with that for β1-subunit yielded significant inward sodium current (Fig.5a), as previously described (Auld et al., 1988; Stühmer et al., 1989). Application of 1.5 μm Lqh III had no effect on the sodium current (Fig. 5a). Because the binding of Lqh III to rat brain synaptosomes is increased at lower pH, sodium currents were measured at pH 6.5 and 7.67 before and after application of the toxin (1–3 μm). However, no difference in the results with Lqh III could be detected (data not shown). To ensure that the absence of effect of Lqh III was effectively caused by the toxin nature, ATX II (0.5–2 μm) was subsequently tested on the same oocytes (without washout). As expected, ATX II strongly inhibited the sodium current inactivation (Fig. 5a; Wallner et al., 1993;Chahine et al., 1996; Rogers et al., 1996; Warmke et al., 1997). The inactivation of sodium current of oocytes can be described by the sum of two decaying time constants: a fast component τ1 (∼1 msec) and a slow component τ2 (∼7 msec). Incubation with up to 3 μm of Lqh III has no effect on either one of these components (Fig. 5b). In contrast, incubation with high concentration of the sea anemone toxin ATX II altered the sodium current inactivation, which was then fitted by only one time constant (∼4.5 msec), as previously mentioned (Chahine et al., 1996), that falls in between the two values described for the control current (Fig.5b). With intermediate concentrations of ATX II (200–400 nm), the inhibition of sodium current inactivation can still be described by two components (τ1 and τ2; Chahine et al., 1996), but the fraction of the slow component became larger (the slow component is 60–80% of the current, as compared to the control, where it is 20–40%; data not shown; see Chahine et al., 1996). Lqh III had no effect on the ratio of these time constants and did not prevent the ability of ATX II to induce its effect, when present simultaneously (Fig. 5). Lqh III has also no effect on the recovery from inactivation, whereas ATX II induced the previously described effect on the slope factor in the same oocyte (Fig. 5a, inset; Warmke et al., 1997). Similarly to ATX II, the classical α-toxin Lqh II at 10 nm induced important inhibition of sodium current inactivation accompanied by 10% increase in sodium current in the same oocytes (data not shown). These results indicate that RIIA sodium channels are not a target of Lqh III in the brain.

Lqh III has no effect on the inactivation kinetics of the RIIA/β1 sodium channel subunits coexpressed in Xenopus oocytes. a, Sodium currents of control oocyte (with no toxin treatment), 1.5 μm Lqh III- treated oocyte, and the same oocyte after application of 2 μm ATX II. Sodium currents were measured with a two-electrode voltage clamp (see Materials and Methods); holding potential was set to −90 mV, currents were measured at −10 mV for 20 msec before (control) and 5 min after application of Lqh III/ATX II, as indicated. Inset, Normalized inward current versus membrane potential of untreated oocyte, Lqh III-, and ATX II-treated oocytes. Steady state inactivation at −10 mV triggered by 250 msec prepulses from −100 mV at 20 mV increments. The solid curve indicates the best fit by a Boltzmann distribution I/Imax = 1/{1 + exp[(v −v1/2)/kv]} where v1/2 is the half-maximal voltage andkv is the slope factor.v1/2 = −50 mV for control and Lqh III-treated oocytes, and −42.5 mV for ATX II-treated oocytes.kv = 7.64 for control and Lqh III-treated oocytes, and 9.5 for ATX II-treated oocytes.b, Inactivation kinetics of the sodium currents before and after application of toxins Lqh III and ATX II, as indicated by thearrows. Protocol as in a. τ1 and τ2 describe the time constant of inactivation before any treatment and after the application of Lqh III, whereas τA describes the time constant of inactivation after the treatment with ATX II. The data represent the average of three oocytes from two separate experiments (with SE).
DISCUSSION
We have shown that the α-like toxin Lqh III, although highly toxic to both insects and mice, binds with high affinity to cockroach neuronal membranes but not to mice and rat brain synaptosomes. Lqh III competes only at high concentration for the high-affinity binding sites of Lqh II in rat brain synaptosomes, suggesting a very low-affinity interaction with receptor site 3. Furthermore, Lqh III has no effect on either sodium channels of cultured embryonic chick central neurons or rat brain sodium channel subtype II expressed in Xenopusoocytes whereas the classical α-toxin Lqh II and the site 3 toxin ATX II are very active. In contrast, Lqh III strongly inhibits sodium current inactivation of rat CA1 pyramidal neurons in acute hippocampal slices, whereas Lqh II has only weak or no effects. Our results suggest differential sodium channel subtype specificity of Lqh II and Lqh III in mammalian CNS.
Implication for receptor site 3 structure
Scorpion α-toxins that are highly active on mammals and insects as well as other α-like toxins and sea anemone toxin ATXII all compete for 125I-Lqh III binding to receptor site 3, suggesting that Lqh II and Lqh III bind to overlapping sites in insect sodium channels (Fig. 1; Gordon et al., 1996). In contrast, in vertebrates, either one or the other toxin binds to the sodium channels of a given preparation. Rogers et al. (1996) have localized a major part of rat brain receptor site 3 in the extracellular linker between transmembrane segments S3 and S4 of domain IV. Strikingly, the corresponding amino acid sequence is highly conserved between insects (Drosophila and cockroach,Loughney et al., 1989; Dong, 1997) and mammalian sodium channels (Goldin, 1995). Other parts of the channel may therefore contribute to receptor site 3 and notably to the specific binding of α-toxins (ScαTxs) and/or α-like toxins (αLTxs). Those parts may reside in the long, highly variable extracellular loops of the sodium channel proteins, previously suggested to be part of receptor site 3 (Thomsen and Catterall, 1989).
Sodium channel subtype selectivity of α- and α-like toxins
Our results demonstrate for the first time that Lqh II and Lqh III (by extension ScαTxs and αLTxs) discriminate between sodium channel subtypes. Indeed, Lqh III targets some sodium channels on rat CA1 pyramidal cells that are not sensitive to Lqh II. This ScαTx, Lqh II, as the sea anemone toxin ATX II, strongly inhibits inactivation of sodium channel subtype II/IIA expressed in oocytes, contrary to Lqh III, which has no effect. Furthermore, Lqh II binds with high affinity to rat brain synaptosomes (Fig. 2B; Little et al., 1998) contrary to Lqh III (Fig. 2) and Bom IV, another αLTx (Cestèle et al., 1999). As rat brain synaptosomes were shown to contain at least the sodium channel subtypes II/IIA (RII; ∼80% of TTX-sensitive sodium channels) and rat I (RI; ∼20%) (Gordon et al., 1987), αLTxs probably target neither RII nor RI subtype. The absence of specific binding of Bom IV on rat caudal brain regions and spinal cord membranes (Cestèle et al., 1999) shown to contain a high level of RI (Gordon et al., 1987; Beckh et al., 1989) reinforces the deduction that RI is probably not a target for αLTxs in rat CNS.
Experiments with rat CA1 pyramidal cells indicate however that at least one sodium channel subunit sensitive to Lqh III and to TTX but not to Lqh II must exist. On this basis, the potential targets for Lqh III might be rat sodium channel subtypes III (RIII), PN1 and NaCh6 (Kayano et al., 1988; Sangameswaran et al., 1997;Toledo-Aral et al., 1997; Dietrich et al., 1998). The high sequence homology of RIII subunit with RI and RII, especially on the extracellular region were scorpion toxins are supposed to bind (Kayano et al., 1988; Rogers et al., 1996) may exclude this subunit. Concerning PN1 subtype, too little information is presently available (sensitivity to TTX and controversial presence in rat spinal cord and brain; Sangameswaran et al., 1997; Toledo-Aral et al., 1997; Felts et al., 1997) to argue for or against its possible involvement in αLTxs effects. By contrast, several considerations point to the NaCh6 subtype as a putative target for Lqh III action. NaCh6 is TTX-sensitive (Dietrich et al., 1998) and highly expressed in many neural groups in rat brain. These include cerebellar granule cells, where Bom III and Bom IV were shown to be active (Gordon et al., 1996) and pyramidal and granule cells in the hippocampus (Schaller et al., 1995; Felts et al., 1997), where Lqh III is active in inhibiting sodium current inactivation at the CA1 pyramidal cells (Fig. 3). Furthermore, little, if any expression of NaCh6 was detected in the white matter or nerve tracts (Vega-Saenz de Miera et al., 1997; Dietrich et al., 1998), and αLTxs do not bind to rat brain synaptosomes (Fig. 2; Vargas et al., 1987; Gordon et al., 1996; Cestèle et al., 1999), the corresponding neuronal membrane fraction, because it probably contains axon and nerve terminal membranes but is thought to be devoid of cell body contamination (Gray and Whittaker, 1962).
Different subcellular localization for Lqh II and Lqh III targets
Our results may provide a rational explanation for the long-lasting riddle of the way by which αLTxs kill mice by direct injection into the brain while having no specific binding in rat and mouse brain synaptosomes (Fig. 2; Vargas et al., 1987; Gordon et al., 1996; Cestèle et al., 1999). Indeed, αLTxs must affect sodium channel subtypes present essentially, or exclusively, on neuronal somata and therefore probably absent from synaptosomes, contrary to ScαTx targets. Inhibition of sodium current inactivation by αLTxs was observed in rat CA1 pyramidal cells (Fig. 3) and cultured cerebellar granule cells (Gordon et al., 1996) using the patch-clamp technique in the whole-cell configuration. Because of space clamp limitations, this method allows only the recording of the electrophysiological properties of the soma and proximal processes. This consideration argues in favor of a somatic localization of αLTx-sensitive channels. Similarly, an axonal localization of the ScαTx-sensitive sodium channels and an electrophysiological access more or less extended may explain why some rat CA1 pyramidal cells were slightly responsive to Lqh II and some other not. These cells express the RII sodium channel subtype (Black et al., 1994), and an axonal, but not somatic, localization of this Lqh II target has been already reported by Westenbroek et al. (1989). These authors have also reported a somatic localization of RI subtype on hippocampal pyramidal cells. In the light of our results, this may suggest that this subtype, which is probably not a target for Lqh III since it is present in synaptosomes, is not a target for Lqh II either, unless the antibody used cross-reacts with an Lqh III-sensitive sodium channel subtype.
A new insight into the scorpion toxin selectivity issue
Scorpion toxins affecting sodium current have been long known to reveal animal group selectivity (Table 1; Zlotkin et al., 1978;Martin-Euclaire and Couraud, 1995; Gordon et al., 1998). The αLTxs that are highly active on both mice and insects (Gordon et al., 1996;Sautière et al., 1998) have been considered to be not selective in this respect. Our present study, however, elucidates a new aspect in the selectivity issue, namely selectivity of scorpion toxins to distinct sodium channel subtypes. This “fine tuning” of certain scorpion toxins is rather surprising, because the α- and α-like toxins are suggested to interact with receptor site 3 on sodium channels in both mammals and insects. The unexpected strong effect of Lqh III on the pyramidal cells as opposed to the lack of effect of the classical α-toxin Lqh II is remarkable because it is the first demonstration of a selective interaction of a scorpion toxin with a sodium channel subtype or subtypes in a discreet subcellular region. The selectivity of Lqh II to chick central neurons, as opposed to the lack of effect of Lqh III, further emphasizes this issue.
Neurons of the mammalian brain express multiple subtypes of sodium channels that are the products of at least six distinct genes (Goldin, 1995; Schaller et al., 1995; Sangameswaran et al., 1997; Toledo-Aral et al., 1997). These channels differ in subcellular localization (Westenbroek et al., 1989), developmental pattern of expression, and abundance in different brain regions (Gordon et al., 1987; Beckh et al., 1989; Beckh, 1990; Black et al., 1994; Schaller et al., 1995;Felts et al., 1997). These variations are suggestive of functional differences, but there is no direct information about different physiological roles of particular sodium channel subtypes. Our results suggest that the multiple sodium channel subtypes in mammalian brain can be pharmacologically discriminated by their sensitivity to certain toxins, such as ScαTxs and αLTxs and to some extent, by the previously described μ-conotoxin PIIIA (Shon et al., 1998). This should provide new tools to study the functional role and distribution of various sodium channel subtypes.
Footnotes
Part of this work was supported by a grant from the German-Israeli Foundation no. I 0375–172.01/94 to I.L. and D.G. This work was also supported by the Swiss National Science Foundation and L’office Fédéral des Sciences de l’Education grants to D.B. We are grateful to Dr. Pierre Sautière, Lille, France for the generous and kind gift of Lqh toxins, and to Dr. M.-F. Martin-Eauclaire, Marseille, France for the gift of Bom III and Bom IV toxins.
Drs. Gilles and Blanchet contributed equally to this work.
Correspondence should be addressed to Dr. Dalia Gordon, c/o Prof. M. Gurevitz, Department of Plant Sciences, Tel-Aviv University, Ramat-Aviv 69978 Tel Aviv, Israel.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}