

Abstract
Binding of neurotransmitter triggers gating of synaptic receptor channels, but our understanding of the structures that link the binding site to the channel is just beginning to develop. Here, we identify an intersubunit triggering element required for rapid and efficient gating of muscle nicotinic receptors using a structural model of the Torpedo receptor at 4 Å resolution, recordings of currents through single receptor channels, measurements of inter-residue energetic coupling, and functional consequences of disulfide trapping. Mutation of the conserved residues, αTyr 127, εAsn 39, and δAsn 41, located at the two subunit interfaces that form the agonist binding sites, markedly attenuates acetylcholine-elicited channel gating; mutant cycle analyses based on changes in the channel gating equilibrium constant reveal strong energetic coupling among these residues. After each residue is substituted with Cys, oxidizing conditions that promote disulfide bond formation attenuate gating of mutant, but not wild-type receptors. Gating is similarly attenuated when the Cys substitutions are confined to either of the binding-site interfaces, but can be restored by reducing conditions that promote disulfide bond breakage. Thus, the Tyr–Asn pair is an intersubunit trigger of rapid and efficient gating of muscle nicotinic receptors.
- acetylcholine receptor
- channel gating
- single channel kinetics
- intersubunit trigger
- coupling energy
- binding
Introduction
Gating of synaptic receptor channels is an allosteric process in which binding of nerve-released transmitter opens an ion-selective channel remote from the binding site. This allosteric process originates from local motions of the protein caused by bound neurotransmitter, which initiate global motions that open the channel and allow ion flow. For the Cys-loop family of synaptic receptors, both intrasubunit and intersubunit motions have been inferred from agonist-dependent changes in electron density (Unwin et al., 2002), changes in solvent accessibility of cysteine-substituted residues (Karlin and Akabas, 1998; Bera et al., 2002), and functional consequences of disulfide trapping (Horenstein et al., 2001; Kash et al., 2003). Motions have also been inferred from molecular dynamics simulations of models of the receptor ligand-binding domain (Henchman et al., 2003), and of the joined binding and pore domains (Law et al., 2005; Cheng et al., 2006). In the related acetylcholine binding protein (AChBP), agonist-dependent motions have been demonstrated by molecular dynamic simulations and changes in tryptophan accessibility to a fluorescence quencher (Gao et al., 2005), changes in hydrogen-deuterium exchange (Shi et al., 2006), and changes in chemical shift by nuclear magnetic resonance (Gao et al., 2006). However, insight into structures that relay the binding event to the channel requires structural perturbations combined with direct measurements of channel gating.
Over the past decade, the wealth of primary sequences of receptor subunits (Le Novere and Changeux, 2002) allowed identification of conserved residues potentially essential for function. This one-dimensional information can now be transposed into three dimensions using high-resolution structures of AChBP from several species (Brejc et al., 2001; Celie et al., 2005; Hansen et al., 2005), a 4 Å resolution structural model of the Torpedo acetylcholine receptor (Unwin, 2005), and homology models based on both AChBP and Torpedo receptor templates (Le Novere et al., 2002; Molles et al., 2002; Schapira et al., 2002; Sine et al., 2002a; Cheng et al., 2006). Thus, conserved residues that congregate in three-dimensional space emerge as candidate contributors to agonist-dependent channel gating. Here, we describe the contribution to gating of one such aggregate of residues conserved in all muscle nicotinic receptors: Tyr 127 in each of the two α-subunits and the juxtaposed Asn 39 in the ε-subunit and the equivalent Asn 41 in the δ-subunit.
Measurements of channel gating combined with mutant cycle determinations of inter-residue energetic coupling (Horovitz and Fersht, 1990) disclosed two clusters of interdependent residues conserved among α-subunits, one proximal to the binding site (Mukhtasimova et al., 2005), and the other at the junction of the extracellular and pore domains (Lee and Sine, 2005). Here, we quantify channel gating after structural perturbations of key residues at interfaces between subunits, and use mutant cycle analyses to assess whether these residues contribute to channel gating in an interdependent manner. Substitution of cysteine followed by disulfide trapping is then used to test for physical proximity of the key residues, and to determine how covalent linkage of the intersubunit residues affects the gating process.
Materials and Methods
Construction of wild-type and mutant AChRs.
cDNAs encoding human α-, β-, δ-, and ε-subunits were obtained and subcloned into the cytomegalovirus-based expression vector pRBG4 as described previously (Ohno et al., 1996). Site-directed mutations were introduced using the Quick-Change mutagenesis kit (Stratagene, Cedar Creek, TX). The presence of each mutation and the absence of unwanted mutations were confirmed by dideoxy sequencing of the entire coding region. Cells from the BOSC 23 cell line (Pear et al., 1993), a variant of the human embryonic kidney (HEK) 293 cell line, were transfected with mutant or wild-type cDNAs using calcium phosphate precipitation as described (Bouzat et al., 1994). Patch-clamp recordings and measurements of 125I-α-bungarotoxin binding were conducted 2 and 3 d after transfection, respectively.
Patch-clamp recording.
Single-channel recordings were obtained as described previously (Lee and Sine, 2004; Mukhtasimova et al., 2005). Briefly, recordings were obtained in the cell-attached patch configuration (Hamill et al. 1981) with a specified concentration of ACh in the patch pipette, a membrane potential of −70 mV and a temperature of 21°C. ACh (Sigma, St. Louis, MO) was dissolved in patch pipette solution [containing (in mm) 142 KCl, 5.4 NaCl, 1.8 CaCl2, 1.7 MgCl2, 10 HEPES, pH 7.4] and kept as a 100 mm stock solution at −80°C until use. The bath solution was identical to the pipette solution. Single-channel currents were recorded using an Axopatch 200B (Molecular Devices, Union City, CA), low-pass filtered at 100 kHz and recorded to hard disk at 200 kHz using the program Acquire (Bruxton, Seattle, WA). Data were obtained over a range of ACh concentrations from 3 μm to 1 mm, spaced at half-log unit intervals of the concentration. Data were accepted for analysis only if channel activity in the patch was low enough to ensure that each series of closely spaced channel openings originated from one channel; for some mutant receptors, this condition restricted the range of ACh concentrations to 30 μm to 1 mm. Data from two to four patches were obtained for each experimental condition, and included 4000 to 8000 open and closed intervals per patch.
Single-channel kinetic analysis.
Estimation of rate constants in a kinetic scheme describing receptor activation was done as described previously (Lee and Sine, 2004; Mukhtasimova et al. 2005). Briefly, currents were filtered at 10 kHz using a digital Gaussian filter, events were detected using a half-amplitude threshold criterion, a dead time of 10 μs was imposed, and dwell times exceeding the detection threshold were corrected for digital filtering using cubic-spline interpolation (Colquhoun and Sigworth, 1983). Dwell times corresponding to the activation process were separated from those caused by desensitization by defining a closed time that distinguishes closings within clusters of closely spaced openings from closings between clusters (Lee and Sine, 2004) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). For each wild-type or mutant receptor, the kinetic scheme below was fitted simultaneously to dwell times obtained over a wide range of ACh concentrations using maximuminterval likelihood (MIL) software (Qub suite; State University of New York, Buffalo, NY).
 Here, A is ACh, R is the receptor, k+n and k−n are ACh association and dissociation rate constants, β and α are channel opening and closing rate constants, and k+b and k−b are channel blocking and unblocking rate constants for Ach, respectively. To determine the scheme rate constants, MIL computes the likelihood or the joint probability of the experimental series of dwell times given the kinetic scheme and trial rate constants, and varies the rate constants until the likelihood is maximized. Error estimates of the fitted rate constants were determined using MIL (Qin et al., 1996). The resulting estimates of β and α yielded the channel gating equilibrium constant β/α.
Here, A is ACh, R is the receptor, k+n and k−n are ACh association and dissociation rate constants, β and α are channel opening and closing rate constants, and k+b and k−b are channel blocking and unblocking rate constants for Ach, respectively. To determine the scheme rate constants, MIL computes the likelihood or the joint probability of the experimental series of dwell times given the kinetic scheme and trial rate constants, and varies the rate constants until the likelihood is maximized. Error estimates of the fitted rate constants were determined using MIL (Qin et al., 1996). The resulting estimates of β and α yielded the channel gating equilibrium constant β/α.
Identification of kinetic modes.
Receptors with multiple Cys substitutions invariably exhibited distinct kinetic modes, or clusters of closely spaced openings that differed qualitatively in their kinetics of activation. To sort the clusters into homogeneous kinetic modes, we first imposed a closed time that discriminated between intracluster and intercluster closed dwell times, and then inspected the distributions of the resulting population of clusters as described previously (Milone et al., 1998). Briefly, we inspected the distributions of four parameters computed for each cluster: mean closed time (τc), mean open time (τo), mean open probability (Po), and relative cluster mean (log[(τccluster × τorecording)/(τcrecording × τocluster)]). Distinct peaks in these distributions were then used to define homogeneous kinetic modes in terms of mean cluster Po, τc, and τo.
Oxidation with hydrogen peroxide (H2O2).
Cell-attached patches were established to cells expressing receptors with Cys substitutions of residues at opposing faces of the α-, ε-, and δ-subunits. Control single-channel currents evoked by ACh in the patch pipette were recorded, and then 50 μl of H2O2 (Sigma), freshly diluted in H2O, was added to the bath solution (700 μl) to establish a final concentration of 4.4 mm. After 5 min, when the change in current kinetics appeared complete, a second recording was obtained.
Reduction with DTT.
Cells were incubated for 5 min in bath solution containing 4.4 mm H2O2, and then washed free of oxidizing reagent using bath solution. After establishing a gigaohm seal, control ACh-evoked single-channel currents were recorded and a freshly prepared solution of dithiothreitol (DTT; Sigma) in calcium-free Ringer's solution [containing (in mm) 115 NaCl, 2.5 KCl, 1.8 MgCl2, 10 HEPES, pH 7.5] was added to the bath to establish a final concentration of 0.02 mm. After 5 min of incubation in DTT, a second recording was obtained.
ACh Binding Measurements.
The total number of [125I] α-bungarotoxin (Btx) binding sites on the cell surface of transfected BOSC 23 cells and ACh competition against the initial rate of [125I]α-Btx were determined as described previously (Sine, 1993). ACh competition measurements were analyzed according to the following form of the Hill equation: 1 − Y = 1/[1 + ([ACh]/Kapparent)nH], where Y is fractional occupancy by ACh, nH is the Hill coefficient, and Kapparent is the apparent dissociation constant.
Results
Intersubunit contributions to ACh-evoked channel gating
Global motions that underlie ACh-evoked channel gating likely include intersubunit displacements. To identify residues that potentially contribute to such intersubunit displacements, we examined interfaces between subunits in the structural model of the Torpedo receptor at a resolution of 4 Å (Unwin, 2005) in light of multiple sequence alignments (Fig. 1). In the extracellular domains that form the agonist binding sites, a conserved Tyr 127 from each of the two α-subunits juxtaposes a conserved Asn 39 or Asn 41 from the neighboring ε- or δ-subunits, respectively. Given their physical proximity and conservation across species, these two pairs of residues become candidate contributors to intersubunit displacements associated with ACh-evoked channel gating.

Sequence conservation and locations in three dimensions of key intersubunit residues. Segments of the primary receptor sequences are shown with the conserved Tyr and Asn residues highlighted. Secondary structural rendering of two subunits from the Torpedo acetylcholine receptor (Protein Data Bank code 2BG9) (Unwin, 2005) that form an ACh binding site is shown with the key Tyr and Asn residues displayed in stick representation. For reference, the approximate location of the ACh binding site is indicated by Trp 149.
Intersubunit contributions to receptor function assessed by ACh binding
To determine whether the pair of interface residues contributes to AChR function, we generated point mutant subunits, coexpressed each with complementary wild-type subunits in HEK 293 cells and measured competition of ACh against the initial rate of radio-labeled α-bungarotoxin binding. The ACh competition assay has served as a useful screen for functionally important residues because most mutations that altered ACh binding were subsequently found to alter elementary steps underlying ACh-dependent channel activation (Engel et al., 1996; Milone et al., 1997; Wang et al., 1997; Sine et al., 2002b). Substitution of Thr for αTyr 127 diminishes steady-state ACh affinity by 18-fold relative to that of the wild-type receptor, whereas substitution of Ala for both εAsn 39 and δAsn 41 diminishes affinity by 84-fold (Table 1). However, when all three mutations are combined in a single receptor, ACh affinity diminishes only 50-fold, far less than the 1500-fold decrease expected from independent contributions of the individual mutations, indicating that these residues contribute to steady-state affinity in an interdependent manner. However, substitution of Phe for αTyr 127 enhances affinity 12-fold, but when αY127F is combined with εN39A and δN41A, affinity diminishes eightfold, indicating roughly independent contributions of the individual mutations. Thus, measurements of steady-state binding of ACh show that αTyr 127, εAsn 39, and δAsn 41 contribute to some facet of receptor function (agonist binding, channel gating, or desensitization) and that the contributions are likely to be interdependent.
ACh competition against 125I-α-bungarotoxin binding for mutant receptors
Contributions of αTyr 127, εAsn 39, and δAsn 41 to channel gating
To determine whether the pair of interface residues contributes to channel gating, we recorded ACh-evoked single-channel currents from cell-attached patches containing the mutant receptors. The initial recordings were obtained with a saturating concentration of ACh in the patch pipette, which allows direct assessment of channel gating. In wild-type receptors, channel openings lasting on the order of milliseconds are interspersed by channel closings on the order of tens of microseconds (Fig. 2), indicating efficient and rapid gating of the doubly occupied receptor channel (Ohno et al., 1996; Hatton et al., 2003; Sine and Engel, 2006). Substitution of Thr for αTyr 127 strongly impairs channel gating, as shown by single channel openings flanked by long, well resolved closings, whereas substitution of Ala for εAsn 39 and δAsn 41 impairs gating to a similar extent. However, when all three mutations are combined in a single receptor, channel gating is more efficient than in receptors containing either mutation alone. Thus, as suggested by measurements of ACh binding, single-channel recordings show that αTyr 127, εAsn 39, and δAsn 41 contribute to channel gating, and that the contributions are interdependent.
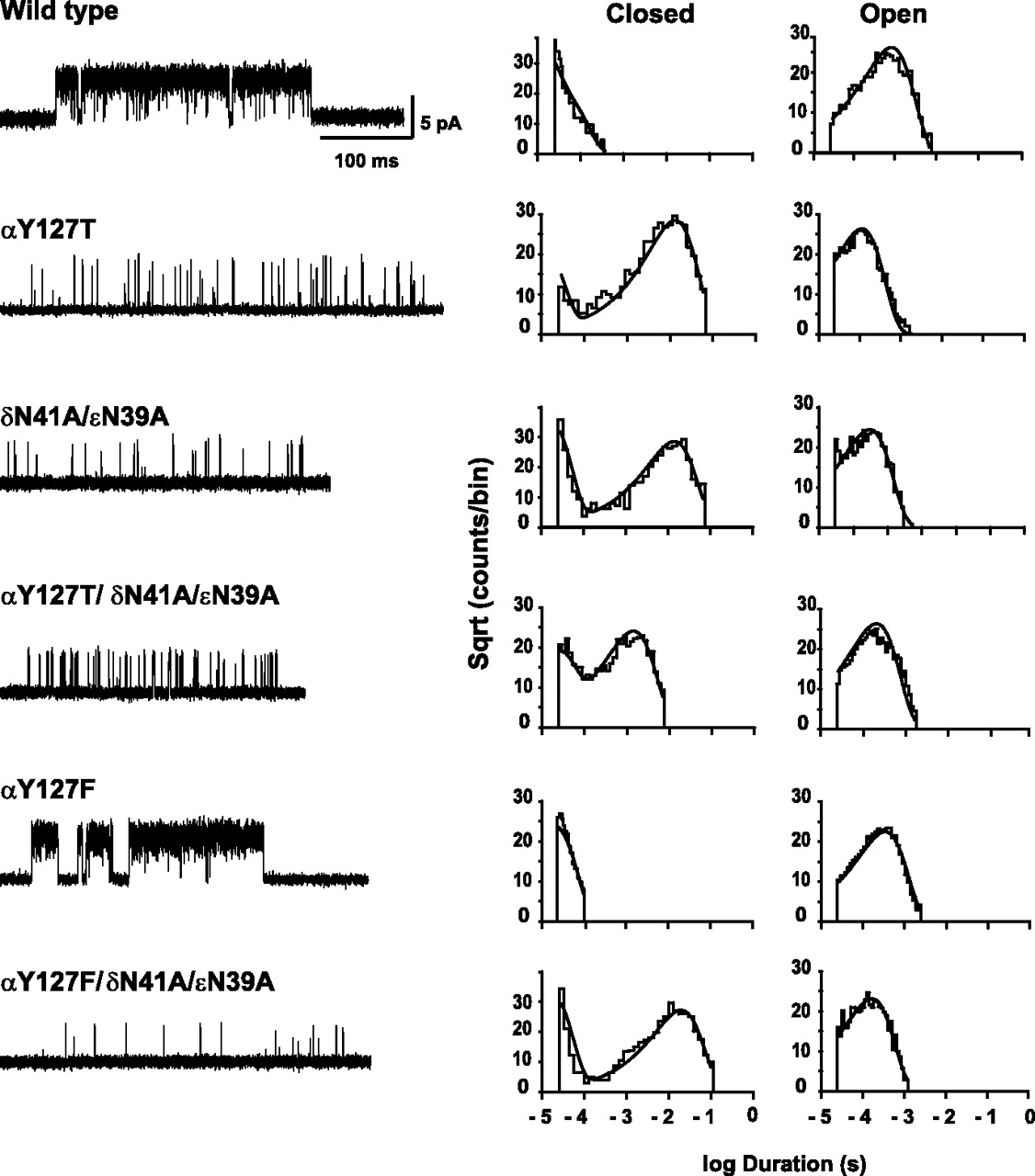
Kinetics of channel gating of wild-type and mutant receptors cloned from human skeletal muscle. Single-channel currents recorded in the cell-attached patch configuration with 1 mm ACh in the patch pipette are displayed with openings as upward deflections (membrane potential, −70 mV; bandwidth, 10 kHz). The displayed segments represent activation episodes from which long flanking closed periods have been removed (see Materials and Methods). For each type of receptor, the current traces are displayed next to the corresponding dwell time histograms with probability density functions generated by kinetic fitting superimposed (see Materials and Methods) (Table 2).
Kinetics of activation of wild-type and mutant AChRs
In contrast, substitution of Phe for αTyr 127 enhances channel gating, but when αY127F is combined with εN39A and δN41A, gating is impaired similarly to that for receptors containing only εN39A and δN41A. Thus, Phe effectively replaces the native αTyr 127, indicating that efficient channel gating requires an aromatic residue at position 127 of the α subunit.
To quantify channel gating for each mutant receptor, we recorded unitary currents over a wide range of ACh concentrations and analyzed the resulting set of open and closed dwell times according to a kinetic scheme in which ACh binds sequentially followed by channel opening and block of the open channel by ACh (see Materials and Methods). To obtain dwell times corresponding to the activation process, we removed long closed intervals caused by desensitization (Lee and Sine, 2004) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), and fitted the scheme to the remaining dwell times by maximum likelihood. For all mutant receptors, the probability density functions generated from the kinetic scheme and fitted rate constants well describe the distributions of closed and open dwell times (Fig. 2). The fitted rate constants reveal that, compared with the wild-type receptor, both αY127T and the εN39A/δN41A pair profoundly slow the channel opening step and modestly accelerate the channel closing step, producing up to a 1500-fold decrease of the channel gating equilibrium constant (Table 1). However, when all three mutations are combined, the channel gating equilibrium constant decreases by only 100-fold, indicating that these proximal residues contribute to channel gating in an interdependent manner.
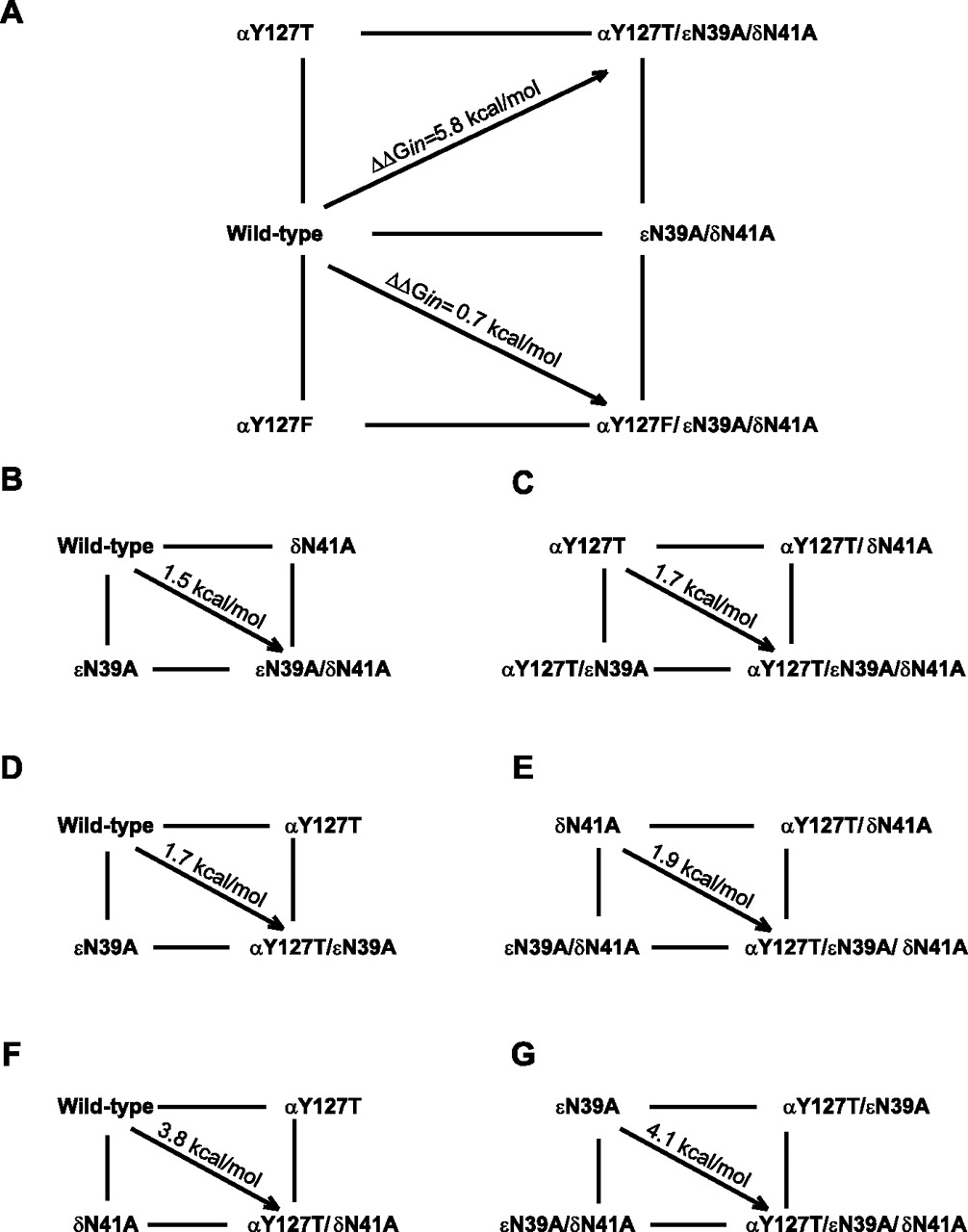
To quantify the inter-residue coupling energy, we generated thermodynamic mutant cycles (Horovitz and Fersht, 1990) using the channel gating equilibrium constants obtained from the fitted rate constants (Lee and Sine, 2005). The cycle corresponding to the triple mutant receptor αY127T/εN39A/δN41A reveals an overall coupling free energy of 5.8 kcal/mol, indicating that these residues are strongly interdependent (Fig. 3A). However, the cycle corresponding to the triple mutant receptor αY127F/εN39A/δN41A reveals a coupling free energy of 0.7 kcal/mol, which approaches RT (0.6 kcal/mol), indicating near independence and confirming that Phe can substitute for the native Tyr.

A–G, Mutant-cycle analysis for receptors with the indicated mutations. For each cycle, coupling free energy (ΔΔGin) was computed from the gating equilibrium constants generated by kinetic fitting according to: ΔΔGin = −RT ln[(Θww × Θmm)/(Θwm × Θmw)], where Θ is the gating equilibrium constant (Table 1) for wild-type (ww), single-mutant (wm, mw), or double-mutant (mm) receptors.
To determine the origin of the large coupling energy for the αY127T/εN39A/δN41A mutant cycle, we measured channel gating equilibrium constants for receptors containing single, double, and triple mutations and generated a series of two-dimensional mutant cycles. The cycle generated from receptors bearing one or both of the mutations εN39A and δN41A reveals a moderate coupling free energy of 1.5 kcal/mol (Fig. 3B); a second cycle in which αY127T is present in each receptor reveals a similar coupling free energy between εN39A and δN41A (Fig. 3C). Thus, although εN39 and δN41 are located at different subunit interfaces, the residues are interdependent in contributing to channel gating.
To determine whether the two subunit interfaces contribute differently to the overall coupling energy, we compared mutant cycles in which αY127T was paired against either εN39A or δN41A. For the two cycles that pair αY127T and δN41A (Figs. 3F,G), the coupling-free energies are more than twice those obtained for the two cycles that pair αY127T and εN39A (Figs. 3D,E). Thus, the Tyr and Asn at both subunit interfaces contribute to the large inter-residue coupling energy, but the coupling energy for the αδ interface is about twice that of the αε interface.
Disulfide trapping
A key question is whether the large inter-residue coupling energy originates from physical association between αTyr 127 and the proximal εAsn 39 or δAsn 41. To address this issue, we replaced each residue with Cys, subjected receptors bearing all three mutations to oxidizing conditions that promote disulfide bond formation, and recorded ACh-evoked single currents. After forming a cell-attached patch and recording control currents elicited by ACh within the patch pipette, we applied the oxidizing reagent hydrogen peroxide (H2O2) to the bath solution and looked for changes in the pattern of unitary currents. Before H2O2 application, the triple mutant receptors activate in several kinetic modes, ranging from high to very low channel opening probability (Fig. 4, Table 3). The distinct kinetic modes could result from receptors in which a disulfide bond spontaneously formed at zero, one, or two subunit interfaces, giving four possible modes, or possibly from kinetic modes independent of disulfide bonding. Within 5 min after H2O2 application, all unitary currents exhibited low probability of channel opening, corresponding to kinetic modes with low and ultra-low opening probability (Fig. 4, Table 3). In this application method, H2O2 presumably crosses the cell membrane or the gigaohm seal to gain access to the engineered Cys residues. The method is advantageous because a control recording is obtained from the same patch of membrane subsequently subjected to oxidation, and the cell-attached patch configuration provides an inherently very stable gigaohm seal. In contrast, application of H2O2 to wild-type receptors does not affect single channel open probability (Fig. 4, Table 3), suggesting that after oxidation the two Cys-substituted residues forms a covalent bond across the subunit interface that suppresses channel gating.

Disulfide trapping of receptors with cysteine substitutions. Cell-attached patches were established to cells expressing wild-type or the indicated mutant receptors, and single-channel currents evoked by 30 μm ACh were recorded at a membrane potential of −70 mV and displayed at a bandwidth of 10 kHz. Five minutes after adding H2O2, a second recording was obtained from the same patch. Dwell-time histograms corresponding to each receptor type and treatment condition are displayed, with the smooth curves generated by fitting the sum of exponentials to the dwell times. Addition of H2O2 has no effect on wild-type receptors, but markedly suppresses channel gating of the mutant receptors.
Modal kinetics of Cysteine-substituted receptors
To further test the interpretation of disulfide bond formation, we asked whether the changes in single-channel kinetics are reversible. Thus, we applied H2O2 to cells expressing receptors bearing all three Cys substitutions, formed a cell-attached patch, recorded control single-channel currents evoked by ACh within the patch pipette, and then applied the reducing agent DTT to the bath solution. After oxidation of receptors and formation of the gigaohm seal, ACh elicited unitary currents with very low probability of channel opening, as just described (Fig. 5, Table 3). After application of DTT, kinetic modes with increased channel open probability appeared, one with intermediate and another with high open probability, similar to the kinetic modes before oxidation. Application of DTT to wild-type receptors had no effect on single channel kinetics (Fig. 5, Table 2). The reversal of kinetics in the triple mutant receptor appeared partial, however, as the two kinetic modes with high open probability comprised a smaller fraction of the total than in the untreated triple mutant, and the mean open duration for one of the modes was twofold to fourfold briefer (Table 3). These quantitative differences could have arisen from incomplete reduction because a minimal concentration of 0.02 mm DTT was used to avoid reduction of native disulfide bonds; 0.02 mm was found to be the maximum DTT concentration that did not affect the gating kinetics and unitary conductance of wild-type receptors. Thus, the impaired channel gating caused by oxidation of the Cys-substituted receptors is at least partially reversible.

Reversal of the effects of disulfide trapping. After applying H2O2 to cells expressing the indicated receptors and establishing a cell-attached patch, control single-channel currents evoked by 30 μm ACh were recorded at a membrane potential of −70 mV and displayed at a bandwidth of 10 kHz. Five minutes after adding DTT, a second recording was obtained. Dwell-time histograms corresponding to each receptor type and treatment condition are displayed, with the smooth curves generated by fitting the sum of exponentials to the dwell times. Addition of DTT has no effect on wild-type receptors, but restores efficient channel gating of the mutant receptors previously treated with H2O2.
To determine whether disulfide bond formation at only one of the two subunit interfaces affects channel opening probability of ACh-evoked unitary currents, we generated receptors with dual Cys substitutions at either the α-ε or the α-δ subunit interface, and compared ACh-induced currents before and after application of H2O2. After obtaining a gigaohm seal, each of the untreated Cys-substituted receptors exhibited predominant kinetic modes with high open probability and a minor mode with low open probability (Fig. 6, Table 3). After H2O2 application, all unitary currents exhibited low probability of opening, as observed when the Cys substitutions were present at both subunit interfaces. As controls, we compared single channel currents from receptors containing Cys substitutions of only αTyr 127, or only εAsn 39 and δAsn 41, before and after application of H2O2. For these mutant receptors, probability of channel opening was indistinguishable before and after H2O2 application (Table 3). Thus, oxidation diminishes probability of channel opening when the Cys substitutions are present at either or both subunit interfaces, but oxidation has no functional consequences when Cys is present in only one subunit of the interface.

Disulfide trapping of receptors with cysteine substitutions confined to either of the two subunit interfaces that form ligand binding sites. Cell-attached patches were established to cells expressing wild-type or mutant receptors, and single-channel currents evoked by 30 μm ACh were recorded at a membrane potential of −70 mV and displayed at a bandwidth of 10 kHz. Five minutes after adding H2O2, a second recording was obtained from the same patch. Dwell-time histograms corresponding to each receptor type and treatment condition are displayed, with the smooth curves generated by fitting the sum of exponentials to the dwell times. Addition of H2O2 suppresses channel gating of receptors with Cys substitutions at either the α-ε or α-δ subunit interfaces.
Discussion
We demonstrate profound contributions to channel gating by the conserved residues αTyr 127, εAsn 39, and δAsn 41, located at the two subunit interfaces that form the agonist binding sites in the human muscle AChR. These contributions satisfy several criteria expected of a structure that triggers channel gating in response to agonist binding. First, the Tyr–Asn pair is conserved across muscle AChRs, and the two residues are proximal in the structural model of the Torpedo receptor at a resolution of 4 Å. Second, the trans-subunit specificity of disulfide trapping demonstrates physical proximity in the human receptor. Third, each residue of the pair contributes approximately equally to channel gating, as shown by the similar gating equilibrium constants when Thr is substituted for αTyr 127 or Ala is substituted for εAsn 39 and δAsn 41. Fourth, the functional contribution of each residue of the pair depends on the other residue, as shown by strong inter-residue energetic coupling. Fifth, disulfide trapping of receptors with trans-subunit Cys substitutions causes a reversible suppression of channel gating, implying that the Tyr and Asn do not associate on rapid formation of a stable open state. Thus, the overall results support a physical association between αTyr 127, εAsn 39, and δAsn 41 that functions as a trigger for rapid and efficient channel gating.
The main functional contribution of the Tyr–Asn pair is to promote a rapid rate of channel opening, a quantity that depends on the free energy difference between the diliganded closed state and the transition state immediately preceding channel opening. Single-channel kinetic analyses show that this free energy barrier increases substantially when Thr is substituted for αTyr 127 or Ala is substituted for εAsn 39 and δAsn 41. This barrier could increase through stabilization of the diliganded closed state, destabilization of the transition state, or perturbation of both states. To determine the open- versus closed-state structure of the pair of residues in the transition state (Grosman et al., 2000), we examined rate-equilibrium free energy relationships (REFER). For the mutant receptors in Table 1, linear regression of a log–log plot of channel opening rate constant versus channel gating equilibrium constant reveals a linear relationship (r2 = 0.98) and a slope φ of 0.81, indicating that in the transition state, the Tyr–Asn pair adopts a predominantly open state structure. REFER analyses applied to multiple AChR structural domains reveal a gradient of φ values, ranging from unity at the agonist binding site to zero at to the bottom of the M2 domain. (Grosman and Auerbach, 2000; Cymes et al., 2002; Mitra et al., 2004), in good agreement with the value of 0.81 measured here for the lower portion of the extracellular domain.
Because in the transition state the Tyr–Asn pair achieves a predominantly open state configuration, and the pair dissociates on formation of a stable open state, we conclude that the Tyr–Asn interaction contributes little to the stability of the open or the transition state. Thus, in promoting rapid channel opening, the Tyr–Asn pair primarily contributes to the free energy of the diliganded closed state.
A seemingly simple explanation for the closed-state specificity of the Tyr–Asn pair is that the two residues associate in the closed receptor before agonist binding, but when the agonist binds, the pair dissociates rapidly, enabling the channel to open. However, if dissociation of the pair enabled rapid channel opening, substitution of Thr for αTyr 127 or Ala for εAsn 39 and δAsn 41 should weaken the trans-subunit complex and speed rather than slow channel opening, contrary to the observations. Alternatively, instead of associating before agonist binding, the Tyr and Asn may associate after formation of the diliganded closed state, possibly because of structural rearrangements propagated from αTrp149 at the center of the binding site via the disulfide linked αCys142/Cys128 that borders αTyr 127. Once associated, the Tyr–Asn pair may represent an activated trigger that creates strain within or between subunits, raising the free energy of the diliganded closed state. Once the trigger is activated, the Tyr–Asn pair should dissociate rapidly given a dwell time of ∼20 μs for the diliganded closed state, releasing strain that initiates a further chain of events that opens the channel. One subsequent event may be the rotation of the inner core of β-strands in the α-subunit observed in electron microscopic images of Torpedo receptors after pulse application of ACh (Unwin et al., 2002). Thus, the overall data suggest that the Tyr–Asn association occurs transiently after binding of agonist to the closed state.
The state dependence of the Tyr–Asn association suggests that the distance between αTyr 127 and εAsn 39/δAsn 41 changes during agonist-evoked channel opening, which might be expected to affect the ability to covalently link the subunits when Cys is substituted for the two residues. We find that channel opening is impaired to similar degrees when the oxidizing reagent is applied in the absence or the presence of agonist, implying that, although the inter-residue distance may change during channel opening, the change is not large enough to prevent covalent linkage of the subunits. An additional consideration is desensitization, which predominates during our agonist application and may allow alignment of the two residues. In any case, the aromatic–amino interaction expected of the Tyr–Asn pair should be highly sensitive to distance and orientation and not require large distance changes to form and dissociate.
The free energy of stabilization by the twin aromatic–amino interactions is not likely sufficient to account for an inter-residue coupling free energy approaching 6 kcal/mol detected by mutant-cycle analysis. By comparison, charge–charge pairs in the protein barnase (Schreiber and Fersht, 1995) and in the muscle AChR (Lee and Sine, 2005) show similar or lower coupling energies. Thus, the intersubunit coupling energy probably includes contributions from both the aromatic–amino interactions and global interactions from αY127, εN39, and δN41 that propagate across subunit interfaces not involved in agonist binding. Evidence for global coupling between the two triggering elements is the moderate coupling free energy observed for the mutant cycle comprising the mutations εN39A and δN41A (Fig. 3B).
Both the present and previous studies indicate that multiple structures couple agonist binding to channel gating, suggesting that the overall coupling process comprises a sequence of triggering steps. An initial triggering step is mediated by a triad of residues near the agonist binding site: αTyr 190 and αAsp 200 within the C-loop linking β-strands 9 and 10, and αLys 145 in β-strand 7 (Mukhtasimova et al., 2005). When an agonist binds, the C-loop undergoes a capping motion that encloses the agonist (Celie et al., 2004, Hansen et al., 2005; Gao et al., 2005), potentially causing rearrangement of β-strands 7, 9, and 10. This residue triad is conserved among all nicotinic receptors, but diverges in other members of the Cys-loop receptor family, indicating that the triad is structurally adapted for different neurotransmitters. Rearrangements initiated by agonist binding likely propagate to the intraprotein interface dividing the extracellular and pore domains, activating a second triggering step mediated by αArg 209 in β-strand 10, αGlu 45 in the linker spanning β-strands 1 and 2, and Pro 272 in the M2–M3 linker (Lee and Sine, 2005). This second triggering structure has been called the principal coupling pathway because of the high conservation of αArg 209 and αGlu 45, and the location suggests it is the immediate actuator of the pore-lining M2 domain that gates ion flow. Like the first two triggering structures, the third structure described here is contiguous with the binding site, with the α-carbon of αTyr 127 connected through a short chain of covalent bonds to αTrp 149 at the center of the binding site. However, this triggering structure is intersubunit rather than intrasubunit, and is conserved only in muscle nicotinic receptors, indicating it is specialized for the functional requirements of the motor endplate. Its significance lies in the potential for mediating global rearrangements required for rapid and efficient gating of the endplate receptor channel. Our findings raise the possibility that homologous intersubunit triggering structures are present in other receptors of the Cys-loop receptor family, and possibly additional triggering structures remain to be identified that contribute to the structural rearrangements that underlie channel gating.
Footnotes
-
This work was supported by National Institutes of Health Grant R37 NS31744 (S.M.S). We thank Chris Free for excellent technical contributions.
- Correspondence should be addressed to Steven M. Sine, Departments of Physiology and Biomedical Engineering and Neurology, Receptor Biology Laboratory, Mayo Clinic College of Medicine, Rochester, MN 55905. sine.steven{at}mayo.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}