

Abstract
Adenosine A2A receptor antagonists provide a promising nondopaminergic approach to the treatment of Parkinson's disease (PD). Initial clinical trials of A2A antagonists targeted PD patients who had already developed treatment complications known as l-3,4-dihydroxyphenylalanine (l-DOPA)-induced dyskinesia (LID) in an effort to improve symptoms while reducing existing LID. The goal of this study is to explore the effect of A2A antagonists and targeted A2A receptor depletion on the actual development of sensitized responses to l-DOPA in mouse models of LID in PD. Hemiparkinsonian mice (unilaterally lesioned with 6-OHDA) were treated daily for 3 weeks with a low dose of l-DOPA (2 mg/kg) preceded by a low dose of selective A2A antagonist (KW-6002 [(E)-1,3-diethyl-8-(3,4-dimethoxystyryl)-7-methyl-3,7-dihydro-1H-purine-2,6-dione] at 0.03 or 0.3 mg/kg, or SCH58261 [5-amino-7-(2-phenylethyl)-2-(2-furyl)-pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine] at 0.03 mg/kg) or vehicle intraperitoneally. In control mice, contralateral rotational responses to daily l-DOPA gradually increased over the initial week before reaching a persistent maximum. Both A2A antagonists inhibited the development of sensitized contralateral turning, with KW-6002 pretreatment reducing the sensitized rotational responses by up to threefold. The development of abnormal involuntary movements (a measure of LID) as well as rotational responses was attenuated by the postnatal depletion of forebrain A2A receptors in conditional (Cre/loxP system) knock-out mice. These pharmacological and genetic data provide evidence that striatal A2A receptors play an important role in the neuroplasticity underlying behavioral sensitization to l-DOPA, supporting consideration of early adjunctive therapy with an A2A antagonist to reduce the risk of LID in PD.
- 6-hydroxydopamine
- Parkinson's disease
- basal ganglia
- conditional A2A knock-out
- KW-6002
- A2A receptor antagonist
Introduction
l-3,4-Dihydroxyphenylalanine (l-DOPA) therapy is commonly prescribed to improve the motor symptoms of Parkinson's disease (PD). However, with prolonged use of l-DOPA (over years), patients often develop involuntary movements called l-DOPA-induced dyskinesia (LID) (including chorea, athetosis, ballism, and dystonia), which can be disruptive and for some disabling (Chase, 1998; Obeso et al., 2000). Recently, adenosine A2A receptors have been directly linked to l-DOPA-induced dyskinesia in humans and nonhuman primates (Zeng et al., 2000; Calon et al., 2004) and to models of LID in rodents (Fredduzzi et al., 2002; Tomiyama et al., 2004). CNS A2A receptors are predominantly expressed in the basal ganglia, particularly in the striatum (Schiffmann et al., 1991). Their unique distribution and colocalization with dopamine D2 receptors as well as the peptide enkephalin in the indirect pathway have implicated A2A receptors in basal ganglia function (Ferre et al., 1997; Kase, 2001; Fredholm et al., 2003) and presage their targeting as a nondopaminergic therapy for the treatment of dyskinesia in PD (Chen et al., 2003).
Assessment of the effect of chronically pairing l-DOPA with an A2A antagonist in a hemiparkinsonian rodent models of LID suggested that the early adjunctive use of an A2A antagonist might prevent or avoid the development of LID, at least indirectly (Pinna et al., 2001). This study indicated that the acute symptomatic (motor stimulant) effect of A2A antagonists may allow for chronic use of lower doses of l-DOPA, which are associated with a reduced risk of developing dyskinesia (Grandas et al., 1999; Fahn et al., 2004). (E)-1,3-Diethyl-8-(3,4-dimethoxystyryl)-7-methyl-3,7-dihydro-1H-purine-2,6-dione (KW-6002), a relatively specific A2A receptor antagonist, reduces parkinsonian symptoms without appreciably inducing or exacerbating l-DOPA-induced dyskinesia in nonhuman primates (Grondin et al., 1999; Kanda et al., 2000; Jenner, 2003, 2005; Kase et al., 2003; Xu et al., 2005). Based on these preclinical findings, initial clinical trials with A2A antagonists have targeted relatively advanced PD patients who have already developed LID (Bara-Jiminez et al., 2003; Hauser et al., 2003).
Our previous study in constitutive A2A knock-out (KO) mice (Fredduzzi et al., 2002) suggested that A2A receptors may also be directly involved in the development or maintenance of LID because the sensitization of l-DOPA-induced rotation and stereotyped behaviors did not develop or persist in mice lacking the A2A receptor. Here, we investigate the effects of low doses of different A2A antagonists coadministered with a low dose of l-DOPA on the development of sensitized rotational responses to repeated treatment in hemiparkinsonian mice. Using a conditional (Cre/loxP system) knock-out of postnatal forebrain A2A receptors, we also address the precise role played by CNS A2A receptors in two complementary models of LID in PD, the development of abnormal involuntary movements (AIMs) as well as rotational responses to repeated l-DOPA. Together, these experiments explore a molecular rationale for the possible prophylactic use of adjuvant A2A antagonists early in PD to reduce the risk of developing LID.
Materials and Methods
Forebrain-specific A2A conditional KO mice.
Adult conditional A2A KO mice (A2A cKO, n = 12; cre, A2A flox/flox , n = 14) were generated using a Cre/loxP system (in which the temporal, regional, and cellular pattern of A2A receptor depletion was specified by the CaMKIIα promoter-driven expression of cre transgene) and genotyped by PCR analysis of tail DNA as described previously (Bastia et al., 2005; Kachroo et al., 2005). Recombination of the A2A receptor gene was first detected at postnatal day 15 and was maximal by day 50 (Rapp et al., 2005). Analysis of A2A receptor transcripts by in situ hybridization at 12–27 weeks of age in a randomly selected subgroup of the cKO mice used in the present study revealed a significant 70 ± 2% reduction of striatal A2A mRNA expression in cKO mice (n = 6) compared with controls (A2A flox/flox ) (n = 5) (p < 0.05, Student's t test). The greater residual transcript (compared with protein) level possibly reflects dysfunctional mRNA, or glial expression with reduced protein production or stability. Protein levels assessed by Western blots were further reduced (95 ± 2% loss) (see Fig. 2 A, labeled as cre+; n = 8 for cKO; n = 7 for controls). A mouse monoclonal anti-A2AR antibody (catalog #05-717; Upstate, Lake Placid, NY) (raised against the third intracellular loop, which is encoded by exon 3) was used at a 1:500 dilution. Our initial characterization of these forebrain cKO mice demonstrated loss of detectable striatal A2A receptor in autoradiograms of coronal brain sections (Bastia et al., 2005).
6-OHDA lesion of striatum.
Male 3-month-old C57BL/6 mice [25–30 g for 5-amino-7-(2-phenylethyl)-2-(2-furyl)-pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine (SCH58261) and KW-6002 experiments] and 2- to 5-month-old conditional A2A KO mice and their nontransgenic floxed littermate controls (20–42 g) were maintained in home cages with a 12 h light/dark cycle. All experiments were performed in accordance with the Massachusetts General Hospital and National Institutes of Health guidelines on the ethical use of animals. On the day of surgery, mice were anesthetized with avertin-HCl (2% 2,2,2-tribromoethanol and 1% amyl alcohol; 15 ml/kg, i.p.). Seven to 10 μg of freshly prepared 6-OHDA bromide salt (5 μg/μl in 0.05% ascorbic acid and shielded from light) were delivered by a microinfusion pump (0.5 μl/min) into the left striatum at the following coordinates (from bregma: 0.5 mm anterior, 2 mm lateral, and 2.8 mm ventral) (Franklin and Paxinos, 1997). To minimize damage to noradrenergic neurons, the mice were pretreated with desipramine hydrochloride (25 mg/kg, i.p.).
Behavioral analysis.
Ten days after the 6-OHDA infusion, the mice were randomly divided into groups that received daily treatment with either l-DOPA (2 mg/kg, i.p.) plus vehicle [10% DMSO, 15% Alkamuls EL-620 (ethoxylated caster oil), and 75% water], l-DOPA plus SCH58261, a xanthine-based A2A antagonist (0.03 mg/kg, i.p.) (a gift from E. Ongini, Schering-Plough Research Institute, Milan, Italy) or l-DOPA plus KW-6002 (0.03 mg/kg and 0.3 mg/kg, i.p.) (a gift from Jacques Petzer and Neal Castagnoli, Jr, Virginia Tech, Blacksburg, VA) (Chen et al., 2001) for 3 weeks. SCH58261 was administered 10 or 20 min before l-DOPA; KW-6002 was injected 25 min before l-DOPA. Benserazide (2 mg/kg, i.p.) was given 20 min before l-DOPA in all animals. In the A2A cKO experiment, both control and cKO groups were given l-DOPA (2 mg/kg, i.p.), preceded by benserazide (2 mg/kg, i.p.). Both contralateral and ipsilateral turns were recorded immediately after l-DOPA administration every 10 min for 60 min total, using an automated rotometry system (San Diego Instruments, San Diego, CA). Each mouse was placed at the center of 1 of 12 opaque glass flat-bottom bowls (10 cm diameter base, 13 cm high wall, with opening diameter of 26 cm) and connected to the lower end of a 45 cm customized cable tether by a rubber band snugly fitted around the chest. The upper end of the cable is attached to a swivel box, which in turn is linked to a computer interface.
AIM testing was conducted on days alternating with the rotometry except on days 1, 5, and 9. Dyskinetic behaviors were assessed and scored by an observer blind to treatment based on each of the following AIM subscale: 1, forelimb dyskinesia (i.e., jerky movements or purposeless fluttering movement of the paws); 2, axial AIMs (i.e., twisted posture of the neck or the upper body toward the contralateral side); 3, orolingual dyskinesia (i.e., twitching of orofacial muscles, empty jaw movements, and tongue protrusion); 4, locomotive AIMs, tight circular locomotion toward the unlesioned side, as previously established and validated (Lundblad et al., 2004). The AIM subscales were evaluated together 15, 30, 45, and 60 min after l-DOPA injection, and each mouse was observed for 1 min.
Biochemical assessments.
Twenty-four hours after the last l-DOPA injection, mice were killed by rapid cervical dislocation, and their striata were dissected out, frozen on dry ice, and later assayed for dopamine (DA) and DOPAC by HPLC with electrochemical detection as described previously (Chen et al., 2001). In the A2A cKO experiment, mice of each genotype were divided into two groups (balanced for their extent of AIM increase and contralateral rotational sensitization). One group (n = 6 for A2A cKO; n = 5 for controls) was used for the quantitation of preproenkephalin (PPE) mRNA and A2A receptor mRNA by in situ hybridization histochemistry as described previously (Benn et al., 2004). The probe sequence (5′ to 3′) for PPE mRNA was as follows: ATC TGC ATC CTT CTT CAT GAA ACC GCC ATA CCT CTT GGC AAT GAT CTC (refer to Mus musculus preproenkephalin 1, Penk 1, BC049766). The other group's striata were cut in half while still frozen, with one-half used for catechol assay by HPLC, and the other one-half for Western blotting for the A2A receptor to confirm the striatal phenotype of the A2A cKO mice (n = 8 for A2A cKO; n = 7 for controls) (Fig. 2 A).
Statistical analysis.
All data are expressed as group average ± SEM. The significance between different treatments was evaluated using nonmatching two-way ANOVA followed by Bonferroni's test (Prism CX3.0; GraphPad, San Diego, CA). Student's t test was used for the remaining statistical analyses. A value of p < 0.05 was considered to be significant. Mice with <90% of dopamine loss (∼15% of the lesioned mice) in the lesioned striatum were excluded from all data analysis.
Results
Chronic pretreatment with selective A2A antagonists partially blocks l-DOPA-induced sensitization
To address whether pharmacological antagonism of the adenosine A2A receptor, like its constitutive depletion (Fredduzzi et al., 2002), attenuates l-DOPA-induced rotational sensitization, we tested the effect of daily coadministration of KW-6002 in this mouse model of LID. Unilaterally 6-OHDA-lesioned mice were treated daily with a low dose of l-DOPA (2 mg/kg, i.p.) plus vehicle or low doses of KW-6002 for 3 weeks. KW-6002, at a dose of 0.03 mg/kg, intraperitoneally (which is below its threshold for motor stimulation) (Bastia et al., 2005) partially blocked the rotational sensitization induced by daily injection of l-DOPA (p < 0.05, two-way ANOVA). Similarly, at a higher but still low dose of 0.3 mg/kg (just above its threshold for motor stimulation) (Bastia et al., 2005) KW-6002 attenuated the development of l-DOPA-induced sensitization (p < 0.05, drug effect, two-way ANOVA; p < 0.05 on days 14 and 20 compared with the vehicle treatment with Bonferroni's post hoc test) (Fig. 1 A,B). Mice treated with the either dose of KW-6002 did not differ significantly from the vehicle-treated animals during the first week of successive treatments. However, starting from day 9, the response to l-DOPA was gradually reduced to about one-third of the response of vehicle-treated animals. Note that the initial (day 1) response to l-DOPA plus 0.3 mg/kg KW-6002 entailed significantly more contralateral rotations than the initial response to l-DOPA plus vehicle (Fig. 1 B) (p < 0.05, Student's t test), consistent with previous findings of acute potentiation of l-DOPA effects by A2A antagonists (Pinna et al., 1996; Fenu et al., 1997). Although the sensitized behavioral response to l-DOPA was significantly attenuated by daily coadministration with KW-6002, by the last day of combined treatment with KW-6002 (at either 0.03 or 0.3 mg/kg) the response was still higher than on day 1 (p < 0.05 at 0.3 mg/kg; p = 0.05 at 0.03 mg/kg, Student's t test) (Fig. 1 B).

Effect of daily KW-6002 pretreatment on l-DOPA-induced behavioral sensitization in 6-OHDA-lesioned mice. Mice were daily treated with KW-6002 (0.03 and 0.3 mg/kg, i.p.) 25 min before l-DOPA (2 mg/kg, i.p.) for 3 weeks. Contralateral and ipsilateral turning was evaluated for 1 h immediately after the administration of l-DOPA. Data are expressed as mean ± SEM. A , Contralateral turns in mice treated with l-DOPA plus vehicle and l-DOPA plus KW-6002. p < 0.05, Drug effect comparing chronic daily KW-6002 treatment between vehicle plus l-DOPA versus KW-6002 plus l-DOPA groups, two-way ANOVA with Bonferroni's post test (*p < 0.05). **p < 0.05, Student's t test. B , Acute and chronic effect of KW-6002 on rotational responses to l-DOPA.
We also assessed the effect of another A2A antagonist SCH58261, which represents a structural class distinct from xanthine-based A2A antagonists like KW-6002. We used a low dose of SCH58261 (0.03 mg/kg, i.p.), which is subthreshold for motor stimulation on its own, for potentiating motor stimulation by amphetamine (Bastia et al., 2005) and for potentiating the rotational response to the initial low dose of l-DOPA administered 10 min later (Table 1, experiment A, day 1). Pairing daily l-DOPA with SCH58261 (0.03 mg/kg, i.p.; 10 min prior) partially attenuated the l-DOPA-induced contralateral turning (F (1,235) = 4.7; p = 0.031, two-way ANOVA) (Table 1, experiment A). To address the possibility that previous pretreatment with low-dose A2A antagonist may enhance its interaction with l-DOPA, the experiment was repeated with SCH58261 administered 20 rather than 10 min before each l-DOPA administration. Under these conditions, SCH58261 significantly potentiated the acute (day 1) rotational response to l-DOPA, suggesting that a doubling of the pretreatment time could in fact enhance the extent of interaction between SCH58261 and l-DOPA (Table 1, experiment B). Despite the acute potentiation, previous pretreatment with SCH58261 reduced l-DOPA-induced contralateral turning compared with controls with even greater statistical significance (F (1,349) = 5.7; p = 0.017, two-way ANOVA).
The effect of SCH58261 on the contralateral turning (turns/60 min) sensitized by chronic l-DOPA treatment in 6-OHDA-lesioned mice
Depletion of postnatal forebrain A2A receptors partially blocks the development of rotational sensitization and dyskinesia in hemiparkinsonian mice
We previously found that global genetic elimination of A2A receptors in A2A KO mice prevents sustained l-DOPA-induced rotational sensitization in hemiparkinsonian mice (Fredduzzi et al., 2002). This global KO approach, however, cannot distinguish between developmental and adult A2A receptor actions, or between its CNS and peripheral actions. We therefore investigated the effect on l-DOPA-induced behavioral sensitization of a recently developed conditional KO (cKO) mouse line (Bastia et al., 2005) in which Cre-mediated recombination driving forebrain A2A receptor depletion is not apparent until after postnatal day 5 (Dragatsis et al., 2000) and reaches a maximum at postnatal day 50 (Rapp et al., 2005).
Control mice gradually increased their contralateral rotational responses to daily l-DOPA administration, reaching a maximum response on day 9 that remained stable throughout the remaining days of treatment (Fig. 2 B). In contrast, within the first week of treatment, conditional A2A KO littermates displayed increased contralateral turning that failed to develop further during the following 2 weeks of treatment and indeed started to diminish toward basal levels (F (1,216) = 27.5; p < 0.0001, two-way ANOVA; p < 0.05 on day 11 compared with the control mice with Bonferroni's post hoc test). Interestingly, the contralateral response to l-DOPA on the last day of treatment is still greater than the initial response on day 1 (p < 0.05, Student's t test), a finding similar to that in the A2A antagonist-treated mice.
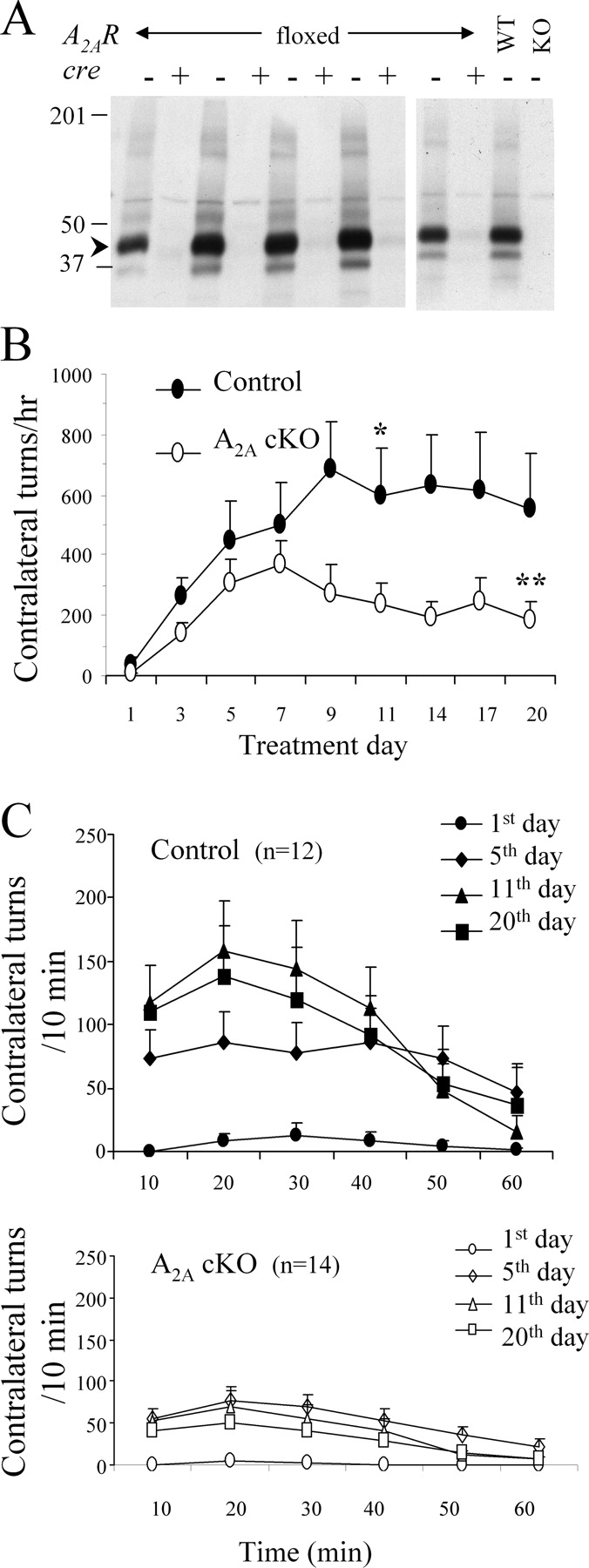
Deficiency of forebrain A2A receptor attenuates l-DOPA-induced behavioral sensitization. A , Western blot analysis of A2A receptor immunoreactivity was performed on striata from conditional A2A cKO (lanes labeled to indicate striata from the mice possessing a homozygous “floxed” A2AR gene alleles, and “+” for the presence of the cre transgene) and their littermate controls [lanes labeled for “floxed” A2AR mice without (“−”) the cre transgene]. Striata from a fully wild-type (WT) mouse and a global A2A KO mouse were run in parallel (in the indicated lanes) as a positive and negative control, respectively. The presence of a band at ∼45 kDa corresponding to the A2A receptor protein (arrowhead) in the controls, its marked reduction (by 95%) (see Materials and Methods) in cKO (cre+, A2ARflox/flox ) mice, and its absence in the global A2A KO confirms the near depletion of striatal A2A receptor protein in the cKO line. B , Conditional A2A KO mice and their littermate controls were treated with l-DOPA (2 mg/kg, i.p.) once a day for 3 weeks. Contralateral rotational behavior was recorded for 1 h immediately after the administration of l-DOPA. Two-way ANOVA showed no interaction between day of treatment and genotype but was significant for both genotype (F (1,216) = 27.5; p < 0.0001) and days of treatment (F (8,216) = 3.7; p < 0.05), in particular on day 11 by Bonferroni's post test (*p < 0.05). **p < 0.05 versus day 1 of same genotype (Student's t test). Data are expressed as mean ± SEM. C , Top, Time course for contralateral turning induced by l-DOPA in control mice over the 3 weeks of treatment. Bottom, Time course for contralateral turning induced by l-DOPA in conditional A2A KO mice over the 3 weeks of treatment.
l-DOPA-induced behavioral sensitization was associated with a progressively shortened onset and duration of action. As shown in Figure 2 C, top panel, the time to reach the maximum rotational response shortened in control mice (from 30 min on day 1, to 20 min on days 11 and 20). In A2A cKO mice, the peak response did not shift over the course of the experiment (staying at 20 min on all days of treatments) (Fig. 2 C, bottom panel), consistent with our findings in global A2A KO mice (Fredduzzi et al., 2002) and paralleling A2A antagonist effects in a rat model of l-DOPA-induced “wearing off” motor complications (Bibbiani et al., 2003).
Given the inherent limitations of rotational sensitization as a model of dyskinesia development, we sought to simultaneously compare for the development of dyskinetic movements per se in response to repeated l-DOPA in these cKO mice and their control littermates (Fig. 3). Once established, episodic dyskinesia as assessed through AIM scoring started 5–10 min after the low dose of l-DOPA that we used and reached a peak response at 15 min, and then gradually declined (data not shown). Consistent with previous findings (Lundblad et al., 2004), axial dyskinesia was found to be proportionally higher than limb or oroligual dyskinesia (Fig. 3, compare B, C).

Deficiency of forebrain A2A receptor attenuates the AIMs induced by l-DOPA. A , Total AIMs (axial, locomotive, limb, and orolingual) were recorded over 1 h after the daily l-DOPA injections in control (no cre, A2A flox/flox ) mice (closed circles) and conditional A2A KO (cre, A2A flox/flox ) mice (open circles). Data are expressed as mean ± SEM; two-way ANOVA followed by Bonferroni's analysis (F (1,192) = 62.99, p < 0.0001, genotype; F (7,192) = 9.034, p < 0.0001, day of treatment; *p < 0.05). B , Axial, limb and orolingual AIMs are shown (F (7,192) = 51.3; p < 0.0001). C , Nonrotational (limb plus orolingual AIMs) subscales are shown (F (7,167) = 53; p < 0.0001).
The AIM scores in A2A cKO and control mice over the 3 week period of the sensitization paradigm (Fig. 3) paralleled the above findings for rotational sensitization. Increasing total (composite) AIM responses in cKO and control mice initially developed in parallel (during the first week of l-DOPA treatment), but diverged thereafter as the response gradually diminished from day 9 in the cKO but not the control mice (F (1,192) = 63; p < 0.0001) (Fig. 3 A) (p < 0.05 on days 9, 16, 19, and 21 compared with the control mice with Bonferroni's post hoc test). This attenuation of sensitized AIMs in cKO mice was also observed when the total AIMs were split out into axial, limb, and orolingual dyskinesias, which share common anatomic substrates of LID in rodents (Lundblad et al., 2002, 2004) (F (7,192) = 51.3; p < 0.0001, two-way ANOVA) (Fig. 3 B) and when assessing only the two subscales devoid of rotational features (nonrotational dyskinesia): limb and orolingual dyskinesias (F (7,161) = 53; p < 0.0001, two-way ANOVA) (Fig. 3 C).
Striatal dopamine loss after 6-OHDA is unaffected by A2A receptor blockade or depletion
Degeneration of dopaminergic nigrostriatal neurons appears to be an important determinant if not a prerequisite for the development of LID (Jenner, 2000; Linazasoro, 2005). Because A2A receptors have been implicated in the pathophysiology of neuronal cell death in general and dopaminergic neurodegeneration in particular (Xu et al., 2005), it is important to consider whether the observed attenuation of l-DOPA-induced behavioral sensitization in 6-OHDA-lesioned mice might simply reflect an attenuation of dopaminergic denervation (or an enhancement of compensatory reinnervation). However, we found no evidence that chronic daily treatment with KW-6002 or SCH58261 (Table 2), or the depletion of forebrain A2A receptors altered the reduced (or control) levels of striatal dopamine measured ipsilateral (or contralateral) to intrastriatal 6-OHDA injection ∼1 month earlier (Table 3). These data indicate that A2A receptor inactivation likely attenuated the development of sensitization without altering the functional extent of the 6-OHDA lesion or any subsequent compensatory changes (e.g., sprouting), consistent with our previous findings of unaltered striatal dopamine transporter density as well as dopamine content in global A2A KO mice after 6-OHDA lesions (Fredduzzi et al., 2002).
Neurochemical measure of nigrostriatal innervation in C57BL/6 mice chronically treated with vehicle, SCH58261, or KW-6002 after a unilateral 6-OHDA lesion
Neurochemical measure of nigrostriatal innervation in wild-type and conditional A2A KO (cKO) mice chronically treated with l-DOPA after a unilateral 6-OHDA lesion
Preproenkephalin expression correlates with LID
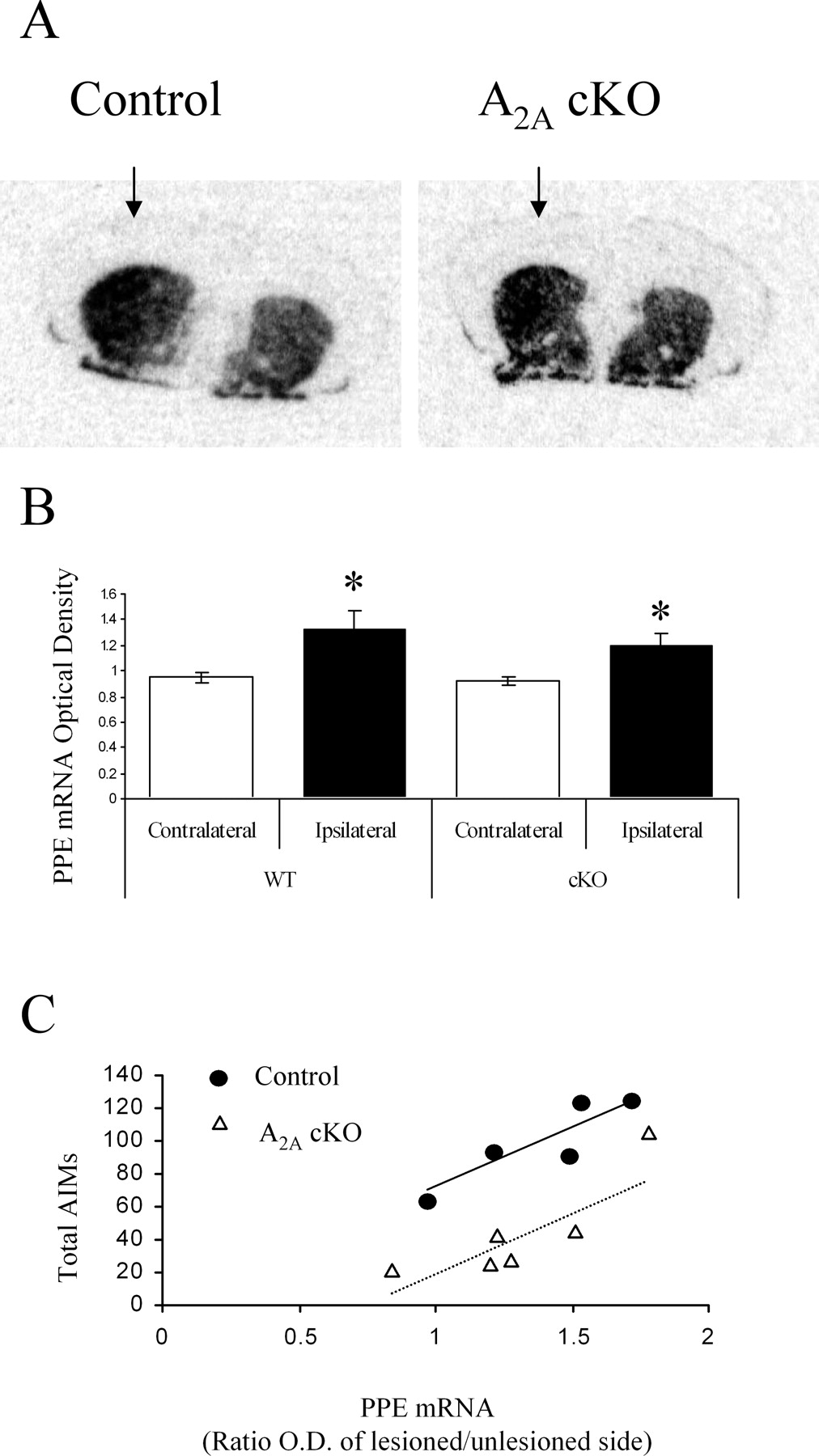
We explored whether alterations of PPE mRNA levels induced by repeated l-DOPA are also attenuated by the absence of postnatal forebrain A2A receptors. Consistent with previous findings (for review, see Xu et al., 2005), striatal PPE mRNA was increased ipsilateral to the 6-OHDA lesion compared with the unlesioned side after daily l-DOPA treatment in control mice (n = 5 for controls; p < 0.05, paired Student's t test) (Fig. 4 A,B). Their A2A cKO littermates also showed a significant elevation of ipsilateral PPE mRNA (n = 6) (Fig. 4 A,B). Pearson's correlation analysis showed that higher AIM scores significantly correlated with greater PPE elevation in both control and cKO mice (p < 0.05; r = 0.90 and 0.85, respectively) (Fig. 4 C).

mRNA expression of preproenkephalin in 6-OHDA-lesioned control and conditional A2A KO mice after chronic l-DOPA treatment. A , Representative autoradiograms of coronal brain sections at the level of the striatum in A2A cKO (right) versus control (left) mice. Arrows show the 6-OHDA-lesioned side. B , The level of striatal PPE mRNA ipsilateral to the lesion was significantly higher than on the contralateral side (*p < 0.05) in both control and A2A cKO mice (p > 0.05, comparing ipsilateral levels between genotypes; n = 5 for control and n = 6 for cKO mice). C , PPE mRNA was significantly correlated with total AIMs (the sum from days 1 to 21) in control (r = 0.90) and A2A cKO (r = 0.85) mice.
Discussion
We demonstrated that the adenosine A2A receptors expressed in postnatal forebrain neurons are required for the full development of l-DOPA-induced behavioral sensitization and dyskinesia in hemiparkinsonian mice. Complementary pharmacological and genetic data substantiate a role for A2A receptors in the adaptive behavioral responses to chronic l-DOPA treatment. In our previous studies, we showed that global elimination of the A2A receptors (in all cells from development through adulthood) attenuates l-DOPA-induced behavioral sensitization (Fredduzzi et al., 2002). However, this phenotype cannot distinguish between a role for the receptor in or out of the CNS and during development or in adulthood. Using a CaMKIIα promoter-driven conditional A2A KO approach (Dragatsis and Zeitlin, 2000; Bastia et al., 2005; Rapp et al., 2005), we could achieve both “spatial” (forebrain) and “temporal” (postnatal) selectivity of A2A receptor depletion. The present cKO findings argue strongly against compensatory prenatal or early postnatal developmental modifications and peripheral effects as the basis for the global A2A KO phenotype, and clarify that A2A receptors expressed in postnatal forebrain neurons likely underlie l-DOPA-induced neuroadaptations. Furthermore, a pharmacological (antagonist) approach inactivated the A2A receptor acutely and suggested that the attenuation effect was not attributable to the preceding absence of A2A receptor postnatally but attributable to the pairing of A2A antagonism with l-DOPA in adult mice.
Adenosine A2A receptor plays a critical role in l-DOPA-induced neuroplasticity
The development of l-DOPA-induced contralateral turning in hemiparkinsonian mice was attenuated by the daily coadministration of low doses of either KW-6002 or SCH58261, two structurally distinct A2A antagonists likely acting through acute inactivation of the A2A receptor. However, the sensitized rotational responses to l-DOPA were only partially reduced, and on the last day of treatment remained significantly greater than the initial response on day 1. Similarly, in the forebrain A2A cKO mice, l-DOPA-induced rotational sensitization was attenuated but only partially. The residual sensitization after either pharmacological blockade or genetic depletion of A2A receptors may reflect the involvement of other receptors and neurotransmitters in the sensitization to repeated dopaminergic stimulation (e.g., adenosine A1 receptors, glutamatergic receptors, and acetylcholine receptors) (Brotchie, 2005). Alternatively, the incomplete attenuation of sensitization in the present study may simply reflect incomplete inactivation of the A2A receptor. As noted above, both A2A antagonists were used at very low doses, and striata from the cKO mice showed evidence of residual A2A receptor expression (see Fig. 2 A). Considering our previous findings in global A2A KO mice (with complete elimination of A2A receptor) in which the persistent sensitization to l-DOPA is completely blocked (Fredduzzi et al., 2002), incomplete A2A receptor inactivation in the present study likely contributed to the residual sensitization observed in the A2A antagonist-treated and cKO mice.
A distinctive feature of the attenuation of sensitized behavioral responses to daily l-DOPA by A2A antagonists or by conditional or global A2A KO (Figs. 1 ⇑–3, Table 1) (Fredduzzi et al., 2002) was its manifestation generally only after sensitization began. The A2A receptor does not appear to be required for the initial induction by l-DOPA of sensitized rotational behaviors in 6-OHDA-lesioned hemiparkinsonian mice. Collectively, the findings point to a subsequent phase of rotational sensitization, such as its maintenance or expression, in which the A2A receptor plays a pivotal role. Interestingly, consistent with an A2A role in maintenance, Kanda et al. (2000) noted that previously induced LID in parkinsonian marmosets showed a gradual trend toward attenuation after subsequent initiation of KW-6002 coadministration with daily l-DOPA. If A2A receptors were indeed required to maintain rather than induce a dyskinetic state, A2A antagonists might be useful therapeutically even after dyskinesias have developed and chronic A2A receptor blockade might lead to a reduction in previously established LID.
However, when we analyzed a nonrotational measure of sensitization perhaps most relevant to LID, namely the nonrotational (orolingual and limb) subscores of the AIM scale (Fig. 3 C), forebrain A2A receptor depletion appeared to attenuate even the earliest manifestations of induction. Similarly, we previously noted that, although the global depletion of KO of A2A receptors did not prevent initial induction of rotational sensitization, it did completely prevent l-DOPA-induced excessive grooming, a more complex abnormal nonrotational behavior (Fredduzzi et al., 2002). Consistent with data from a nonhuman primate model of LID (Bibbiani et al., 2003), these preclinical rodent findings support a role for A2A receptors in the induction of dyskinesia.
In contrast, in the AIM-based rat model of LID developed by Cenci and colleagues, coadministration of KW-6002 (at antiparkinsonian doses substantially higher than those used here) did not attenuate LID. Although, their data (Lundblad et al., 2003) support the concept of early pairing of A2A antagonists with l-DOPA to indirectly reduce the risk of LID (Kanda et al., 1998; Pinna et al., 2001), they found no evidence for a direct role A2A receptors in LID (Lundblad et al., 2003). Similarly, Bové et al. (2002) found that pairing an A2A antagonist CSC [8-(3-chlorostryryl) caffeine] with l-DOPA in 6-OHDA-lesioned rats did not prevent (but could reverse) the characteristic shortening in duration of the rotational response to l-DOPA, a model of the development of problematic wearing off of l-DOPA benefit between doses in PD patients. In contrast, Chase and colleagues (Bibbiani et al., 2003) using the same model found that KW-6002 could prevent the shortening of rotational response. Thus, depending on the model of LID, its behavioral readout, dose of antagonist, and species used, preclinical studies have variably suggested that A2A antagonists may serve to avoid, prevent, mask, or reverse the development of LID. Nevertheless, these studies have highlighted the antidyskinetic therapeutic potential of A2A antagonists and the importance of investigating the potential mechanisms involved.
Potential mechanisms of facilitative A2A role in l-DOPA-induced dyskinesia
The neurochemical mechanisms by which basal ganglia A2A receptors facilitate dopaminergic behavioral sensitization are poorly understood. Presynaptic and postsynaptic enhancement of dopaminergic neurotransmission (attributable to greater dopamine release and greater responsiveness to dopamine, respectively) can produce this sensitization to repeated dopaminergic stimulation, regardless of whether by l-DOPA in a dopamine-deficient state or by psychostimulants in unlesioned animals. Although some evidence suggests that A2A receptors can facilitate CNS dopamine release (Okada et al., 1996; Dassesse et al., 2001), this mechanism would be indirect because A2A receptors are not appreciably expressed on dopaminergic neurons. Moreover, the fact that LID strengthens as the remaining number of presynaptic dopaminergic nerve terminals dwindles in PD makes an A2A-dependent enhancement of dopamine release seem a counterintuitive explanation for LID. In the present study, our demonstration that an A2A antagonist had no effect in dopamine-deficient mice on the initial ipsilateral turning response to l-DOPA (supplemental figure, available at www.jneurosci.org as supplemental material), an index of presynaptic dopamine release contralateral to the lesion (Weiss et al., 2003), also argues against A2A involvement based solely on presynaptic dopamine release.
Our finding that conditional forebrain A2A KO mice, lacking A2A receptors in striatal and cortical neurons, showed an attenuation of behavioral sensitization to l-DOPA supports a mechanism by which the A2A receptors on corticostriatal or striatopallidal neurons might contribute to LID. Although corticostriatal neurons express relatively low levels of A2A receptors, these appear be functionally active in facilitating glutamate release into the striatum (Quarta et al., 2004; Rodrigues et al., 2005; Ciruela et al., 2006). Thus, A2A antagonists might attenuate the development of LID by acting directly on corticostriatal neurons to reduce their facilitative glutamatergic role in LID.
Alternatively and also consistent with our cKO findings, A2A antagonism may prevent the full development of LID by acting directly on the A2A receptors expressed at high levels on striatopallidal neurons. These GABAergic output neurons of the indirect pathway also express the D2 dopamine receptor and the neuropeptide enkephalin, and increased expression of the latter has been linked to the development of LID (Bedard et al., 1999; Calon et al., 2002; Winkler et al., 2002; Morissette et al., 2006). Elevated striatal enkephalin expression may reflect excessive activity of striatopallidal neurons attributable to the loss of dopamine and attendant D2 receptor-mediated inhibition of striatopallidal neurons in a parkinsonian state. The increased enkephalin expression may also reflect a pathological imbalance between a persistently overactive indirect pathway and a direct pathway whose hypoactivity has been selectively normalized by l-DOPA treatment (for review, see Brotchie, 2005). In either case, the ability of A2A antagonists to reduce the overactivity of the striatopallidal neuron could account for their attenuation of l-DOPA-induced sensitization. That PPE mRNA was not significantly reduced in cKO might reflect a lack of PPE involvement in the A2A receptor dependence of AIM development in mice. Alternatively, the small sample size available for PPE determinations in this study may have precluded identification of a role for PPE expression.
Translational significance for Parkinson's disease
A2A antagonists are currently being investigated in human clinical trials for advanced PD patients who have developed motor complications of standard dopaminergic therapies. Our findings strengthen the contention that introducing A2A receptor antagonism earlier in the disease on initiation of traditional l-DOPA treatment may attenuate the ensuing basal ganglia plasticity that underlies LID. They encourage consideration of future A2A antagonist trials in PD that are aimed at reducing the development rather than the expression of dyskinesia.
Footnotes
-
This work was supported by the Michael J. Fox Foundation, National Institutes of Health Grants DA013508, NS054978 (M.A.S.), NS41083 (J.-F.C.), NS045242, and NS038106, and the Glendorn Foundation (J.-H.J.C.). We thank Allison Curley, Angela Scibelli, and Robert Caiazzo Jr for excellent technical assistance, and Lianna Orlando and Angela Cenci for helpful discussion.
- Correspondence should be addressed to Michael A. Schwarzschild, Department of Neurology, Massachusetts General Hospital, 114 16th Street, Charlestown, MA 02129. MichaelS{at}helix.mgh.harvard.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}